
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceConversion of methanol on rutile TiO2 (110) and tungsten oxide clusters: 1. population of defect-dependent thermal reaction pathways†
Lars
Mohrhusen
 * and
Katharina
Al-Shamery
* and
Katharina
Al-Shamery
Institute of Chemistry, Carl von Ossietzky University of Oldenburg, Carl-von-Ossietzky Strasse 9-11, D-26129 Oldenburg, Germany. E-mail: lars.mohrhusen@uol.de
First published on 17th May 2021
Abstract
Tungsten oxide clusters deposited on rutile TiO2 (110) single crystals were used as a model system for heterogenous oxide-oxide bifunctional catalysts. The population of different thermal reaction routes in methanol conversion in the presence of preadsorbed oxygen was probed under UHV conditions. By temperature programmed reaction spectroscopy, we have identified three thermal reaction channels, namely the deoxygenation under formation of methane, the partial oxidation forming formaldehyde and the condensation route under desorption of ethane and dimethyl ether. The specific local reaction environment at the oxidic surface was found to be key for the population of the different reaction channels as exhibited by the introduction of Lewis acidic and basic sites (especially (WO3)n clusters) and available charge carriers such as Ti3+. Especially the amount of bulk Ti3+ interstitials, that can partially transfer charge towards the tungsten oxide clusters at the TiO2 surface, was found to be a key parameter that enables a relatively high methanol conversion in thermal reactions. It turned out that the deoxygenation is by far the most dominant reaction followed by the partial oxidation. The condensation is observed only in low amounts under special conditions, but is an interesting example for reactivity at defect sites.
1. Introduction
Transition metal oxides, often as mixed compounds, are widely used as non-toxic and well accessible heterogeneous (photo-) catalysts. However, there is little understanding of underlying processes at bifunctional oxide–oxide catalysts. In order to understand key parameters ruling the catalytic activity of such systems, UHV studies on tungsten oxide clusters (WO3)n deposited on rutile TiO2 (110) surfaces are presented in this work as a model study. Supported (WO3)n clusters appear as relevant candidates for thermal and photochemical reactions due to their relatively high Lewis acidity at the W6+ centers, combined with the presence of strong Brønsted bases in form of the terminal W![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) Ot groups.1,2 Such clusters have been subject to experimental and theoretical studies of dehydration, dehydrogenation and condensation reactions of alcohols,3–6 partial oxidations,2 aldehyde and ketone polymerization7–9 as well as the decomposition of toxic compounds such as organophosphates.10
Ot groups.1,2 Such clusters have been subject to experimental and theoretical studies of dehydration, dehydrogenation and condensation reactions of alcohols,3–6 partial oxidations,2 aldehyde and ketone polymerization7–9 as well as the decomposition of toxic compounds such as organophosphates.10
To study the conversion of alcohols on such systems, we follow the reaction of methanol as a test molecule. Methanol is an important compound in the chemical industry and therefore an interesting model reactant for catalytic conversion.11,12 The chemistry of methanol and other alcohols on stoichiometric rutile TiO2 (110) was already in the focus of several studies, especially by the groups of Henderson and Dohnálek.13–23 In addition, from reports of the group of Madix and our own work it is known that the chemistry of methanol is sensitive to the presence of defects, such as oxygen vacancies or Ti3+ interstitials.24–26 Therefore, methanol is a perfect probe molecule for testing the conversion in different reaction pathways involving reduced transition metal oxide centers. In detail, methanol is hardly converted to any products on the defect free stoichiometric rutile TiO2 (110) surface by simple thermal reactions. In contrast, at least two additional thermal reaction pathways will be populated, if bulk and surface defects are present: the deoxygenation pathway forming mainly methane and, in the presence of oxygen, the partial oxidation to formaldehyde. In consensus with earlier studies,22,24,25,27 we have recently shown in a temperature programmed reaction spectroscopy (TPRS) and Fourier-transformation infrared reflection–absorption spectroscopy (FT-IRRAS) based study, that the methane formation proceeds via a methoxy intermediate, while formaldehyde desorbs from a dioxomethylene-like structure at the surface.26
While the chemistry of alcohols on pristine TiO2 is in the focus of a number of studies, alcohol conversion at tungsten oxide clusters on top of rutile TiO2 surfaces has not been studied so intensively in comparison. However, several parameters relevant for alcohol conversion at tungsten oxide clusters on rutile TiO2 (110) have been investigated, predominantly by the group of Dohnálek. This includes effects by the aliphatic chain,4,28 binding to supports and cluster deactivation thereby,5,6,29 as well as the impact of altered Lewis acidic sites on activation barriers.3 Nevertheless, in case of rutile TiO2, point defects such as Ti3+ interstitials drastically influence chemical reactions, such as O2 activation,30–32 C–C coupling33–35 or alcohol (photo-) conversion.24–26,36,37 Therefore it is surprising, that the role of defects in oxide-oxide systems involving tungsten oxide at TiO2 has not been addressed before. That is one aim of this work.
Recently, we demonstrated that Ti3+ point defects can induce a temperature dependent charge transfer between (WO3)n clusters and an underlying rutile TiO2 (110) substrate.38 As surface charges are known to have a substantial impact on the chemical reactivity we present a systematic study of thermal methanol conversion with oxygen as a function of the Ti3+ content as well as the (WO3)n cluster coverage as a model system to understand the conversion of alcohols on defect rich oxide-oxide systems.
2. Experimental
All results were obtained using a home-built UHV chamber (base pressure below 10−10 mbar) equipped with a commercial low energy electron diffraction (LEED) setup (OCI Vacuum Microengineering, BDL800IR-LMX-ISIJ), a quadrupole mass spectrometer equipped with a Feulner-cup for temperature programmed reaction spectroscopy (TPRS) (Pfeiffer Vacuum Prisma TM QMA 200) and an argon ion source. Moreover, the system allows adsorption of high purity compounds via a directional pinhole doser or chamber backfilling via a leak valve. In this work, oxygen (Air liquide, 99.999%), argon (Air liquide, 99.999%) and methanol (Fisher Scientific, 99.99% (HPLC grade)) were used. Methanol was cleaned before use by at least five freeze–pump–thaw cycles. The deposition of tungsten oxide clusters from WO3 powder (Sigma Aldrich, purity 99.995%) was done at room temperature in an individual evaporation chamber (allowing in vacuo sample transfer at a base pressure below 10−9 mbar) using a Focus EFM3 electron beam evaporator equipped with a molybdenum crucible with an alumina inset with identical devices and conditions as described elsewhere.38 Since the sample preparation was identically conducted using the same conditions, devices, parameters and evaporating materials, the detailed surface characterization with respect to thermal and coverage effects involving charge transfer with Ti3+ point defects may be directly transferred to the experiments within this work. For clarity, we present a collection of central aspects in the ESI† (Fig. S1 and S2). After deposition and before use in TPRS experiments, the clusters were initially heated to 880 K (2 K s−1, keeping 880 K for 1 minute) to remove possible residual adsorbates from the cluster evaporation process.We have used a home-built sample holder,39 which can be transferred in vacuo to a liquid nitrogen cooled stage, allowing experiments between 110 K and 900 K. The sample temperature was measured via a K-type thermocouple (CHAL-005, Omega Engineering, approx. error 0.75%) glued inside a tiny hole at the side of the sample (Ceramabond 569, Aremco). For all experiments, rutile TiO2 (110) single crystals (10 × 10 × 1 mm3, Surface Net GmbH) were used, which were initially cleaned by sonification in isopropanol before use.
Clean (110)(1 × 1) reconstructed surfaces were produced by repeated cycles of argon ion bombardment (20 minutes at 300 K, 1 keV, 2 μA cm−2 at 5 × 10−5 mbar argon background pressure) and subsequent annealing at 880 K for 15 minutes in UHV. This procedure leads to a partial reduction of the titania sample. Hence, this will be called one “reduction cycle” in the following. One should note, that a certain amount of argon (typically <0.9 at% based on X-ray photoelectron spectroscopy (XPS)) is present in titania surfaces from this very common sample preparation protocol.40 The surface quality was frequently checked by LEED. All TPR spectra were recorded using a temperature ramp of 2 K s−1. Based on earlier recorded XPS data38 and comparison of different m/z = 18 TPRS traces, we exclude a relevant impact of surface hydroxylation prior to adsorption of methanol by water adsorption from the chamber background (ESI,† Fig S3).
3. Results
In the following, we present the conversion of methanol with oxygen preadsorption (75 L O2 at 110 K) in thermal reactions. All experiments were performed as a function of the tungsten oxide cluster coverage as well as the amount of bulk Ti3+ interstitials. To clearly differentiate between the TiO2 surfaces with and without tungsten oxide clusters, we shall refer to the “pristine TiO2” surface in the following for absence of tungsten oxide, even if (sub-)surface defects of course are already a variation compared to stoichiometric TiO2.In order to produce mixed oxide systems, tungsten oxide clusters can be prepared by evaporation of bulk WO3 for example from effusion cells at temperatures around 1120–1300 K. By this way, (WO3)n clusters with n = 1, 2, 3,… are formed, with n = 3 being the dominant compound.41–45 In our case, an electron beam evaporator is used to deposit such clusters onto the rutile (110) surface. We have characterized the surface of such a model system for a bifunctional oxidic catalyst by means of mainly XPS but also LEED and probe molecule temperature-programmed desorption (TPD) spectroscopy in a recently published report.38 Here, we use the identical devices under the same conditions with comparable materials as this paper is the second in a series of three consecutive papers.37,38 Therefore, in this case, the surface characterization may be directly transferred from our previously published results. Since we do not want to reproduce all details here, we will give a brief overview and refer to central aspects in the original publication38 and the ESI† (Fig. S1 and S2):
1. Oxidation state of the produced clusters: stoichiometric (WO3)n clusters are the dominant compound in the oxide vapor, but in addition substoichiometric, oxygen deficient (WO3−x)n clusters with 0 < x < 1 are produced. By electron transfer from these clusters towards the TiO2 substrate, the first layer of clusters becomes reoxidized under formation of Ti3+ sites in the near subsurface. Hence, coverages up to 7.1 WO3 nm−2 lead to a layer of stoichiometric (WO3)n clusters in direct interaction with the surface possibly via Ti-O–W like features. On TiO2, substoichiometric (WO3−x)n clusters were only observed for coverages above 7.1 WO3 nm−2.
2. Accessibility of Ti5c sites: the amount of accessible five-fold coordinated Ti5c sites is decreased with increasing cluster coverage, but a significant part is still available at higher coverage based on water and CO TPD. Thus, the clusters appear as an unordered, open layer growing in a three-dimensional motif.
3. Equilibrium-like Ti3+ content: the Ti3+ content reaches an equilibrium around 6–7% Ti3+/Ti4+ based on Ti2p XPS for deposition of a monolayer of clusters (up to 7.1 WO3 nm−2) roughly independent of the initial Ti3+ content. Thus, Ti3+ formed by electron transfer from substoichiometric clusters stays in the near subsurface in case of low bulk Ti3+ density, while such states partially disappear into the bulk likely by diffusion in case of high Ti3+ bulk content.
4. High-temperature accumulation of Ti3+ and electron transfer towards the clusters: in temperature dependent experiments using X-ray photoelectron spectroscopy (XPS), the clusters appeared very stable on TiO2 up to 900 K. However, for titania samples with a large amount of bulk Ti3+, an accumulation of Ti3+ interstitials near the surface was observed between 500 and 800 K. By the appearance of a new W4f species in this temperature range, we have suggested the possible formation of an anionic [(WO3)n]z−-like species by electron transfer from Ti3+ in these cases. This is also in line with several predictions from DFT-based studies.1,8,46,47
3.1 Defect-dependent thermal conversion of methanol
At this point, the thermal chemistry of methanol at a mixed oxide-oxide catalyst consisting of tungsten oxide clusters at rutile TiO2 shall be presented. To examine the impact of defects, all experiments were carried out using TiO2 substrates with a small (light blue, 10–15 reduction cycles, Ti3+/Ti4+ ratio <4.2%) and large Ti3+ content (dark blue, >80 reduction cycles, Ti3+/Ti4+ ratio >7.5%). While we have not measured XPS here, we calculated the Ti3+/Ti4+ ratio from Ti2p spectrum deconvolution of samples that were analogously prepared with a similar number of reduction cycles under the same conditions in our group.38The TPR spectra obtained after preadsorption of 75 L O2 and a saturation coverage of methanol at 110 K are shown in Fig. 1, left for the pristine rutile TiO2 (110) surface with a small amount of bulk Ti3+, right for a cluster coverage of 3.5 WO3 nm−2 on top.
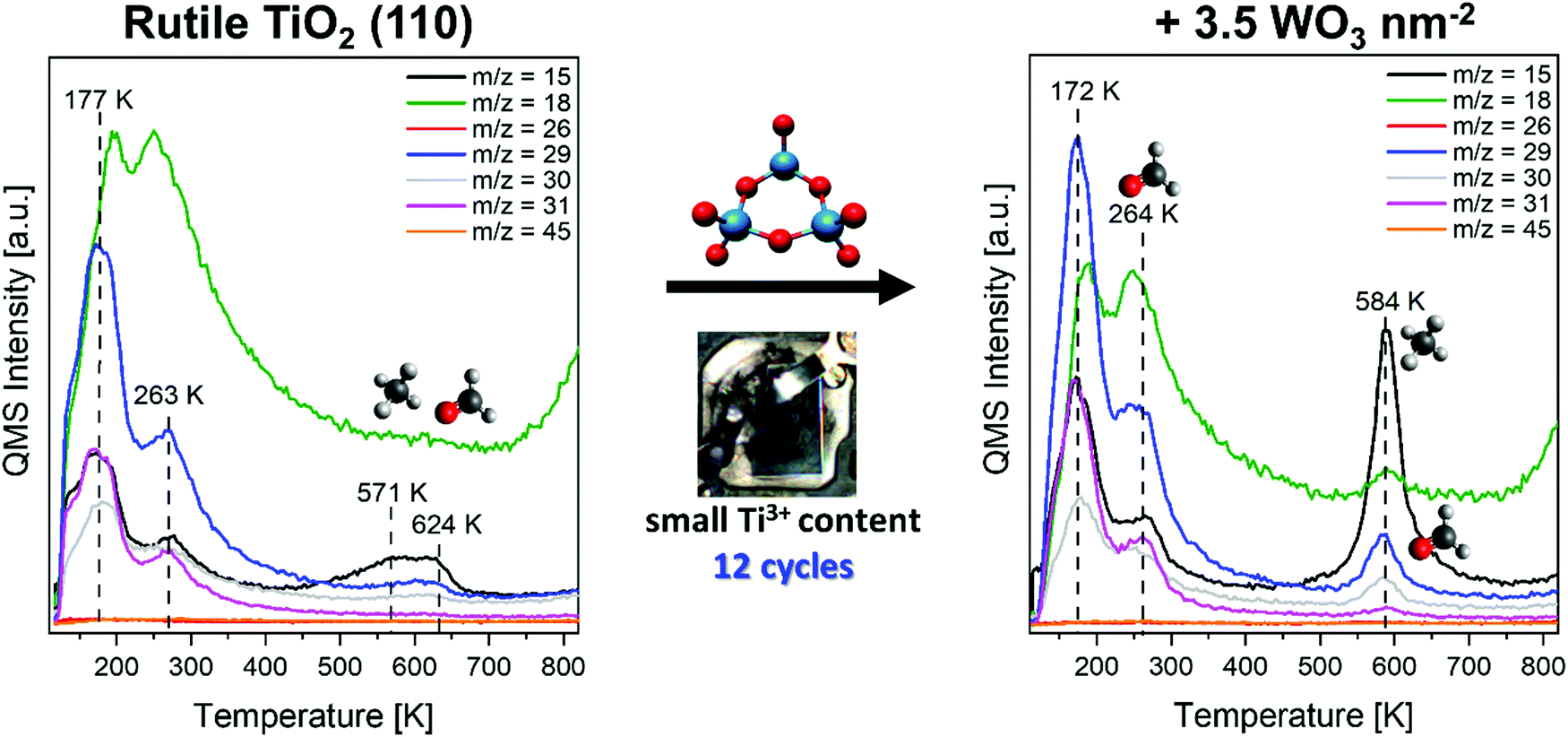 | ||
| Fig. 1 Temperature programmed reaction spectra (TPRS) of a saturation coverage of methanol with preadsorption of 75 L O2 at 110 K. Left, obtained from the pristine rutile TiO2 (110) surface. Right, obtained from the same surface after deposition of 3.5 WO3 nm−2 flashed to 880 K before adsorption of methanol and oxygen at 110 K. A rutile sample with a small amount of Ti3+ was used. | ||
For the pristine titania surface, four relevant desorption features can be observed at 144 K, 177 K, 263 K and in the range of 571–624 K. In accordance with earlier publications, we attribute all three low temperature features (144 K, 177 K and 263 K) to the molecular desorption of unconverted methanol. With respect to the fragmentation pattern48 (Fig. S4 in the ESI†), we consequently observe signals at these temperatures in m/z = 15, 29, 30 and 31, but not in 26 or 45 (Table 1).
| m/z | Major contributor | Fragments | Minor contributor | Fragments |
|---|---|---|---|---|
| a H2O is not a direct fragment of methanol, but can be detected in traces likely arising from recombination in the mass spectrometer. | ||||
| 15 | Methane | CH3, (13CH2) | Ethane | CH3, (13CH2) |
| Methanol | CH3, (13CH2) | Formaldehyde | 13CH2 | |
| Dimethyl ether | CH3, (13CH2) | |||
| 18 | Water | H2O | Methanol | H2Oa |
| 26 | Ethene | HCCH, H2CC | — | — |
| Ethane | HCCH, H2CC | |||
| 29 | Methanol | HCO | Ethene | H213CCH2 |
| Formaldehyde | HCO | |||
| Ethane | H3CCH2, (H213CCH2) | |||
| Dimethyl ether | HCO | |||
| 30 | Formaldehyde | H2CO | Dimethyl ether | H2CO |
| Methanol | H2CO, HCOH | |||
| Ethane | H3CCH3 | |||
| 31 | Methanol | H3CO, (H2COH) | Dimethyl ether | H3CO |
| 45 | Dimethyl ether | H2COCH3 | — | — |
Especially the low temperature desorption signals are already well discussed in the literature,16,24,26 so we can easily assign them to methanol desorption from Ti5c centers peaking at 263 K, methanol desorption from bridging oxygen atoms adsorbed via hydrogen bonds at 177 K and weak multilayer desorption at 144 K. One should note, that recombination of dissociated methanol (methoxy fragments) with the corresponding surface hydroxyl groups also contributes to the desorption of methanol at 263 K. The coverage shown here is the lowest coverage, that exhibits a saturated species at 177 K and hence first little shoulders for the multilayer desorption. In this situation, all adsorption sites at the surface (Ti5c and Obr sites) are covered by methanol. Despite the fact, that traces of a methanol multilayer may be present, we shall call this a saturation coverage of methanol in the following. Thus, we will not further discuss this little shoulder, as it is assigned to methanol multilayer desorption at similar temperatures (141–145 K) in all cases of appearance. While in the low temperature regime no significant methanol conversion can be observed, desorption of methane and formaldehyde is detected in small amounts in two overlapping features at 571 K and 624 K (m/z = 15 and 29, 30 respectively, while signals in m/z = 31 are absent). This is in accordance with results from earlier reports.26
Since the desorption of water is the most important way to remove hydrogen from the surface yielding the reactive methoxy-species, we further emphasize the m/z = 18 trace. This turned out to be a superposition of different contributions. First, methanol itself can contribute to the m/z = 18 fragment according to the fragmentation pattern (Table 1 and Fig. S4 in the ESI†). More important, water can be formed from surface hydroxyls arising from OH dissociation under formation of methoxy fragments. This water desorbs in well-known desorption features around 180 K (water hydrogen bonded to Obr) and 265 K (desorption from Ti5c sites). Accordingly, the desorption features around 175 K (arising from methanol) are shifted in comparison to the m/z = 18 trace (water). In addition, water is released during the formation of formaldehyde due to the necessary C–H dissociation (for example around 585 K after (WO3)n cluster deposition). Finally, at the high-temperature end of our experiments, frequently also water desorption from parts of our equipment due to partial heatup may occur (e.g.Fig. 1 above 750 K). It is noteworthy that surface hydroxylation from water adsorption from the chamber background does not appear as a relevant issue based on earlier XPS experiments38 and comparison of different m/z = 18 TPRS traces (ESI,† Fig. S3).
After the deposition of 3.5 WO3 nm−2 at this low reduced titania surface, we still observe the desorption of unreacted methanol from Ti5c at 264 K as well as from bridging oxygen atoms at 172 K. The desorption temperatures appear roughly unchanged for both features. However, methanol desorption from the bridging oxygen atoms of the tungsten oxide clusters may appear at a similar temperature as for bridging oxygens at titania. But, compared to pristine TiO2, more methanol is converted to other products, especially for the high-temperature reactions: here, the two features observed on pristine TiO2 are replaced by a more intense formation of methane (m/z = 15, dominant) and formaldehyde (m/z = 29 and 30, less than methane) exhibiting a relatively narrow desorption feature at 584 K. One should note, that this is significantly shifted to lower temperatures especially compared to the second high temperature desorption feature peaking at 624 K on pure TiO2. A very weak signal for m/z = 31 at this temperature indicates, that a little fraction of methanol also desorbs during this reaction.
In brief, the TiO2 substrate appears more reactive after cluster deposition, similar to a higher reduced one. This suits well with our earlier finding, that Ti3+ sites are generated by deposition of tungsten oxide and subsequent charge transfer from substoichiometric tungsten oxide clusters towards the TiO2 crystal.38 We will discuss this at a later point in more detail.
The titania surface with a high defect density appears much more reactive. In Fig. 2, left side, TPR spectra are shown recorded after adsorption of oxygen followed by a saturation coverage of methanol on a TiO2 substrate with a high Ti3+ density. Additional to the typical desorption features of unreacted methanol from bridging oxygens (177 K) and Ti5c sites (here 295 K), a significant amount of formaldehyde is formed mainly in a low-temperature reaction peaking at 263 K (no signal at m/z = 15 and 31) as well as to some extend in a high-temperature reaction around 640 K (signals at m/z = 30 and 29, but not at 31). However, the dominant product is methane formed in a deoxygenation reaction at the same temperature (m/z = 15). This is in agreement with a recent publication of our group demonstrating the defect dependent formation of methane from a methoxy-intermediate, while a dioxomethylene-like species was suggested for the formaldehyde formation.26 In contrast, here the desorption of methanol from Ti5c is somewhat shifted in temperature from 263 K to 295 K (+32 K). However, as the position of the onset of multilayer desorption (144 K) as well as desorption from bridging oxygens (177 K) is almost the same for low and highly reduced TiO2, we exclude an error of the thermocouple readout and suggest, that due to the high defect density, the desorption energy for methanol from Ti5c may be increased in this case.
 | ||
| Fig. 2 Temperature programmed reaction spectra (TPRS) of a saturation coverage of methanol with preadsorption of 75 L O2 at 110 K. Left, obtained from the pristine rutile TiO2 (110) surface and right obtained from the same surface after deposition of 3.5 WO3 nm−2 flashed to 880 K before adsorption of methanol and oxygen at 110 K. A rutile sample with a large concentration of defects was used. | ||
After deposition of 3.5 WO3 nm−2 on this surface, the TPR spectra still exhibit the three desorption features of unreacted methanol as described above. It is noteworthy, that the desorption of methanol from Ti5c sites is shifted back to 268 K, which is close to the value for low reduced TiO2. Again, the presence of tungsten oxide clusters has a bigger impact on the high-temperature (HT) reaction. Here, still methane is the dominant product (m/z = 15), but the desorption peak is narrowed (FWHM: −24 K), while the peak temperature is shifted significantly to 612 K (−28 K compared to highly reduced TiO2). Besides, an enhanced formation of formaldehyde at 639 K (m/z = 29 and 30, not 31) was detected, again accompanied by the desorption of traces of methanol (m/z = 31) while the low temperature formation of formaldehyde is slightly reduced.
Remarkably, this relatively rich chemistry was exclusively observed in case of oxygen preadsorption (here, 75 L O2 at 110 K). For comparison, the corresponding TPRS spectra in absence of additional oxygen are given in the ESI,† Fig. S5 and S6. There, the overall methanol conversion appears much less. Similar molecular desorption features (around 180 K and 265–300 K) were observed in absence of additional oxygen, but no partial oxidation or condensation products were recorded. The formation of methane in a deoxygenation reaction is observed, but drastically diminished in absence of oxygen which is in accordance with our earlier results.26 Although a certain CO2 formation is detected throughout our experiments especially for tungsten oxide on reduced TiO2, total oxidation does not appear dominant and is therefore neglected. The TPRS spectra including m/z = 28 and 44 are given in the ESI,† Fig. S7 and S8.
4. Analysis and discussion
4.1 Trends in product distributions: population of deoxygenation, partial oxidation and condensation routes
In the following, we shall analyze and discuss the trends for three different reaction pathways (deoxygenation, partial oxidation and condensation) of methanol in coadsorption with oxygen in more detail. To be correct, we want to point out that a large amount of methanol desorbs unconverted in our experiments. Anyway, significant amounts of products were observed, predominantly methane from the deoxygenation reaction. Deposition of 3.5–7.0 WO3 nm−2 on highly reduced TiO2 leads to the most reactive surfaces shown here (ESI:† Tables S1 and S2). This coverage is in line with other publications reporting the highest catalytic activity for coverages around 7 W nm−2.49 Interestingly, cluster coverages significantly higher than the monolayer coverage of 7.1 WO3 nm−2 do not show a relevant catalytic activity which is indicative that the direct interaction of tungsten oxide clusters and the TiO2 surface is crucial in this case. The product distribution is dependent on the detailed environment of precursor species such as the methoxy fragment. Especially Ti3+ sites as well as Lewis acid or Brøndsted base sites can significantly affect the adsorbates and steer the product distribution. In this work, all reaction pathways required the presence of oxygen (partial oxidation and condensation) or were at least significantly enhanced (deoxygenation) by oxygen preadsorption (see for comparison Fig. 1, 2 and Fig. S5, S6 in the ESI†). One reason for this behavior may be the boosted OH dissociation forming methoxy species, which can be confirmed in this work based on the water desorption from the surface. This is in line with earlier reports based on FT-IRRAS or TPRS experiments consenting that the methoxy-fragments appears much more reactive than molecular methanol.13,24,26 Since no particular desorption feature for molecular oxygen (m/z = 16, 32) can be identified in our TPR spectra, we conclude, that oxygen dissociates at the surface. Furthermore, we cannot rule out that oxygen also reacts with the tungsten oxide clusters, especially for the case of highly reduced TiO2 in which the clusters may exhibit an enriched electron density.38,47 | ||
| Fig. 3 Formation of methane at elevated temperatures as a function of cluster coverage and Ti3+ content. (a) Methane formation as observed in the m/z = 15 trace obtained from TPRS experiments. Note that the traces for small Ti3+ contents (left) are differently scaled for better visibility (by a factor of two in comparison to the traces for large Ti3+ content). (b) Integral peak area obtained from a. Blue columns correspond to small Ti3+ content, red columns to large Ti3+ contents. More details are given in the ESI,† Table S1. | ||
Fig. 3 gives an overview on the methane desorption. For TiO2 with a small Ti3+ content, the methane yield can be significantly increased by the deposition of tungsten oxide clusters. This is in line with the formation of Ti3+ by charge transfer from the clusters.38 However, higher amounts of tungsten oxide (especially above 7 WO3 nm−2) decrease the methane yield in comparison to small WO3 coverages. In case of high Ti3+ contents, the methane yield is drastically larger. The highest methane formation can be found for 3.5 WO3 nm−2, whereas higher coverages slightly decrease the yield again. Notably, by the presence of tungsten oxide clusters, the high-temperature deoxygenation reaction forming methane is substantially shifted with respect to the reaction temperature (approx. −30 K) for TiO2 with a large amount of Ti3+ and significantly narrowed. Hence, we attribute Ti3+ sites to be the dominant species responsible for the deoxygenation reaction. In presence of (WO3)n, the Ti3+ centers appear more localized and pinned to the (WO3)n cluster, leading also to a higher charge density near the surface.38 So, the desorption features are shifted to lower temperatures and appear more narrow. This is in line with an earlier finding of Dohnàlek and coworkers, highlighting that the alkene formation temperature on transition metal oxide surfaces decreases with increasing available electron density, in their case supplied by +I effects of the aliphatic chain.19
 | ||
| Fig. 4 Thermal formation of formaldehyde as a function of cluster coverage and bulk Ti3+ content. (a) Ratio of m/z = 29 and m/z = 31 TPRS spectra for the high temperature regime (500–750 K). (b) and (c) Integral area of the low and high temperature peaks shown in a and Fig. S9 in the ESI.† Note, that the peak area is a measure of the formaldehyde yield. | ||
One can easily see that the formation of formaldehyde in a HT reaction is evidently increased for 3.5 WO3 nm−2 and 7.0 WO3 nm−2 compared to pristine TiO2. Simultaneously, the LT formaldehyde yield drops (for highly reduced TiO2) or stays roughly constant (for low reduced TiO2). One should note, that especially for low reduced TiO2 this LT formation appears as a broad weak peak in the m/z = 29 to m/z = 31 ratio (Fig. S9 in the ESI†). The overall formaldehyde yield is higher for titania with a large Ti3+ content, but in that case the enhancement by tungsten oxide clusters is not so pronounced (see Table S2 in the ESI,† for a detailed calculation of the relative changes). This portends the importance of Ti3+ for the thermal production of formaldehyde, because the deposition of tungsten oxide clusters up to 7.1 WO3 nm−2 regulates the amount of surface-near Ti3+ to an equilibrium-like Ti3+/Ti4+ value of 6–7% as we have shown recently.38 Consequently, the oxide-oxide catalyst from an initially low reduced titania support acts like a stronger reduced simple titania surface. Interestingly, only the high temperature partial oxidation yield is enhanced by the cluster deposition, not the low temperature reaction. Furthermore, the partial oxidation obviously needs a certain amount of available Ti5c sites in the cluster neighborhood, because high coverages (28 WO3 nm−2) do not show a high temperature formaldehyde formation independent of the bulk Ti3+ density. Hence, we suggest, that the partial oxidation of methanol at higher temperatures proceeds at TiO2 adsorption sites, that are affected by the presence of the tungsten oxide clusters nearby as well as by subsurface Ti3+.
Moreover, the role of the tungsten oxide clusters in this reaction is interesting. For the formation of formaldehyde, a C–H bond has to be broken under abstraction of one H atom. As water desorption accompanies the formaldehyde desorption even at higher temperatures, it is likely, that the C–H dissociation itself induces the formaldehyde desorption. Therefore, we rule out, that the high temperature formaldehyde results from a molecular precursor formed at lower temperatures, but forms under direct release at higher temperatures. However, still Lewis base sites are required to accomplish the hydrogen abstraction. Since the W(Ot)2 sites are particularly strongly polarized moieties with a huge basicity, the hydrogen abstraction by such groups appears as the key step that steers the product composition. The basicity of these groups is likely increased by the presence of electronic charge density supplied by defect sites.
For better clarity, we refer to Fig. 5a. Here, the same TPR spectra as in Fig. 2, right side are shown, but now normalized to its maximum each. Thus, the desorption peaks in m/z = 45 and 26 become evident. Together with the similar peak shape and the HT peak in the m/z = 18 trace, which indicates the desorption of water, we can suggest the condensation of methanol forming dimethyl ether and ethane under water desorption. One should note, that as from the desorption peak maximum, the HT formation of formaldehyde (639 K) is not related to this condensation. Due to the overlapping desorption of formaldehyde and methane it is challenging to correctly assign the peak in the m/z = 26 trace, because ethane and ethene both exhibit the m/z = 26 and other common fragments. However, based on the asymmetry of the m/z = 29 and 30 peaks with a small shoulder at 617 K, we assign the m/z = 26 signal to ethane. By comparison with other m/z traces, we have additionally ruled out ethanol, formic acid and carbon dioxide (13CO2) being the source of the m/z = 45 fragment.
 | ||
| Fig. 5 (a) Temperature programmed reaction spectra obtained from a saturation coverage of methanol with preadsorption of 75 L O2 at 110 K on 3.5 WO3 nm−2 on a highly reduced TiO2 surface as in Fig. 2. All spectra have been normalized to their maximum for better clarity, the normalization factor is given with respect to the m/z = 15 trace. (b) m/z = 45 traces (attributed to desorption of dimethyl ether) after adsorption of a saturation coverage of methanol with 75L O2 preadsorption at 110 K on different TiO2 substrates and tungsten oxide coverages. | ||
It is even more interesting, that the formation of ethane and dimethyl ether is sensitive to the cluster coverage as well as the bulk Ti3+ density (Fig. 5b). The described condensation reaction was only found for 3.5 and 7.0 WO3 nm−2 deposited on the titania substrate with a high Ti3+ density, and remarkably here the condensation yield appears roughly equal for both cluster coverages (blue TPR spectra in Fig. 5b). For all other cluster coverages as well as in the absence of tungsten oxide clusters, no evidence for the formation of dimethyl ether around 600 K was found. This also implies, that this particular reaction at least partially takes place at the tungsten oxide clusters itself, but it proves that the interaction with the titania substrate is crucial. On titania with a small Ti3+ content, this condensation reaction was absent independent of the cluster coverage. We have also recognized, that the presence of oxygen is mandatory for the condensation. Additionally, UV irradiation (365 nm, 30 min, >1.5 × 1016 s−1 cm−2) quenched this reaction for 3.5 and 7.0 WO3 nm−2 on highly reduced TiO2 (Fig. S10 in the ESI†). By UV irradiation, methoxy species are consumed for the photochemical formation of low temperature formaldehyde.37 This suits earlier studies by the group of Henderson which gave evidence, that only methoxy species can be photo-oxidized on TiO2 (110), but not undissociated methanol.13 Therefore, we suggest that methoxy intermediates are involved in this special condensation pathway under the simultaneous formation of dimethyl ether and ethane. In accordance, alkoxy species were identified by other groups to be the key intermediates in such reactions on supported and unsupported tungsten oxide clusters.3–5 As the formation of dimethyl ether was previously assigned to the OWO moieties, one may suspect that defect sites such as Ti3+ can be accounted for the ethane formation. In this process, Ti3+ from the bulk may serve as oxygen acceptors and may react with the leftover oxygen to form TiOx islands.30,32,50,51 Besides the involvement of methoxy-intermediates adsorbed at the metal sites, it is challenging to derive more elementary reaction steps from our data. However, it is striking that the condensation yield is approximately equal for 3.5 and 7 WO3 nm−2. One possible hypothesis for this effect may be, that the reaction is not limited by the amount of tungsten oxide clusters but by the charge that is supplied by Ti3+ defects from the bulk. This suits the low overall yield for this reaction route, since the amount of defects can be roughly estimated in the same order of magnitude as the approximate dimethyl ether yield. In a recent publication, we have shown a Ti3+ accumulation in the cluster neighborhood between 500 and 800 K on highly reduced TiO2 and postulated the formation of anionic tungsten oxide clusters ([(WO3)n]z−-like) by electron transfer from Ti3+ towards the clusters.38 Since DFT results47 suggested that molecular oxygen can strongly interact with such partially anionic tungsten oxide clusters under formation of superoxide species (O2−), which can act as strong bases in proton abstraction, one may also consider that species here.52 In turn, the formation of that species is expected to be limited by the amount of available electron charge transferred from defect sites, suiting our finding for the dimethyl ether yield.
5. Conclusion
By a systematic TPRS based study we have elucidated the conversion of methanol as a model reactant in the presence of oxygen on rutile TiO2 (110) as a function of tungsten oxide cluster coverage as well as the amount of bulk Ti3+. Three important thermal conversion routes (deoxygenation, partial oxidation and condensation) were found. The central findings are the following:High bulk Ti3+ contents increase the thermal reactivity in the dominant deoxygenation reaction and the less pronounced partial oxidation. The deposition of tungsten oxide clusters (up to 7.1 WO3 nm−2) on TiO2 substrates under concomitant generation of surface-near Ti3+ activates both of these reaction pathways. Especially the thermal partial oxidation forming formaldehyde (desorption around 640 K) is strongly enhanced on all TiO2 substrates, likely due to a more effective proton abstraction from C–H bonds. The deoxygenation of methanol producing methane is shifted to lower temperature by the presence of tungsten oxide clusters.
In contrast to earlier reports we have shown that the thermal condensation of methanol under the formation of dimethyl ether exhibits an impact of Ti3+ sites. Unlike those previous publications, ethane was identified as an additional product of this conversion route. As apparent from photochemical experiments, methoxy intermediates are involved in this reaction that proceed at tungsten oxide clusters which are directly interacting with a Ti3+ rich TiO2 (sub-)surface. Since a high Ti3+ content in the bulk is mandatory for this reaction, we suggest a reaction limitation by the available charge from defect sites under involvement of partially anionic clusters.
Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
Thanks to Ralf Nustedt and Jessica Kräuter for support with the technical construction. The funding of the DFG research training group GRK 2226 “Chemical Bond Activation” is appreciated. L. M. acknowledges the funding of the German Academic Scholarship Foundation (Studienstiftung des deutschen Volkes).References
- C. Di Valentin, F. Wang and G. Pacchioni, Tungsten Oxide in Catalysis and Photocatalysis: Hints from DFT, Top. Catal., 2013, 56, 1404–1419 CrossRef CAS.
- R. Rousseau, D. A. Dixon, B. D. Kay and Z. Dohnalek, Dehydration, dehydrogenation, and condensation of alcohols on supported oxide catalysts based on cyclic (WO3)3 and (MoO3)3 clusters, Chem. Soc. Rev., 2014, 43, 7664–7680 RSC.
- Z. Li, Z. Fang, M. S. Kelley, B. D. Kay, R. Rousseau, Z. Dohnalek and D. A. Dixon, Ethanol Conversion on Cyclic (MO3)3 (M = Mo, W) Clusters, J. Phys. Chem. C, 2014, 118, 4869–4877 CrossRef CAS.
- Z. Fang, Z. Li, M. S. Kelley, B. D. Kay, S. Li, J. M. Hennigan, R. Rousseau, Z. Dohnálek and D. A. Dixon, Oxidation, Reduction, and Condensation of Alcohols over (MO3)3 (M = Mo, W) Nanoclusters, J. Phys. Chem. C, 2014, 118, 22620–22634 CrossRef CAS.
- X. Tang, D. Bumueller, A. Lim, J. Schneider, U. Heiz, G. Ganteför, D. H. Fairbrother and K. H. Bowen, Catalytic Dehydration of 2-Propanol by Size-Selected (WO3)n and (MoO3)n Metal Oxide Clusters, J. Phys. Chem. C, 2014, 118, 29278–29286 CrossRef CAS.
- Y. K. Kim, R. Rousseau, B. D. Kay, J. M. White and Z. Dohnálek, Catalytic Dehydration of 2-Propanol on (WO3)3 Clusters on TiO2 (110), J. Am. Chem. Soc., 2008, 130, 5059–5061 CrossRef CAS.
- Z. Li, Z. Zhang, B. D. Kay and Z. Dohnálek, Polymerization of Formaldehyde and Acetaldehyde on Ordered (WO3)3 Films on Pt(111), J. Phys. Chem. C, 2011, 115, 9692–9700 CrossRef CAS.
- C. Di Valentin, M. Rosa and G. Pacchioni, Radical versus Nucleophilic Mechanism of Formaldehyde Polymerization Catalyzed by (WO3)3 Clusters on Reduced or Stoichiometric TiO2 (110), J. Am. Chem. Soc., 2012, 134, 14086–14098 CrossRef CAS PubMed.
- J. Kim, B. D. Kay and Z. Dohnálek, Formaldehyde Polymerization on (WO3)3/TiO2 (110) Model Catalyst, J. Phys. Chem. C, 2010, 114, 17017–17022 CrossRef CAS.
- X. Tang, Z. Hicks, G. Ganteför, B. W. Eichhorn and K. H. Bowen, Adsorption and Decomposition of DMMP on Size-Selected (WO3)3 Clusters, ChemistrySelect, 2018, 3, 3718–3721 CrossRef CAS.
- Methanol: The Basic Chemical and Energy Feedstock of the Future, ed. M. Bertau, H. Offermanns, L. Plass, F. Schmidt and H.-J. Wernicke, Springer Berlin Heidelberg, Berlin, Heidelberg, 2014 Search PubMed.
- G. A. Olah, Towards Oil Independence Through Renewable Methanol Chemistry, Angew. Chem., Int. Ed., 2013, 52, 104–107 CrossRef CAS PubMed.
- M. Shen and M. A. Henderson, Role of Water in Methanol Photochemistry on Rutile TiO2 (110), J. Phys. Chem. C, 2012, 116, 18788–18795 CrossRef CAS.
- M. Shen, D. P. Acharya, Z. Dohnálek and M. A. Henderson, Importance of Diffusion in Methanol Photochemistry on TiO2 (110), J. Phys. Chem. C, 2012, 116, 25465–25469 CrossRef CAS.
- J. Kräuter, L. Mohrhusen, F. Waidhas, O. Brummel, J. Libuda and K. Al-Shamery, Photoconversion of 2-Propanol on Rutile Titania: A Combined Liquid-Phase and Surface Science Study, J. Phys. Chem. C, 2021, 125, 3355–3367 CrossRef.
- M. A. Henderson, S. Otero-Tapia and M. E. Castro, The chemistry of methanol on the TiO2 (110) surface: the influence of vacancies and coadsorbed species, Faraday Discuss., 1999, 114, 313–329 RSC.
- L. Chen, Z. Li, R. S. Smith, B. D. Kay and Z. Dohnálek, Conversion of 1,2-Propylene Glycol on Rutile TiO2 (110), J. Phys. Chem. C, 2014, 118, 15339–15347 CrossRef CAS.
- Y. K. Kim, B. D. Kay, J. M. White and Z. Dohnálek, 2-Propanol dehydration on TiO2 (110): The effect of bridge-bonded oxygen vacancy blocking, Surf. Sci., 2008, 602, 511–516 CrossRef CAS.
- Y. K. Kim, B. D. Kay, J. M. M. White and Z. Dohnálek, Alcohol Chemistry on Rutile TiO2 (110): The Influence of Alkyl Substituents on Reactivity and Selectivity, J. Phys. Chem. C, 2007, 111, 18326–18333 CrossRef.
- Z. Zhang, O. Bondarchuk, J. M. White, B. D. Kay and Z. Dohnálek, Imaging Adsorbate O–H Bond Cleavage: Methanol on TiO2 (110), J. Am. Chem. Soc., 2006, 128, 4198–4199 CrossRef CAS PubMed.
- L. Chen, Z. Li, R. S. Smith, B. D. Kay and Z. Dohnálek, Conversion of 1,3-Propylene Glycol on Rutile TiO2 (110), J. Phys. Chem. C, 2014, 118, 23181–23188 CrossRef CAS.
- Z. Li, B. D. Kay and Z. Dohnálek, Dehydration and dehydrogenation of ethylene glycol on rutile TiO2 (110), Phys. Chem. Chem. Phys., 2013, 15, 12180 RSC.
- Y. K. Kim, B. D. Kay, J. M. White and Z. Dohnálek, Inductive Effect of Alkyl Chains on Alcohol Dehydration at Bridge-bonded Oxygen Vacancies of TiO2 (110), Catal. Lett., 2007, 119, 1–4 CrossRef CAS.
- E. Farfan-Arribas and R. J. Madix, Role of Defects in the Adsorption of Aliphatic Alcohols on the TiO2 (110) Surface, J. Phys. Chem. B, 2002, 106, 10680–10692 CrossRef CAS.
- E. Farfan-Arribas and R. J. Madix, Different binding sites for methanol dehydrogenation and deoxygenation on stoichiometric and defective TiO2 (110) surfaces, Surf. Sci., 2003, 544, 241–260 CrossRef CAS.
- M. Osmić, L. Mohrhusen and K. Al-Shamery, Bulk Defect Dependence of Low-Temperature Partial Oxidation of Methanol and High-Temperature Hydrocarbon Formation on Rutile TiO2 (110), J. Phys. Chem. C, 2019, 123, 7615–7626 CrossRef.
- Z. Zhang, R. Rousseau, J. Gong, S.-C. Li, B. D. Kay, Q. Ge and Z. Dohnálek, Vacancy-Assisted Diffusion of Alkoxy Species on Rutile TiO2, Phys. Rev. Lett., 2008, 101, 156103 CrossRef PubMed.
- Y. K. Kim, Z. Dohnálek, B. D. Kay and R. Rousseau, Competitive Oxidation and Reduction of Aliphatic Alcohols over (WO3)3 Clusters, J. Phys. Chem. C, 2009, 113, 9721–9730 CrossRef CAS.
- S.-C. Li, Z. Li, Z. Zhang, B. D. Kay, R. Rousseau and Z. Dohnálek, Preparation, Characterization, and Catalytic Properties of Tungsten Trioxide Cyclic Trimers on FeO(111)/Pt(111), J. Phys. Chem. C, 2012, 116, 908–916 CrossRef CAS.
- E. Lira, S. Wendt, P. Huo, J. Ø. Hansen, R. Streber, S. Porsgaard, Y. Wei, R. Bechstein, E. Laegsgaard and F. Besenbacher, The Importance of Bulk Ti3+ Defects in the Oxygen Chemistry on Titania Surfaces, J. Am. Chem. Soc., 2011, 133, 6529–6532 CrossRef CAS PubMed.
- Z. Zhang, J. Lee, J. T. Yates, R. Bechstein, E. Lira, J. Ø. Hansen, S. Wendt and F. Besenbacher, Unraveling the Diffusion of Bulk Ti Interstitials in Rutile TiO2 (110) by Monitoring Their Reaction with O Adatoms, J. Phys. Chem. C, 2010, 114, 3059–3062 CrossRef CAS.
- E. Lira, J. Hansen, P. Huo, R. Bechstein, P. Galliker, E. Lægsgaard, B. Hammer, S. Wendt and F. Besenbacher, Dissociative and Molecular Oxygen Chemisorption Channels on Reduced Rutile TiO2 (110): An STM and TPD study, Surf. Sci., 2010, 604, 1945–1960 CrossRef CAS.
- P. M. Clawin, C. M. Friend and K. Al-Shamery, Defects in Surface Chemistry-Reductive coupling of Benzaldehyde on Rutile TiO2 (110), Chem. – Eur. J., 2014, 20, 7665–7669 CrossRef CAS PubMed.
- L. Benz, J. Haubrich, R. G. Quiller, S. C. Jensen and C. M. Friend, McMurry Chemistry on TiO2 (110): Reductive C–C Coupling of Benzaldehyde Driven by Titanium Interstitials, J. Am. Chem. Soc., 2009, 2, 15026–15031 CrossRef PubMed.
- S. C. Jensen and C. M. Friend, The Dynamic Roles of Interstitial and Surface Defects on Oxidation and Reduction Reactions on Titania, Top. Catal., 2013, 56, 1377–1388 CrossRef CAS.
- A. S. Crampton, L. Cai, N. Janvelyan, X. Zheng and C. M. Friend, Methanol Photo-Oxidation on Rutile TiO2 Nanowires: Probing Reaction Pathways on Complex Materials, J. Phys. Chem. C, 2017, 121, 9910–9919 CrossRef CAS.
- L. Mohrhusen, J. Kräuter and K. Al-Shamery, Conversion of Methanol on Rutile TiO2 (110) and Tungsten Oxide Clusters: 2. The Role of Defects and Electron Transfer in Bifunctional Oxidic Photocatalysts, Phys. Chem. Chem. Phys., 2021 10.1039/d1cp01176f.
- L. Mohrhusen, M. Grebien and K. Al-Shamery, Electron Transfer in Oxide–Oxide Cocatalysts: Interaction of Tungsten Oxide Clusters with Ti3+ States in Rutile TiO2, J. Phys. Chem. C, 2020, 124, 23661–23673 CrossRef CAS.
- U. Leist, A. Winkler, J. Büssow and K. Al-Shamery, Mobile sample holder applying multiple heating systems with a variable heating and cooling rate, Rev. Sci. Instrum., 2003, 74, 4772–4778 CrossRef CAS.
- L. Mohrhusen, J. Kräuter, M. Willms and K. Al-Shamery, Argon Embedded by Ion Bombardment: Relevance of Hidden Dopants in Rutile TiO2, J. Phys. Chem. C, 2019, 123, 20434–20442 CrossRef CAS.
- J. Kim, O. Bondarchuk, B. D. Kay, J. M. White and Z. Dohnálek, Preparation and characterization of monodispersed WO3 nanoclusters on TiO2 (110), Catal. Today, 2007, 120, 186–195 CrossRef CAS.
- M. Wagner, S. Surnev, M. G. Ramsey, G. Barcaro, L. Sementa, F. R. Negreiros, A. Fortunelli, Z. Dohnalek and F. P. Netzer, Structure and Bonding of Tungsten Oxide Clusters on Nanostructured Cu-O Surfaces, J. Phys. Chem. C, 2011, 115, 23480–23487 CrossRef CAS.
- Z. Li, Z. Zhang, Y. K. Kim, R. S. Smith, F. Netzer, B. D. Kay, R. Rousseau and Z. Dohnálek, Growth of Ordered Ultrathin Tungsten Oxide Films on Pt(111), J. Phys. Chem. C, 2011, 115, 5773–5783 CrossRef CAS.
- J. Berkowitz, W. A. Chupka and M. G. Inghram, Polymeric Gaseous Species in the Sublimation of Tungsten Trioxide, J. Chem. Phys., 1957, 27, 85–86 CrossRef CAS.
- O. Bondarchuk, X. Huang, J. Kim, B. D. Kay, L.-S. Wang, J. M. White and Z. Dohnálek, Formation of Monodisperse (WO3)3 Clusters on TiO2 (110), Angew. Chem., Int. Ed., 2006, 45, 4786–4789 CrossRef CAS PubMed.
- X. Huang, H.-J. Zhai, B. Kiran and L.-S. Wang, Observation of d-Orbital Aromaticity, Angew. Chem., Int. Ed., 2005, 44, 7251–7254 CrossRef CAS PubMed.
- X. Huang, H.-J. Zhai, T. Waters, J. Li and L.-S. Wang, Experimental and Theoretical Characterization of Superoxide Complexes [W2O6(O2−)] and [W3O9(O2−)]: Models for the Interaction of O2 with Reduced W Sites on Tungsten Oxide Surfaces, Angew. Chem., Int. Ed., 2006, 45, 657–660 CrossRef CAS PubMed.
- NIST Mass Spectrometry Data Center and W. E. Wallace, in NIST Chemistry WebBook, NIST Standard Reference Database, Number 69, ed. P. J. Linstrom and W. G. Mallard, National Institute of Standards and Technology, Gaithersburg MD, 2018, accessed February 2019 Search PubMed.
- M. A. Cortes-Jácome, M. Morales, C. Angeles Chavez, L. F. Ramírez-Verduzco, E. López-Salinas and J. A. Toledo-Antonio, WOx/TiO2 Catalysts via Titania Nanotubes for the Oxidation of Dibenzothiophene, Chem. Mater., 2007, 19, 6605–6614 CrossRef.
- S. Wendt, P. T. Sprunger, E. Lira, G. K. H. Madsen, Z. Li, J. O. Hansen, J. Matthiesen, A. Blekinge-Rasmussen, E. Laegsgaard, B. Hammer and F. Besenbacher, The Role of Interstitial Sites in the Ti3d Defect State in the Band Gap of Titania, Science, 2008, 320, 1755–1759 CrossRef CAS PubMed.
- J. Kräuter, L. Mohrhusen, T. Thiedemann, M. Willms and K. Al-Shamery, Activation of Small Organic Molecules on Ti2+-Rich TiO2 Surfaces: Deoxygenation vs. C–C Coupling, Zeitschrift für Naturforsch. A, 2019, 74, 697–707 CrossRef.
- P. Salvador, On the Nature of Photogenerated Radical Species Active in the Oxidative Degradation of Dissolved Pollutants with TiO2 Aqueous Suspensions: A Revision in the Light of the Electronic Structure of Adsorbed Water, J. Phys. Chem. C, 2007, 111, 17038–17043 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Overview on surface characterization including XPS and LEED data for tungsten oxide clusters at rutile TiO2 with a small (Fig. S1) and large (Fig. S2) Ti3+ density; Fig. S3: additional m/z = 18 traces; Fig. S4: fragmentation pattern obtained from NIST database of methanol and possible reaction products; Fig. S5 and S6: additional TPR spectra for methanol on TiO2 with and without tungsten oxide clusters in the absence of additional oxygen; Fig. S7 and S8: additional TPR spectra for m/z = 28 and 44 obtained from methanol on TiO2 with (S7) and without (S8) tungsten oxide clusters; Tables S1 and S2: semi-quantitative analysis changes of the thermal methane (Table S1) and formaldehyde (Table S2) formation; Fig. S9: ratio of m/z = 29 and 31 for the LT formaldehyde formation; Fig. S10: additional TPR spectra for m/z = 45 (dimethyl ether). See DOI: 10.1039/d1cp01175h |
| This journal is © the Owner Societies 2021 |