Electrochemical behavior and electrodeposition of gallium in 1,2-dimethoxyethane-based electrolytes†
Received
10th March 2021
, Accepted 10th May 2021
First published on 18th June 2021
Abstract
The electrochemical behavior and electrodeposition of gallium was studied in a non-aqueous electrolyte comprising of gallium(III) chloride and 1,2-dimethoxyethane (DME). Electrochemical quartz crystal microbalance (EQCM) and rotating ring disk electrode (RRDE) measurements indicate that reduction of gallium(III) is a two-step process: first from gallium(III) to gallium(I), and then from gallium(I) to gallium(0). The morphology and elemental composition of the electrodeposited layer were examined using scanning electron microscopy (SEM) and energy-dispersive X-ray spectroscopy (EDX). Metallic gallium was deposited as spheres with diameters of several hundred nanometers that were stacked on top of each other. X-ray photoelectron spectroscopy (XPS) revealed that each gallium sphere was covered by a thin gallium oxide shell. Electrochemical experiments indicated that these oxide layers are electrically conductive, as gallium can be electrodeposited and partially stripped on or from the layer of spheres below. This was further evidenced by simultaneous electrodeposition of gallium and indium, using indium as a tracer. Electrodeposition of gallium from an O2-containing electrolyte resulted in spheres with smaller diameters. This was due to the formation thicker oxide shells, through which diffusion of gallium atoms that were electrodeposited on the surface, was slower. The concentration of gallium adatoms on top of the gallium spheres to form a new sphere therefore reaches the critical concentration for nucleating a new gallium sphere sooner, leading to smaller spheres.
Introduction
Gallium is utilized for the production of several semiconductors, including gallium arsenide (GaAs), gallium nitride (GaN), gallium phosphide (GaP), gallium antimonide (GaSb) and copper indium gallium selenide (CIGS).1–5 These materials are all indispensable for the electric and electronic equipment industry. More particularly, they serve as components in integrated circuits, mobile phones, light-emitting diodes, photodetectors and photovoltaics devices. Despite the high demand of gallium, its resources are relatively scarce. Although its natural abundance is not extremely low (abundance in the earth's crust is comparable to e.g. lead), gallium occurs exclusively as an impurity in the ores of base metals (mainly aluminum and zinc). It can therefore be generated only in the form of a by-product, typically from bauxite ore, used for the production of alumina via the Bayer process, and to a lesser extent from sulfidic zinc ores and coal.6,7
The electrodeposition of gallium has been extensively studied for: (1) the production of highly-pure gallium metal, (2) the production of gallium-based semiconductors, and (3) the electrowinning of gallium from Bayer liquor.8 In aqueous solutions, the electrodeposition of gallium metal competes with the hydrogen evolution reaction (HER) due to the negative standard reduction potential of the Ga(III)/Ga(0) redox couple (E0 = −0.529 V vs. SHE).9,10 Therefore, the current efficiency of gallium electrodeposition processes is low, especially in acidic solutions in which the hydrogen evolution is more prominent. The HER can also instigate pH changes at the electrode interface, leading to a local increase in the pH, causing the passivation of gallium deposits by gallium oxide.10 Flamini et al. demonstrated that in acidic aqueous media, gallium(III) is reduced to gallium(I) at low current densities, whereas the electrodeposition of gallium(0) and the HER occur at higher current densities.11 Hong et al. also identified gallium(I) in the form of Ga2O in their investigation of the redox behavior of gallium in alkaline solution.12 The standard reduction potential of the Ga2O/Ga redox couple was calculated to be −0.40 V vs. SHE.13 However, gallium(I) is unstable in aqueous environment; it undergoes disproportionation to gallium(III) and gallium metal.14
Electrowinning of gallium from the alkaline Bayer liquor has been extensively studied.15–18 However, the gallium content in Bayer liquor is low, varying between 20 and 80 ppm. Furthermore, the presence of iron(III) and vanadium(V) species complicates the process and lowers the cathodic current efficiency.19 These species adsorb or electrodeposit on the surface of gallium deposits, leading to changes in morphology and enhancement of the HER. Electrowinning is therefore considered as an inefficient extraction method of gallium from Bayer liquor.
Electrodeposition of gallium has also been studied from several non-aqueous electrolytes to circumvent the HER. Verbrugge and Carpenter reported a gallium electrodeposition process in the gallium(III) chloride/1-methyl-3-ethylimidazolium chloride room-temperature molten salt.20 Sun et al. prepared gallium electrodeposits on tungsten electrodes from a 60.0/40.0 mol% aluminum(III) chloride/l-methyl-3-ethylimidazolium chloride (AlCl3/MEIC) melt containing gallium(I).21 Gasparotto et al. studied by in situ scanning tunneling microscopy (STM) the gallium electrodeposition from the air- and water-stable ionic liquid 1-butyl-1-methylpyrrolidinium bis(trifluoromethylsulfonyl)imide which contained gallium(III) chloride as gallium source.22 These authors demonstrated that gallium can be deposited from the ionic liquid on gold electrodes both by electroless deposition and electrodeposition. Freyland et al. investigated gallium electrodeposition on a gold electrode from two Lewis-acidic ionic liquids: a chloroaluminate melt and a chlorogallate(III) melt.23 They observed underpotential deposition, leading to the formation of a gold–gallium alloy, as well as electrodeposition at more cathodic overpotentials, leading to 3-D gallium clusters of nanometer size. Seddon et al. reported that metallic gallium electrodeposits can be obtained from buffered acidic chlorogallate(III) ionic liquids, but not from the corresponding basic and neutral melts.24 This is due to the formation of the anionic [GaCl4]− complex in the basic and neutral metals and this complex is not electroactive within the electrochemical windows of these metals. Dale et al. demonstrated the electrodeposition of gallium, as well as copper–gallium and indium–gallium alloys from the deep-eutectic solvent (DES) Reline, which is a mixture of choline chloride and urea in 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2 molar ratio, as an alternative method to produce CIGS absorber layers for photovoltaic cells.25,26 Bakkar and Neubert studied the electrodeposition of gallium from a chloroaluminate ionic liquid composed of aluminum chloride and 1-ethyl-3-methylimidazolium chloride outside a glove box.27 They added a layer of n-decane on top of the ionic liquid electrolyte, protecting it from moisture in the atmosphere. A sacrificial gallium anode was used and it was shown that gallium was oxidized to gallium(I) at this anode. On the cathode, electrodeposition of gallium was achieved. The morphology of the gallium deposits was found to depend on the cathode material. This was attributed to the formation of alloys between gallium and the substrate.
:2 molar ratio, as an alternative method to produce CIGS absorber layers for photovoltaic cells.25,26 Bakkar and Neubert studied the electrodeposition of gallium from a chloroaluminate ionic liquid composed of aluminum chloride and 1-ethyl-3-methylimidazolium chloride outside a glove box.27 They added a layer of n-decane on top of the ionic liquid electrolyte, protecting it from moisture in the atmosphere. A sacrificial gallium anode was used and it was shown that gallium was oxidized to gallium(I) at this anode. On the cathode, electrodeposition of gallium was achieved. The morphology of the gallium deposits was found to depend on the cathode material. This was attributed to the formation of alloys between gallium and the substrate.
In this paper, the electrochemistry and the electrodeposition of gallium in 1,2-dimethoxyethane (DME) is investigated using a variety of experimental electrochemical techniques. DME has a wide electrochemical window and is therefore a popular solvent for different electrochemical applications.28,29 It exhibits a much lower viscosity than most ionic liquids and water-lean deep-eutectic solvents so that DME-based electrolytes can be applied at room temperature and do not require heating.
Experimental
Chemicals
Gallium(III) chloride (GaCl3 anhydrous beads, 99.99%), tetrabutylammonium perchlorate ([TBA][ClO4], 99.5%), and 1-butyl-1-methylpyrrolidinium bis(trifluoromethylsulfonyl)imide ([BMP][Tf2N], 98%) were purchased from Sigma-Aldrich (Overijse, Belgium). Hydrochloric acid (Analar Normapur, 37%) was purchased from VWR (Leuven, Belgium) and acetone (99 + %) was purchased from Chem-Lab (Zedelgem, Belgium). Tetrabutylammonium perchlorate was dried on a Schlenk line at 70 °C for 48 hours prior to use. All other chemicals were used as received, without any further purification.
Instrumentation
All electrochemical experiments were performed in an argon-filled glovebox with oxygen and moisture concentrations below 1 ppm, and performed using an Autolab PGSTAT 302N potentiostat, controlled by a computer with NOVA2 software. Measurements were carried out using a three-electrode setup. Disk electrodes which served as working electrode (WE) for the recording of CVs were made of glassy carbon (GC) cylinders embedded in glass, and had a diameter of 4 mm. GC plates with exposed surface areas of 20 mm2 were used as WE for deposition experiments. For all experiments, the counter electrode (CE) consisted of a GC block whose area was at least five times larger than that of the WE. The reference electrode (RE) was a ferrocene/ferrocenium electrode, consisting of a glass tube filled with a solution of ferrocene (5 mM) and ferrocenium (5 mM) in 1-butyl-1-methyl-pyrrolidinium bis(trifluoromethylsulfonyl)imide, [BMP][Tf2N], in which a platinum wire with a diameter of 1 mm was immersed. Prior to use, the GC electrodes were ultrasonicated in ethanol, and subsequently washed with hydrochloric acid (35%), rinsed with demineralized water and ethanol, and air-dried. A ceramic crucible was used as electrochemical cell in which 3 mL of electrolyte was added. Cyclic voltammograms (CVs) were always started at the open circuit potential (OCP) and scanned first towards cathodic overpotentials. All presented CVs in this work correspond to the first recorded cycle. Rotating ring disk electrode (RRDE) measurements were carried out using a Metrohm Autolab RRDE. This electrode consisted of a GC disk with a diameter of 5 mm, a gap of 0.375 mm and a platinum ring with a width of 0.5 mm. For RRDE measurements, a larger electrochemical cell was used that contains 7 mL of electrolyte. The same RE and CE were used as for stationary experiments. EQCM measurements were performed using platinum-coated QCM-crystals (1 inch in diameter, 5 MHz AT-cut platinum-coated crystals, INFICON) with an electrochemically active surface of 121 mm2. Experiments were performed using an EQCM device (MaxTek) that was connected to the Autolab PGSTAT 302N potentiostat. CVs, RRDE measurements and EQCM analysis were recorded in tandem. The morphology of the gallium deposits was studied using scanning electron microscopy (SEM) on a Phillips XL-30 FEG SEM and a FEI Nova 600 Nanolab NanoSEM. Energy-dispersive X-ray spectroscopy was used for elemental analysis (EDX; Octane elite super silicon drift detector, Ametek EDAX). The viscosity of the electrolyte was measured using a rolling-ball type viscometer (Anton Paar, Lovis 2000 ME) whereas the density of the electrolyte was determined using a density meter with an oscillating U-tube sensor (Anton Paar, DMA 4500 M). Samples for X-ray photoelectron spectroscopy (XPS) were transferred to a portable load-lock inside an argon-filled glovebox and transported under argon to the XPS instrument and directly attached to the ultra-high-vacuum system (10−9–10−8 bar) to avoid exposure to air. The X-ray line was Al Kα (1487 eV) produced by an XR4 Twin X-ray Anode (Thermo Fisher Scientific) and the electron energy analyzer was an Alpha110 (Thermo Fisher Scientific) with a pass energy of 25 eV. The resolution of the measurements was 1.3 eV. The XPS spectra were calibrated by shifting the C 1s peak (adventitious carbon) to 285.2 eV.
Results and discussion
Redox behavior of gallium in DME
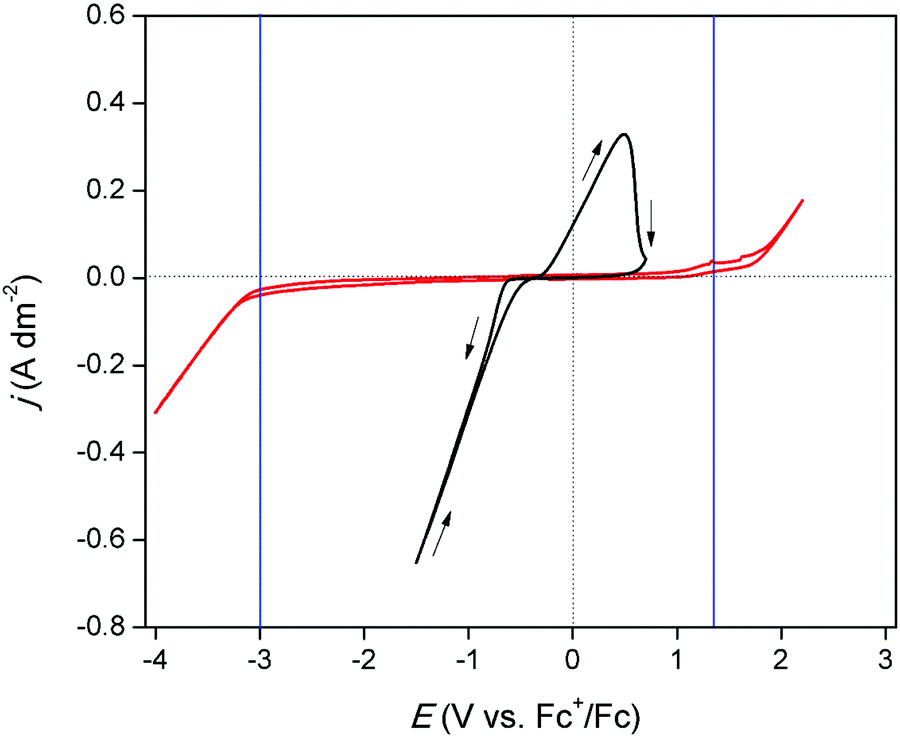
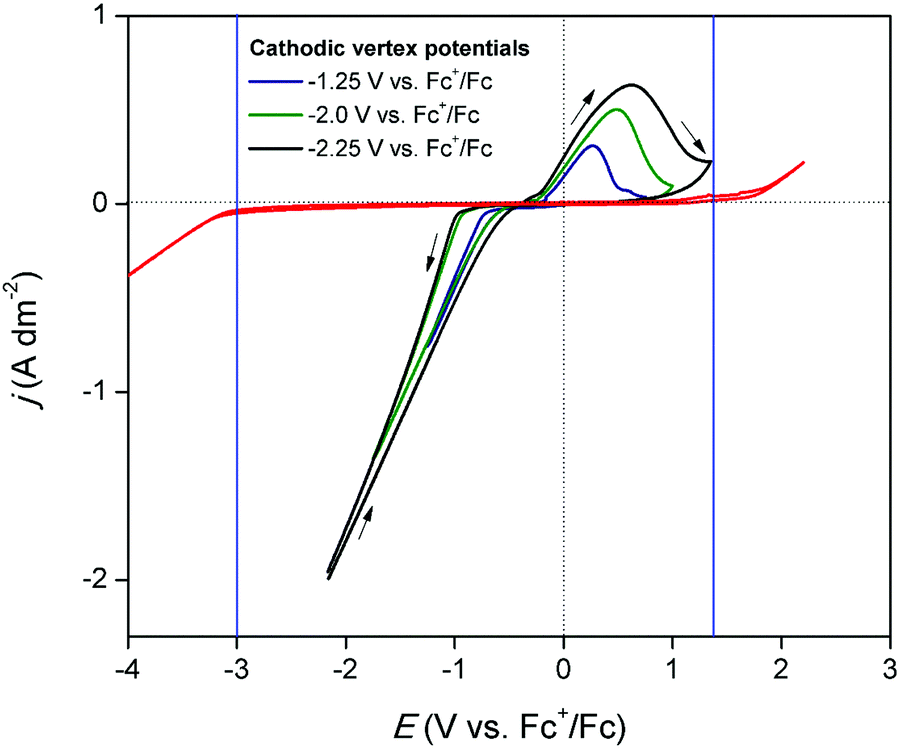
The electrochemical window of DME was measured to identify the potential region in which the redox behavior of gallium can be investigated. Fig. 1 (red) shows the CV of DME containing 0.2 mol dm−3 of tetrabutylammonium perchlorate ([TBA][ClO4]), recorded on a glassy carbon (GC) electrode at 26 °C. Gallium does not form alloys with GC so that GC was the electrode material of choice to study the conditions in which pure gallium is electrodeposited and stripped. The cathodic and anodic limit of DME, marked by the blue lines, are positioned at −3.0 V vs. Fc+/Fc and +1.32 V vs. Fc+/Fc, respectively, indicating that the electrochemical window is 4.32 V wide. Fig. 1 (black) shows the CV of DME with 0.1 mol dm−3 of GaCl3 and 0.2 mol dm−3 of [TBA][ClO4]. In the forward scan, a cathodic wave is observed which starts at −0.72 V vs. Fc+/Fc. This wave involves the reduction of gallium(III), as no other species is electrochemically active at this potential. The standard reduction potentials of the Ga3+/Ga+ and Ga3+/Ga0 equal −0.40 V vs. SHE and −0.53 V vs. SHE, respectively, and thus lie in close proximity of each other. Presumably, reduction of gallium(III) to gallium(0) takes place. However, the involvement of gallium(I) cannot be excluded, as its existence has been observed in several other aqueous and non-aqueous systems.23,27 Formation of gallium(II) is disregarded, as this species has not been identified in any electrochemical studies. The absence of a limiting current indicates that the reduction process is not diffusion-controlled in this potential range. In the reverse scan, the current passes through zero at −0.38 V vs. Fc+/Fc and a clear nucleation loop is observed. Subsequently, an anodic peak occurs which almost drops to zero at +1.00 V vs. Fc+/Fc. This feature is ascribed to the stripping of deposited gallium to gallium(I) or gallium(III). Fig. 2 shows CVs of the same electrolyte scanned to various vertex potentials. Both higher cathodic waves and anodic peaks are observed with increasing vertex potentials. This indicates that the reduction and oxidation reactions are mutually linked, and might therefore indeed comprise of the reduction of gallium(III) to gallium(0), and the oxidation of gallium(0) to gallium(I) or gallium(III), respectively. Yet, there is a distinct discrepancy in the charge consumed during reduction (Qred) and oxidation (Qox). A near 4:1 ratio for Qred/Qox is attained for a single CV, indicating that the processes might not be as straightforward as they appear at first sight (Q vs. t plot of a single CV can be found in Fig. S1 ESI†).
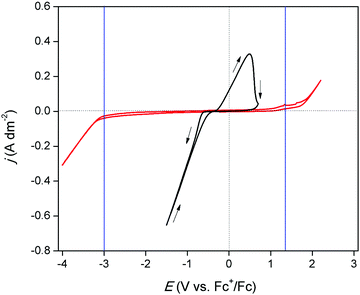 |
| | Fig. 1 CVs recorded on a GC WE (∅ = 4 mm) for DME containing 0.2 mol dm−3 of [TBA][ClO4] (red) and DME containing 0.1 mol dm−3 of GaCl3 and 0.2 mol dm−3 of [TBA][ClO4] (black) at a scan rate of 20 mV s−1 at 26 °C. | |
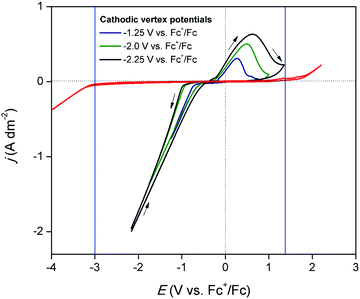 |
| | Fig. 2 CVs recorded on a GC WE (∅ = 4 mm) for DME containing 0.1 mol dm−3 of GaCl3 and 0.2 mol dm−3 of [TBA][ClO4] (black), scanned to different vertex potentials at a scan rate of 20 mV s−1 at 26 °C. | |
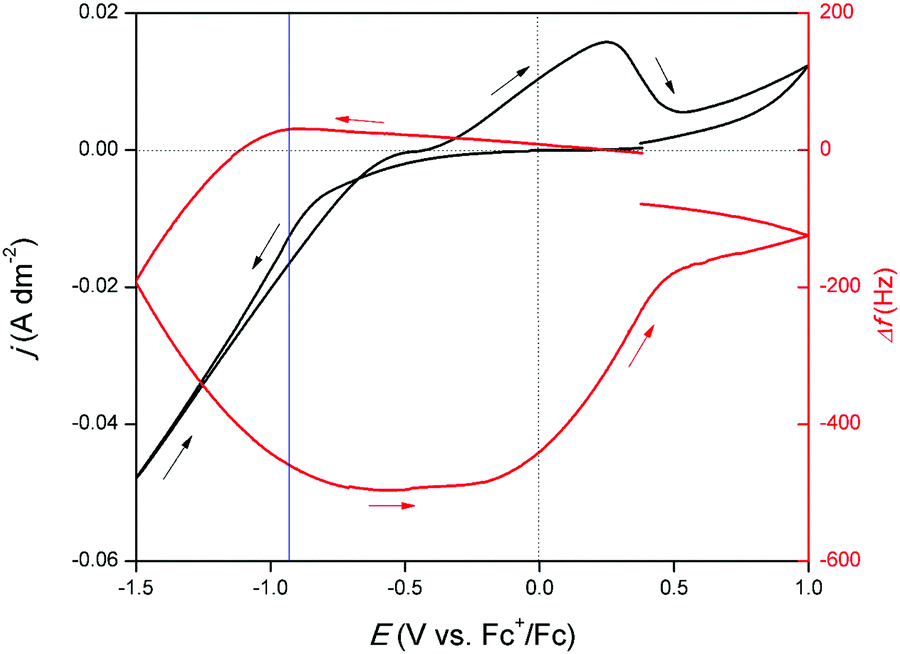
To acquire more insight in the occurring redox reactions, an EQCM experiment was performed. Herein, a CV of the DME-based solution was measured on a platinum-coated quartz crystal. Platinum was selected as, to the best of our knowledge, no glassy carbon EQCM crystals are commercially available. Addition or removal of mass on or from this crystal, which serves as the WE, results in a change in the resonance frequency Δf. By monitoring this frequency, gallium electrodeposition and stripping can be associated to certain potential regions in the CV. The resulting measurement is shown in Fig. 3. Starting from −0.10 V vs. Fc+/Fc, a gradually increasing cathodic wave is observed. Interestingly, upon initiation of this cathodic wave, Δf increases, which in theory implies mass loss. Naturally, no mass can be removed from the crystal at this point. The reduction process is therefore presumed to involve reduction of gallium(III) to gallium(I). Differences in coordination chemistry between the trivalent and monovalent gallium species in solution can lead to local changes in viscosity at the electrode interface, as different amounts of solvent molecules can be bound as ligands. Formation of gallium(I) is presumed to result in a local decrease in viscosity at the interface, leading to the observed minor increase in Δf. This correlation between solution viscosity and frequency change of the EQCM crystal was first demonstrated by Kanazawa et al.31,32 Starting from −0.87 V vs. Fc+/Fc (indicated by the blue line), Δf sharply decreases, indicating gallium electrodeposition due to further reduction of gallium(I) to gallium(0). As a significant cathodic overpotential is required to induce further reduction, this step is presumed to be rate-determining. The resonance frequency continues to decrease until −0.75 V vs. Fc+/Fc in the backward scan is reached. The signal subsequently flattens, implying that gallium is no longer electrodeposited. In this potential region, gallium(III) reduces only to gallium(I). After −0.30 V vs. Fc+/Fc, Δf gradually increases. This evolution in resonance frequency overlaps with the oxidation peak, indicating that the corresponding process involves stripping of gallium metal. After the oxidation peak, a second oxidation wave is present. In parallel with this feature, a small increase in Δf is observed. This is attributed to the stripping of formed gallium–platinum alloy. As Δf does not fully recede back to 0 Hz, not all deposited gallium is stripped from the gallium–platinum alloy in the investigated potential region. Furthermore, from the derived data of the EQCM experiment, a curve of mass/charge (m/z) as a function of the potential was constructed. The resulting graph is shown in Fig. S2, ESI† and demonstrates that the reduction of gallium(III) to gallium(I) takes place followed by further reduction to gallium(0).
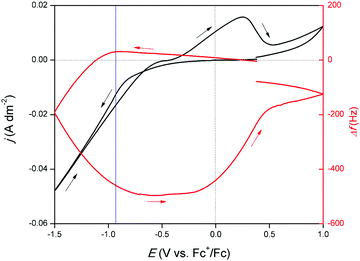 |
| | Fig. 3 CV recorded on a 5 MHz platinum-coated quartz crystal (∅ = 12.4 mm) for DME containing 10 mmol dm−3 of GaCl3 and 0.1 mol dm−3 of [TBA][ClO4] at a scan rate of 10 mV s−1 at 26 °C (full line left axis) with EQCM analysis (right axis). | |
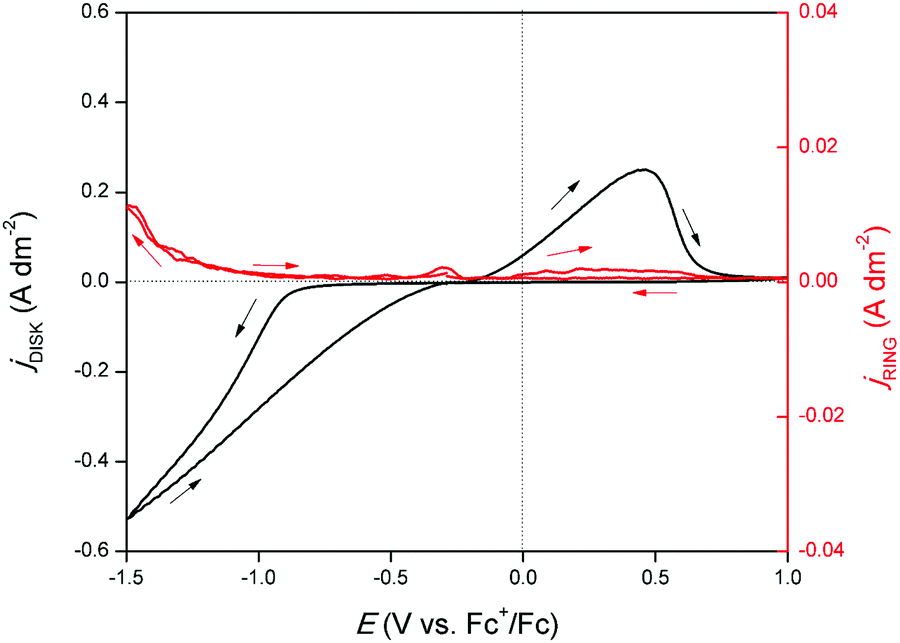
An RRDE experiment was performed to confirm the formation of gallium(I) species during reduction and oxidation. A CV was recorded on the disk and a potential of +1.0 V vs. Fc+/Fc was applied on the ring while rotating at 1000 rpm. In principle, gallium(I) species formed on the disk are spun outwards and oxidized to gallium(III) due to the large positive potential applied to the ring. The corresponding measurement is shown in Fig. 4. Upon initiation of the cathodic wave of the CV, the current response on the ring increases, demonstrating that the reduction of gallium(III) to gallium(I) takes place. This is in agreement with the findings from the EQCM experiment. The current continues to increase until the outer potential of −1.5 V vs. Fc+/Fc is reached. Hence, throughout the entire cathodic wave, gallium(I) is generated. In the backward scan, a second increase in ring current response is observed in parallel with the stripping peak of the CV. Hence, gallium, deposited during the cathodic wave is stripped to gallium(I). A large fraction of generated gallium(I) likely undergoes disproportionation to gallium(III) and gallium(0) before to reaching the ring:
| 3Ga(I) → 2Ga(0) + Ga(III) |
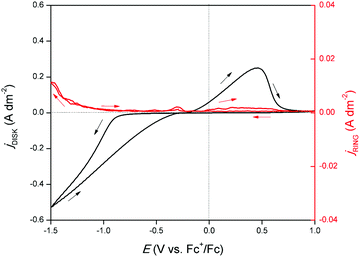 |
| | Fig. 4 CV recorded on a GC disk electrode (∅ = 5 mm) for DME containing 0.1 mol dm−3 of GaCl3 and 0.2 mol dm−3 of [TBA][ClO4] at a scan rate of 10 mV s−1 at 26 °C (black line, left axis) with current response on platinum ring on which +1.0 V vs. Fc+/Fc was applied (red line, right axis). The RRDE was rotated at 1000 rpm. The ring current density data was smoothed by a Savitzky–Golay function with 50 point window size. | |
At a rotation speed of 200 rpm, with all other conditions held the same, no ring current response was observed (Fig. S3 ESI†). This implies a fast reaction rate of the disproportionation reaction of gallium(I), as this species can only be detected on the ring when its mass transfer is increased by convection.
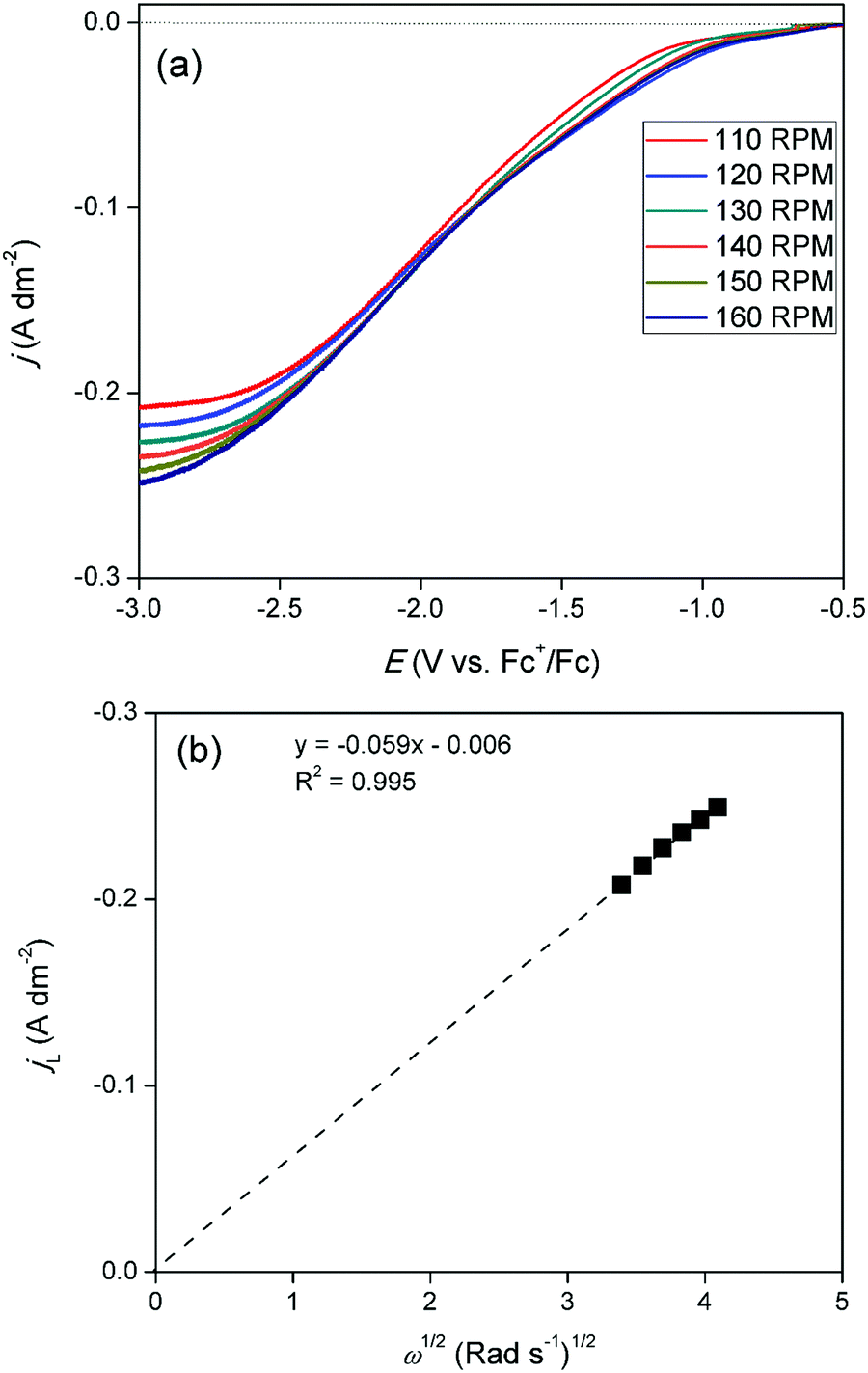
Measurement of diffusion coefficient
Linear sweep voltammograms (LSVs) were recorded for cathodic overpotentials at various rotation rates for a solution of 15 mmol dm−3 of GaCl3 and 0.20 mol dm−3 of [TBA][ClO4] in DME, and are shown in Fig. 5a. The gallium concentration in the electrolyte was lowered to reach a diffusion-limited current during the reduction process. The LSVs were used to construct a Levich plot. This plot is shown in Fig. 5b and depicts a linear relation between the limiting currents and the square root of the rotation rate. The extrapolated line intercepts the vertical axis at zero current. This indicates the system is completely under diffusion control. As the observed limiting currents in the LSVs are attributed to the reduction of gallium(III) to gallium(I)/gallium(0), the slope of the Levich plot can be used to calculate the diffusion constant of the gallium(III) species:
| IL = 0.620nFAD2/3ν−1/6Cω1/2 |
where, IL is the limiting current from the LSV (A), n is the number of electrons transferred (3, dimensionless), F is the Faraday constant (96485.33 C mol−1), A is the surface area of the electrode (19.63 × 10−6 m2), D is the diffusion coefficient of the electroactive species (m2 s −1), ν is the kinematic viscosity of the electrolyte (0.55 × 10−6 m2 s−1), C is the concentration of indium in the electrolyte (0.015 × 10−3 mol m−3) and ω is the rotation rate of the electrode (rad s−1). Using the Levich equation, the diffusion coefficient of gallium(III) in the diluted solution was calculated to be 2.6 × 10−10 m2 s−1 at 26 °C. Reported diffusion coefficients of gallium(III) species in the DES reline at 60 °C, and in the ionic liquid [BMIM][TfO] at 70 °C equaled 8.2 × 10−12 m2 s−1 and 5.57 × 10−12 m2 s−1, respectively.21,27,30 Differences in values can be attributed to varying viscosities of the electrolytes as well as varying hydrodynamic radii of the solvated gallium(III) species in the electrolytes.33
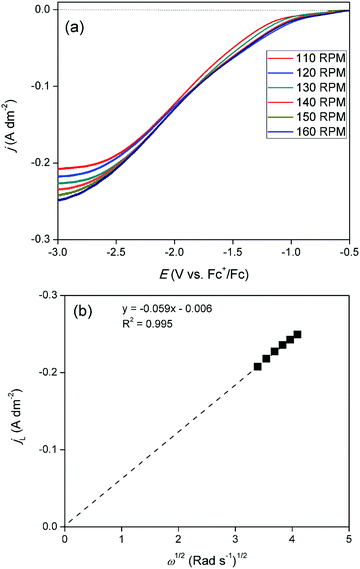 |
| | Fig. 5 (a) LSVs recorded on a GC disk electrode (∅ = 5 mm) for DME containing 10 mmol dm−3 of GaCl3 and 0.2 mol dm−3 of [TBA][ClO4] at various rotation rates, at a scan rate of 10 mV s−1, at 26 °C. (b) Levich plot constructed using the limiting current densities of the LSVs at −3.0 V vs. Fc+/Fc. | |
Electrodeposition of metallic gallium
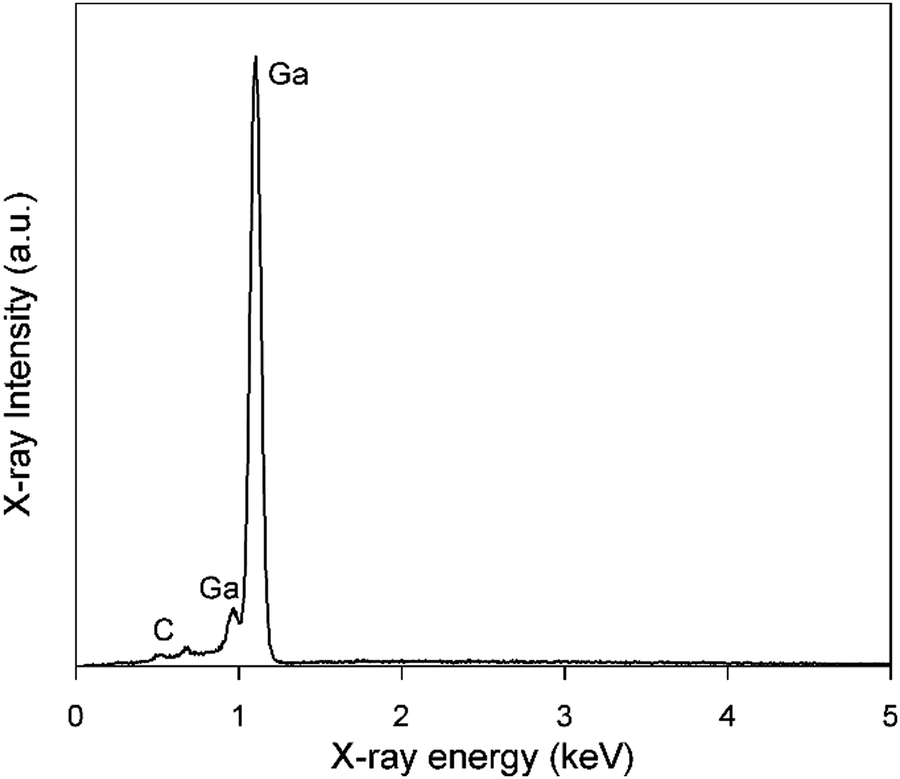
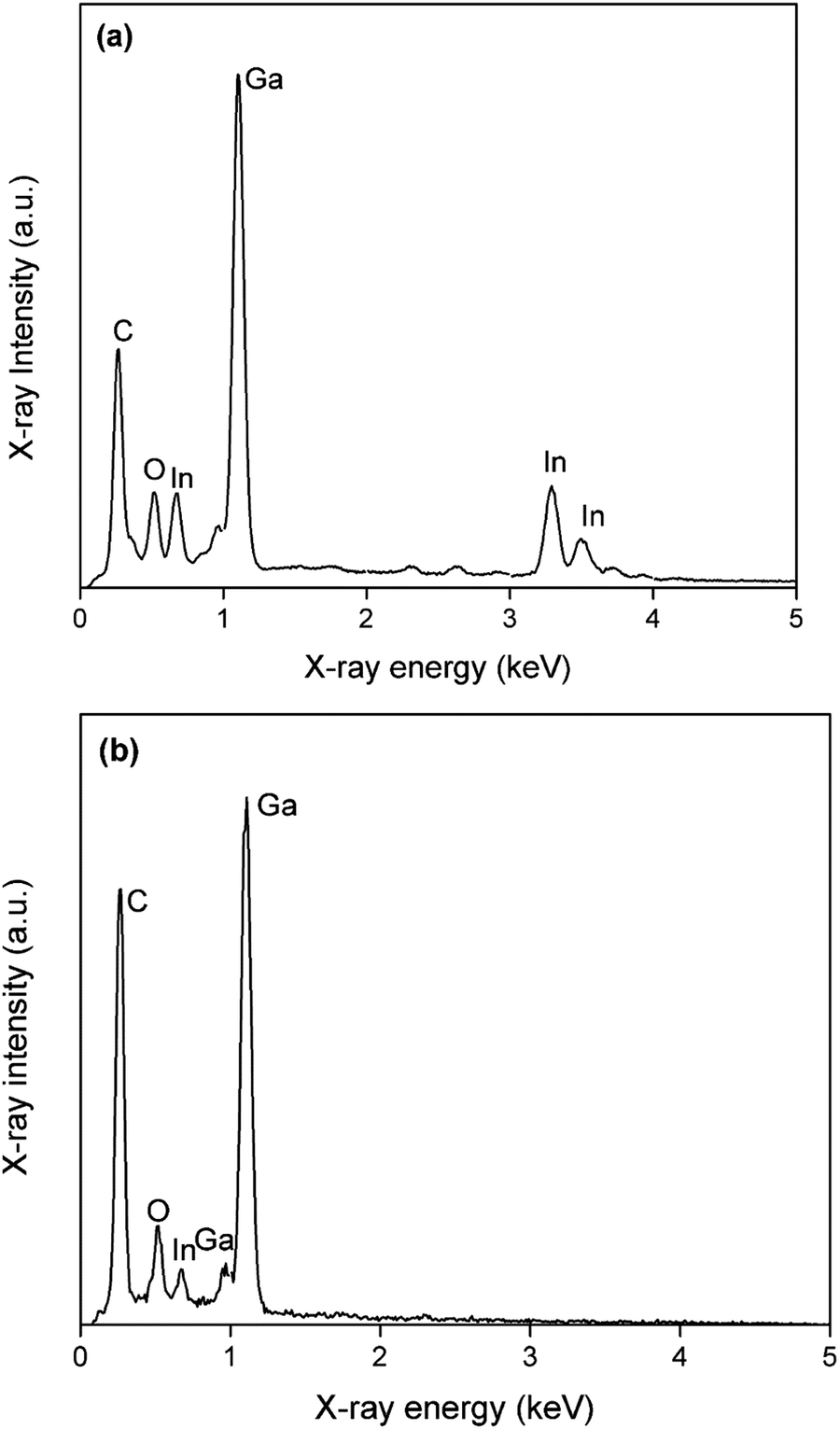
Electrodeposition of gallium was carried out on a GC substrate from a solution of DME containing 0.1 mol dm−3 of GaCl3 and 0.2 mol dm−3 of [TBA][ClO4] by applying −1.5 V vs. Fc+/Fc for 30 min at 26 °C. The resulting deposit is silvery-white in color. The morphology and elemental composition were examined using SEM and EDX, respectively. In Fig. 6, the SEM images show that the deposit is composed of spherical particles with diameters of several hundred nanometers which are stacked on top of each other. A SEM image of the spherical particles and the corresponding histogram of the size distribution of the particles can be found in figure S4 ESI.†Fig. 7 depicts the corresponding EDX spectrum, and shows major peaks for gallium and a small peak for carbon. As carbon can be attributed to the substrate, the spheres presumably consist entirely of gallium metal. However, studies investigating gallium and gallium–indium eutectics for liquid metal applications indicate that observed gallium spheres are typically covered by a gallium oxide shell.34–38 These shells form upon exposure of gallium to even trace amounts of oxygen, and prevent the individual gallium particles from agglomerating to larger-sized entities.39,40 As our recorded EDX spectrum does not show any peak for oxygen, these oxide shells are presumed to be very thin.
 |
| | Fig. 6 SEM images of gallium deposits generated from DME containing 0.1 mol dm−3 of GaCl3 and 0.2 mol dm−3 of [TBA][ClO4] by applying −1.5 V vs. Fc+/Fc for 30 min. on a GC WE at 26 °C. The applied acceleration voltage was 5 keV. | |
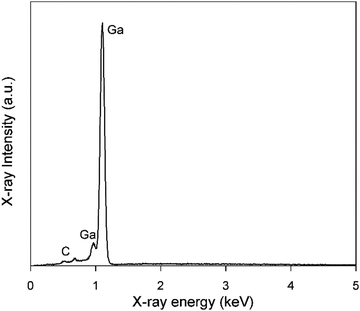 |
| | Fig. 7 EDX spectrum of gallium deposits generated from DME containing 0.1 mol dm−3 of GaCl3 and 0.2 mol dm−3 of [TBA][ClO4] by applying −1.5 V vs. Fc+/Fc for 30 min on a GC WE at 26 °C. The applied acceleration voltage was 10 keV. | |
XPS measurement
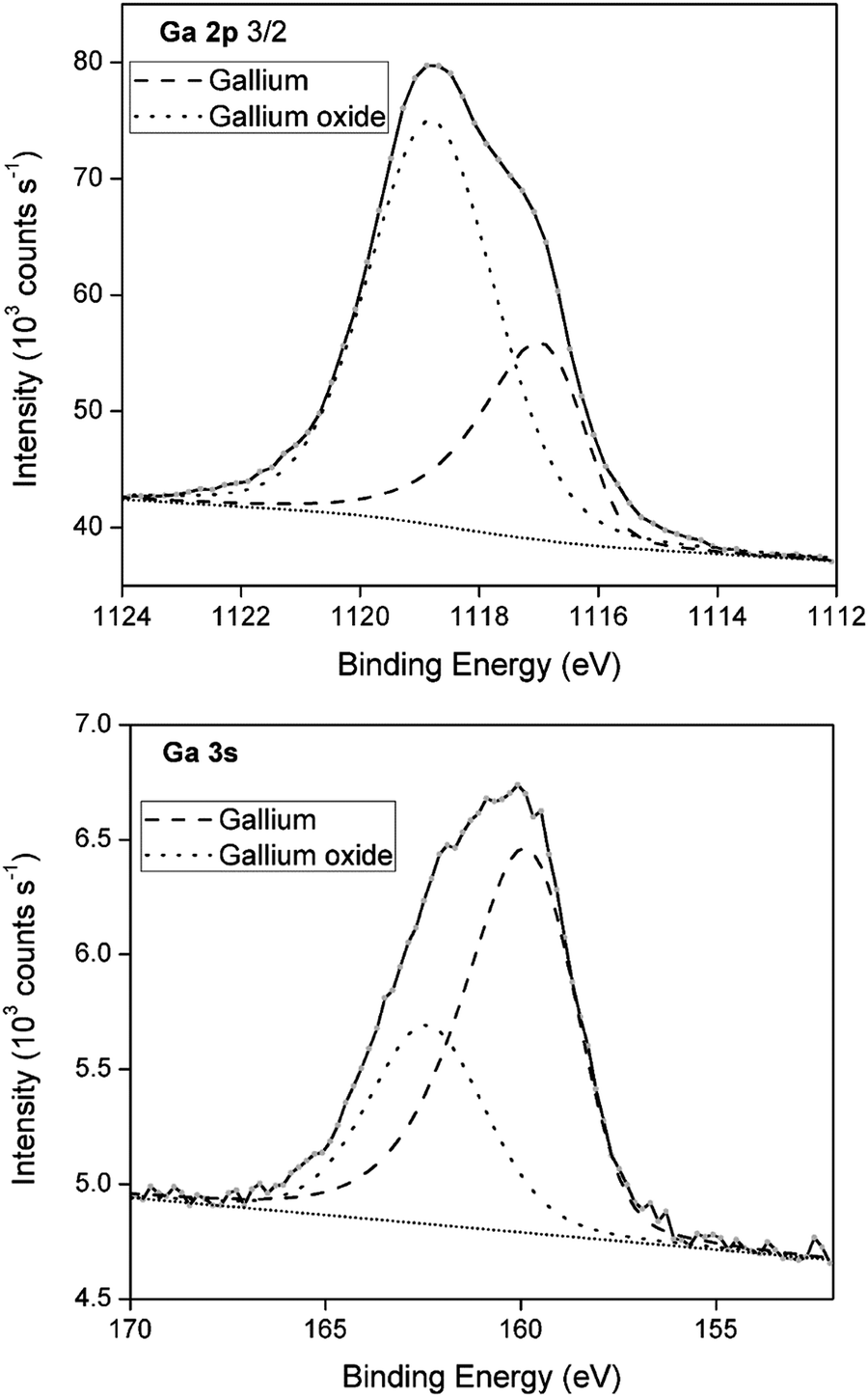
To demonstrate the presence of gallium oxide shells, X-ray photoelectron spectroscopy (XPS) was performed of a deposit generated under conditions analogous to those of the deposit analyzed for SEM/EDX. XPS provides information on the electron binding energies of the investigated elements, and thus their oxidation state. XPS spectra of the Ga 2p and Ga 3s region are shown in Fig. 8. The spectra are deconvoluted into two peaks representing gallium metal (dashed line) and gallium oxide (dotted line). For Ga 2p, the gallium oxide component corresponds to 71% of the multipeak, whereas for Ga 3s, a significantly lower value of 34% is found. Varying contributions of the gallium oxide peak in the two spectra can be attributed to the differing depths at which the signals were recorded. By calculating the electron kinetic energy (kinetic energy = photon energy (1487 eV) − core-level binding energy (1118 eV for Ga 2p and 161 eV for Ga 3s)), the sampling depths from which both spectra originate can be estimated using the universal electron escape depth curve.41 The Ga 2p spectrum originates from a depth of approximately 0.5–1 nm, whereas the Ga 3s spectrum originates from a slightly larger depth of roughly 2 nm. Clearly, a much larger contribution of gallium oxide is found near the surface of the spheres (Ga 2p) as opposed to slightly deeper in the bulk (Ga 3s). This is reminiscent of an oxide shell. As the penetration depth of XPS is at maximum 5 nm, and as signals for both gallium metal and gallium oxide are detected in the Ga 2p and Ga 3s spectra, it can be deduced that the gallium oxide shells have a thickness of only a few nanometers. This oxide layer does not grow very thick as the Pilling–Bedworth (PB) values for α-Ga2O3 and β-Ga2O3, equal 1.23 and 1.35, respectively. Typically, materials with PB values between 1 and 2 grow a metal oxide that provides a protecting coating against further surface oxidation, disabling growth of thick oxides. An XRD measurement was performed on a blank GC substrate and a deposit generated under analogous conditions to those of the samples used for XPS analysis (Fig. S5 ESI†). The diffractogram of the deposit reveals two broad peaks that can be attributed to the GC substrate, as well as one peak for the (002) orientation of Ga2O3. The broadness of this peak might indicate the poor crystallinity of the gallium oxide shells. Interestingly, no characteristic peaks for gallium metal were obtained. This is likely due to the (near)-liquid state of gallium cores.
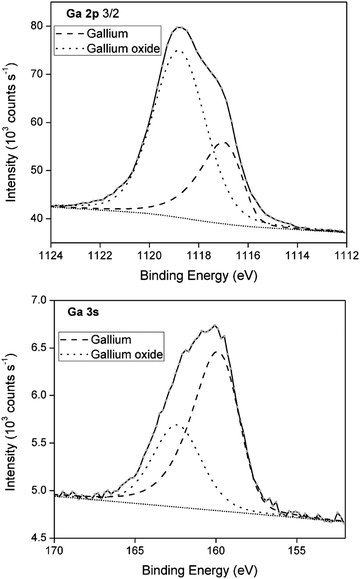 |
| | Fig. 8 Curve-fitted Ga 2p3/2 and Ga 3s XPS spectra from deposits generated from DME containing 0.1 mol dm−3 of GaCl3 and 0.2 mol dm−3 of [TBA][ClO4] by applying −1.5 V vs. Fc+/Fc for 30 min on a GC WE at 26 °C. | |
Deposition mechanism
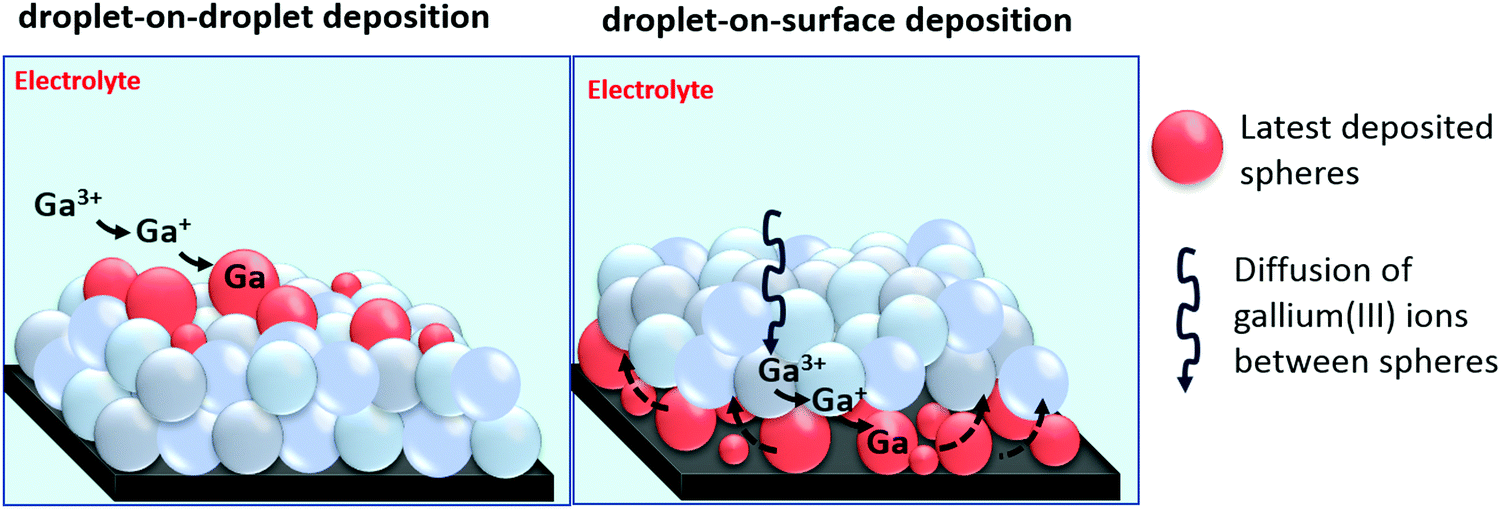
Combined results of SEM, EDX, XPS and XRD establish that gallium is electrodeposited on glassy carbon in the form of spheres that consist of a gallium metal core covered by a thin gallium oxide shell. Determining the exact nature of these oxide shells is not easy. However, it is possible to determine whether the oxide shells are electrically conductive or insulating by performing a series of electrochemical experiments. In case of conductive oxide shells, gallium spheres can be electrodeposited on top of each other, as electrons can pass through the oxide shells, enabling reduction of gallium(III) ions, via gallium(I) to gallium(0), subsequently leading to nucleation and growth of new spheres on top of the ones beneath. This mechanism is referred to here as droplet-on-droplet deposition. Furthermore, conductive oxide shells might enable stripping of electrodeposited gallium. In case of passivation, the oxide shells do not permit transfer of electrons. Consequently, reduction of gallium(III) cannot occur on the surfaces of the gallium spheres. However, as spheres never fully cover the surface of the substrate, gallium(III) ions can still migrate between the gallium spheres towards the substrate, at which they can be reduced to metallic state. The newly growing gallium spheres on the surface of the substrate can subsequently push up the previous deposited spheres, instigating the continuous growth of new layers of gallium spheres. We refer to this mechanism here as droplet-on-surface deposition. A graphical presentation of the two growth mechanisms is shown in Fig. 9.
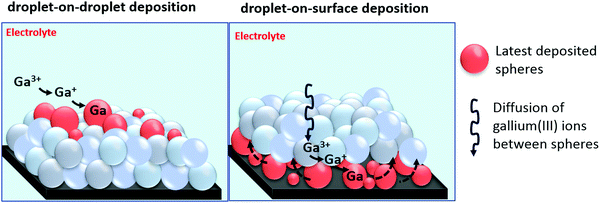 |
| | Fig. 9 Cartoon of the droplet-on-droplet and droplet-on-surface deposition mechanisms. | |
To validate which of the two mechanisms occurs, a first experiment was performed in which gallium was electrodeposited on a GC substrate from DME containing 0.1 mol dm−3 of GaCl3 and 0.2 mol dm−3 of [TBA][ClO4] by applying −1.5 V vs. Fc+/Fc for a short deposition time of 1 min. It was aimed to electrodeposit only a discrete number of spheres, and to observe whether nucleation and growth of new spheres would occur on top of existing ones, or on the GC substrate. Fig. 10 shows SEM images of the attained deposit. As intended, only three layers of spheres were electrodeposited on substrate. Small spheres can be observed on top of bigger entities (indicated by red circles), while none are present on the substrate. These are assumed to be growing gallium spheres, indicating droplet-on-droplet deposition, and accordingly, electrically conductive oxide shells.
 |
| | Fig. 10 SEM images of gallium deposits generated from DME containing 0.1 mol dm−3 of GaCl3 and 0.2 mol dm−3 of [TBA][ClO4] by applying −1.5 V vs. Fc+/Fc for 1 min on a GC WE at 26 °C. The applied acceleration voltage was 5 keV. | |
In a second experiment, gallium was first electrodeposited on a GC substrate from DME containing 0.1 mol dm−3 of GaCl3 and 0.2 mol dm−3 of [TBA][ClO4]by applying −1.5 V vs. Fc+/Fc for 2 min. To determine whether gallium can be stripped, an anodic overpotential of +0.2 V vs. Fc+/Fc was applied for 1 min. on the gallium deposit. Resulting SEM images are shown in Fig. 11. Rough structures can be observed on top of a layer of intact spheres. It is believed that these structures are the result of the partial stripping of the gallium core from the inside of the spheres. The image was focussed on the intact, lower spheres. Due to this height difference, the overall image appears slightly out of focus. The partly stripped structures consequently indicate that the oxide shells must enable the transfer of electrons. The corresponding EDX spectrum (Fig. S6 ESI†) shows a minor peak for oxygen alongside gallium and carbon. This implies that these rough structures consist of gallium oxide.
 |
| | Fig. 11 SEM images of gallium structures generated by first electrodepositing gallium from DME containing 0.1 mol dm−3 of GaCl3 and 0.2 mol dm−3 of [TBA][ClO4] by applying −1.5 V vs. Fc+/Fc for 2 min and then partly stripping the deposited gallium by applying +0.2 V vs. Fc+/Fc for 1 min at 26 °C. The applied acceleration voltage was 10 keV. | |
In a third experiment, gallium was first electrodeposited from DME containing 0.1 mol dm−3 of GaCl3 and 0.2 mol dm−3 of [TBA][ClO4] by applying −1.5 V vs. Fc+/Fc for 30 min., resulting in a thick layer of gallium spheres. Thereafter, gallium with a small amount of indium was electrodeposited on top of the initial layer of gallium spheres, from DME containing 0.1 mol dm−3 of GaCl3, 0.01 mol dm−3 of InCl3 and 0.2 mol dm−3 of [TBA][ClO4] by applying −1.5 V vs. Fc+/Fc for 5 min. Indium is highly soluble in gallium and can therefore act as a tracer, to determine whether indium–gallium alloys form on top of the initial layered gallium spheres (droplet-on-droplet deposition) or on the surface of the GC substrate (droplet-on-surface deposition) by means of EDX. Fig. 12 shows SEM images of (a) the top of the deposit, and (b) the bottom of the deposit which was initially in contact with the GC substrate. The latter was attained by removing the deposit, shown in (a) from the GC substrate by conductive Scotch tape. This exposes the bottom of the initial deposit (images of the deposit on the GC substrate, and of the bottom of the deposit that was removed by the Scotch tape are shown in Fig. S7 ESI†). While both images show multiple layers of spheres, (b) appears much rougher than (a). This is most likely due to the mechanical forces applied when removing the deposit with Scotch tape. EDX spectra of deposits (a) and (b) are shown in Fig. 13. Evidently, distinctive peaks for indium can be observed in the spectrum recorded from the top of the deposit (a) whilst only a single, small peak of indium is present in the spectrum from the bottom of the deposit (b). The clear difference in indium peaks in both spectra reveals that gallium–indium spheres form on top of the initial layer of gallium spheres, and that the droplet-on-droplet deposition is followed. The oxygen peaks in the spectra might suggest that indium–gallium alloys are more prone to oxidation as opposed to pure gallium. The carbon peaks in both spectra can be attributed the the GC substrate (a), and the conductive Scotch tape (b), respectively. The combination of the three experiments clearly indicates that the oxide shells are quite electrically conductive, and allow transfer of electrons, enabling electrodeposition and partial stripping of gallium (and gallium–indium) spheres on the spheres beneath. The gallium oxide covering the gallium sphere is therefore not crystalline Ga2O3, as this material does not exhibit electrical conductivity at room temperature. Undoped Ga2O3 has an ultrawide bandgap of 4.7–4.9 eV and it's electric conductivity at room temperature is extremely low.42,43 Instead, the observed oxide shells are presumed to be amorphous, and possibly non-stochiometric in nature. Nagarajan et al. showed that the electrical conductivity of non-stochiometric gallium oxide with an excess of gallium at room temperature can be as high as 105 Ω−1 m−1, many orders of magnitude higher than that of stochiometric Ga2O3.44
 |
| | Fig. 12 SEM image of (a) deposit generated by electrodeposition of gallium from DME containing 0.1 mol dm−3 of GaCl3 and 0.2 mol dm−3 of [TBA][ClO4] by applying −1.5 V vs. Fc+/Fc for 30 min followed by co-electrodeposition of gallium and indium from DME containing 0.1 mol dm−3 of GaCl3 and 0.2 mol dm−3 of TBAClO4 by applying −1.5 V vs. Fc+/Fc for 5 min at 26 °C. (b) bottom of this deposit, generated by removing the deposit in (a) with conductive Scotch tape and exposing the layer that was in contact with the GC substrate. | |
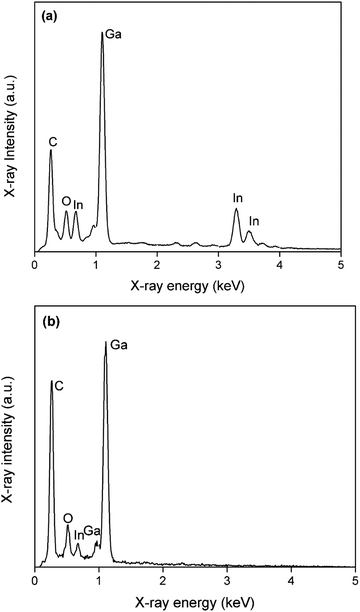 |
| | Fig. 13 EDX spectra of SEM images (a) and (b) in Fig. 11. The applied acceleration voltage was 10 keV. | |
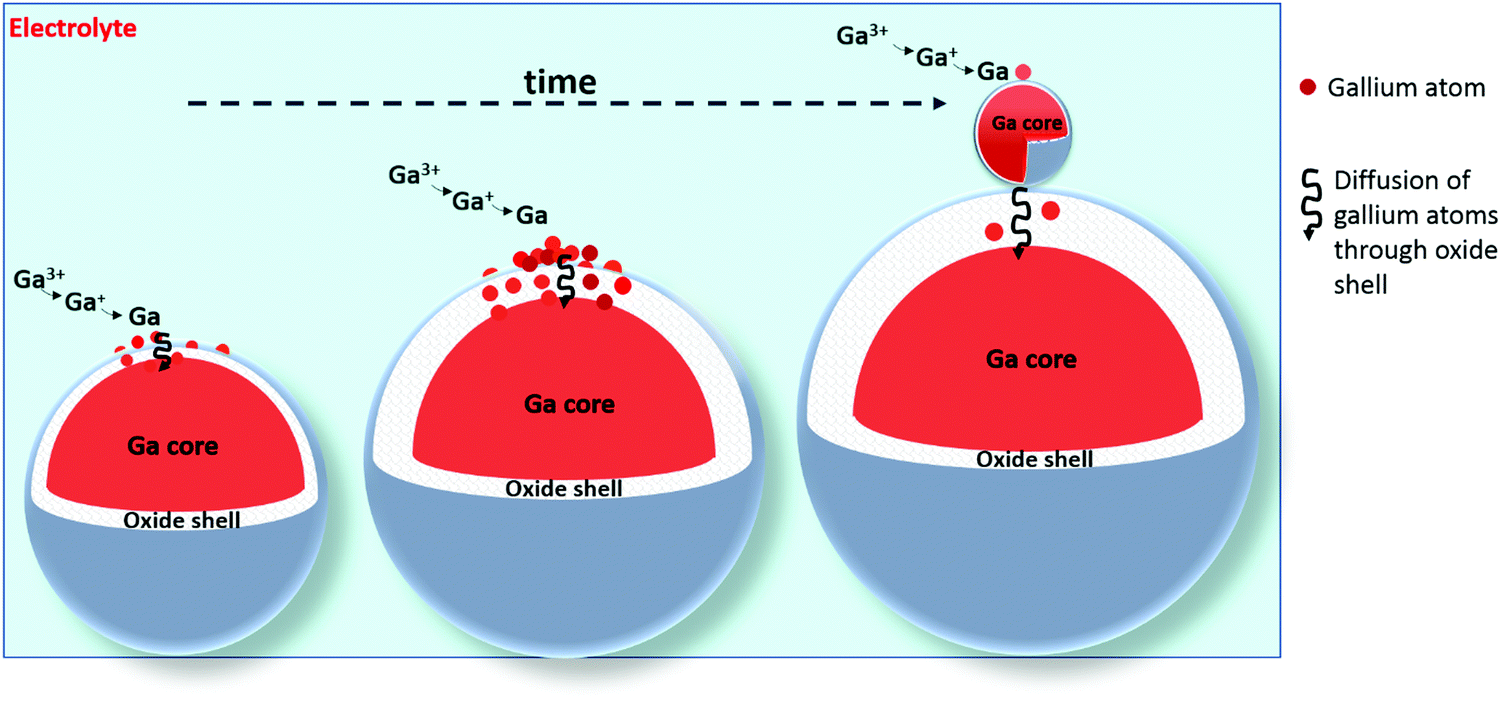
Although the oxide shells are electrically conductive, our data does not demonstrate how the gallium spheres themselves grow and why they stop growing after a certain time, at an approximate diameter of 200 nm. The electrodeposition of gallium spheres can occur according to two possible paths: gallium(III) ions can either reduce to gallium atoms on the surface of the oxide shells, after which the neutral gallium atoms diffuse through the thin shell, or (2) gallium(III) ions can first electromigrate through the thin oxide shell, and subsequently reduce inside the droplet at the gallium–gallium oxide interface. Both mechanisms lead to growth of the gallium droplets. In view of the large electrical conductivity of the oxide shells, the first path is the most likely one. During the growth of the gallium spheres, the thickness of the oxide shell increases and hence the diffusion of gallium atoms through it slows down. At a certain moment, the diffusion of gallium atoms through this oxide layer becomes so slow, that the concentration of gallium adatoms on top of the gallium spheres becomes high enough that a new gallium sphere nucleates. A graphical representation of this growth mechanism is shown in Fig. 14.
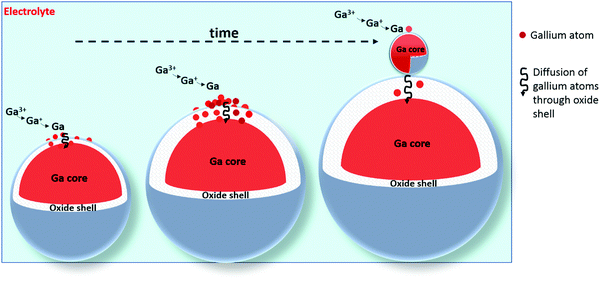 |
| | Fig. 14 Cartoon of the growth mechanism of individual spheres. During the growth of the gallium spheres, the thickness of the oxide shell increases due to exposure to oxygen in the electrolyte. Gallium atoms are electrodeposited, and diffuse through the oxide shell. As time progresses and the oxide shell thickness increases, diffusion of gallium atoms through this oxide layer becomes so slow, that the concentration of gallium adatoms on top of the gallium spheres becomes high enough that a new gallium sphere nucleates. | |
To validate the proposed growth mechanism, electrodeposition of gallium was carried out in an oxygen-rich electrolyte. The presence of oxygen in solution is presumed to increase the thickness of the oxide shells. Diffusion of gallium atoms through these thicker shells is slower. Therefore, the point of nucleation of a new sphere through the build-up of gallium adatoms on top of the shell occurs faster. Consequently, smaller gallium spheres are expected. DME was bubbled with pure oxygen gas for 30 minutes, and an electrolyte composed of this oxygen-rich DME, 0.1 mol dm−3 of GaCl3 and 0.2 mol dm−3 of [TBA][ClO4] was prepared. Gallium was electrodeposited on a GC substrate by applying −1.5 V vs. Fc+/Fc for 30 min. The attained deposit was analyzed using SEM, and compared with deposits generated from a non-bubbled electrolyte. The resulting SEM images are shown in Fig. 15. Clearly, the spheres attained from the O2-rich electrolyte (b) are smaller than those from the normal electrolyte (a), confirming the postulated growth mechanism.
 |
| | Fig. 15 SEM image of (a) deposit generated by electrodeposition of gallium from DME containing 0.1 mol dm−3 of GaCl3 and 0.2 mol dm−3 of [TBA][ClO4] by applying −1.5 V vs. Fc+/Fc for 30 min at 26 °C and (b) deposit generated by electrodeposition of gallium from O2-rich DME containing 0.1 mol dm−3 of GaCl3 and 0.2 mol dm−3 of [TBA][ClO4] by applying −1.5 V vs. Fc+/Fc for 30 min at 26 °C. | |
Conclusions
The electrochemical behavior and electrodeposition of gallium from an electrolyte composed of gallium(III) chloride in 1,2-dimethoxyethane (DME) was investigated. The occurring redox reactions were identified using EQCM and RRDE measurements. The gallium(III) species was first reduced to gallium(I). Further reduction of gallium(I) to gallium(0) required a substantial cathodic overpotential, indicating that this is the rate-limiting step. The diffusion coefficient of the gallium(III) species was determined by means of a Levich plot, equaling 2.6 × 10−10 m2 s−1 at 26 °C. SEM and EDX analysis indicated that metallic gallium is electrodeposited in the form of spherical particles with a diameter of several hundred nanometers. XPS demonstrated that these spheres are covered by thin gallium oxide shells. These shells prevent the gallium spheres from agglomerating and thus forming layers of gallium drops. Electrochemical deposition and stripping experiments of gallium, and of gallium in combination with an indium tracer indicated that a droplet-on-droplet deposition mechanism is followed. Consequently, these thin oxide shells must be electrically conductive. During the growth of the gallium spheres, it is postulated that the thickness of the oxide shell increases and hence the diffusion of gallium atoms that are initially electrodeposited on the surface slows down. After some time, diffusion of gallium atoms through this oxide layer becomes so slow, that the concentration of gallium adatoms on top of the gallium spheres becomes high enough that a new gallium sphere nucleates. The occurrence of this growth mechanism was evidenced by an experiment in which gallium was electrodeposited from an O2-containing electrode which led to gallium spheres with smaller diameters. This was due to the formation of thicker oxide shells, at which the point of formation of a new sphere occurs faster as diffusion of gallium adatoms through the thick shell is very slow.
Conflicts of interest
There are no conflicts of interest to declare.
Acknowledgements
WM thanks the Research Foundation Flanders (FWO) for a PhD grant (1SB8319N). The research was supported by the European Research Council (ERC) under the European Union's Horizon 2020 Research and Innovation Programme: Grant Agreement 694078 – Solvometallurgy for critical metals (SOLCRIMET). The authors also want to thank Merve Kübra Aktan for her assistance with the SEM measurements.
References
- S. F. Fang, K. Adomi, S. Iyer, H. Morkoc, H. Zabel, C. Choi and N. Otsuka, J. Appl. Phys., 1990, 68, R31–R58 Search PubMed.
- S. Strite and H. Morkoç, J. Vac. Sci. Technol., B: Microelectron. Nanometer Struct.–Process., Meas., Phenom., 1992, 10, 1237–1266 Search PubMed.
-
M. J. Scott, J. Li and J. Wang, in IEEE Power and Energy Conference at Illinois (PECI), IEEE, 2013, pp. 1–7 Search PubMed.
- J. Ramanujam and U. P. Singh, Energy Environ. Sci., 2017, 10, 1306–1319 Search PubMed.
- P. S. Dutta, H. L. Bhat and V. Kumar, J. Appl. Phys., 1997, 81, 5821–5870 Search PubMed.
- R. G. Bautista, JOM, 2003, 55, 23–26 Search PubMed.
- F. Lu, T. Xiao, J. Lin, Z. Ning, Q. Long, L. Xiao, F. Huang, W. Wang, Q. Xiao, X. Lan and H. Chen, Hydrometallurgy, 2017, 174, 105–115 Search PubMed.
- Y. Chung and C.-W. Lee, J. Electrochem. Sci. Technol., 2013, 4, 1–18 Search PubMed.
- W. M. Saltman and N. H. Nachtrieb, J. Electrochem. Soc., 1953, 100, 126 Search PubMed.
- M. Pourbaix and J. Burbank, J. Electrochem. Soc., 1964, 111, 14C Search PubMed.
- D. O. Flamini, S. B. Saidman and J. B. Bessone, J. Appl. Electrochem., 2007, 37, 467–471 Search PubMed.
- B. Hong, Y. Wang, X. Wei, Q. Huang, X. Wang, T. Fujita and Y. Wei, Hydrometallurgy, 2020, 105344 Search PubMed.
-
A. J. Bard and L. R. Faulkner, Electrochemical Methods, Fundamentals and Applications, John Wiley and Sons, New York, 1980 Search PubMed.
- R. K. McMullan and J. D. Corbett, J. Am. Chem. Soc., 1958, 80, 4761–4764 Search PubMed.
- A. Kader, Trans. SAEST, 1975, 10, 249–254 Search PubMed.
-
S. Miyake, Method for directly electrochemically extracting gallium from a circulating aluminate solution in the bayer process by eliminating impurities, US pat. US3677918A, 1972 Search PubMed.
- Z. Zhao, Y. Yang, Y. Xiao and Y. Fan, Hydrometallurgy, 2012, 125, 115–124 Search PubMed.
- R. Dorin and E. J. Frazer, J. Appl. Electrochem., 1988, 18, 134–141 Search PubMed.
- L. Liu, M. Wang, Z. Wang and Y. Zhang, Hydrometallurgy, 2014, 146, 76–81 Search PubMed.
- M. K. Carpenter and M. W. Verbrugge, J. Electrochem. Soc., 1990, 137, 123–129 Search PubMed.
- P. Chen, Y. Lin and I. Sun, J. Electrochem. Soc., 1999, 146, 3290–3294 Search PubMed.
- L. H. S. Gasparotto, N. Borisenko, O. Höfft, R. Al-Salman, W. Maus-Friedrichs, N. Bocchi, S. Z. El Abedin and F. Endres, Electrochim. Acta, 2009, 55, 218–226 Search PubMed.
- G.-B. Pan, O. Mann and W. Freyland, J. Phys. Chem. C, 2011, 115, 7656–7659 Search PubMed.
- K. R. Seddon, G. Srinivasan, M. Swadźba-Kwaśny and A. R. Wilson, Phys. Chem. Chem. Phys., 2013, 15, 4518–4526 Search PubMed.
- M. Steichen, M. Thomassey, S. Siebentritt and P. J. Dale, Phys. Chem. Chem. Phys., 2011, 13, 4292–4302 Search PubMed.
- J. C. Malaquias, D. Regesch, P. J. Dale and M. Steichen, Phys. Chem. Chem. Phys., 2014, 16, 2561–2567 Search PubMed.
- A. Bakkar and V. Neubert, J. Electroanal. Chem., 2020, 856, 113656 Search PubMed.
- Y. Choquette, G. Brisard, M. Parent, D. Brouillette, G. Perron, J. E. Desnoyers, M. Armand, D. Gravel and N. Slougui, J. Electrochem. Soc., 1998, 145, 3500 Search PubMed.
- S. Tang and H. Zhao, RSC Adv., 2014, 4, 11251–11287 Search PubMed.
- J. Zhang, M. An, Q. I. Chen, A. Liu, X. Jiang, S. Ji, Y. Lian and X. Wen, Electrochim. Acta, 2016, 190, 1066–1077 Search PubMed.
- K. K. Kanazawa and J. G. Gordon, Anal. Chem., 1985, 57, 1770–1771 Search PubMed.
- K. K. Kanazawa and J. G. Gordon II, Anal. Chim. Acta, 1985, 175, 99–105 Search PubMed.
- J. T. Edward, J. Chem. Educ., 1970, 47, 261 Search PubMed.
- M. A. Creighton, M. C. Yuen, M. A. Susner, Z. Farrell, B. Maruyama and C. E. Tabor, Langmuir, 2020, 36, 12933–12941 Search PubMed.
- S. Catalán-Gómez, A. Redondo-Cubero, F. J. Palomares, L. Vázquez, E. Nogales, F. Nucciarelli, B. Méndez, N. Gordillo and J. L. Pau, Nanotechnology, 2018, 29, 355707 Search PubMed.
- N. J. Morris, Z. J. Farrell and C. E. Tabor, Nanoscale, 2019, 11, 17308–17318 Search PubMed.
- S. Liu, S. N. Reed, M. J. Higgins, M. S. Titus and R. Kramer-Bottiglio, Nanoscale, 2019, 11, 17615–17629 Search PubMed.
- Z. J. Farrell and C. Tabor, Langmuir, 2018, 34, 234–240 Search PubMed.
- M. J. Regan, H. Tostmann, P. S. Pershan, O. M. Magnussen, E. DiMasi, B. M. Ocko and M. Deutsch, Phys. Rev. B: Condens. Matter Mater. Phys., 1997, 55, 10786 Search PubMed.
- D. Schmeisser and K. Jacobi, Surf. Sci., 1981, 108, 421–434 Search PubMed.
- C. J. Powell, J. Electron Spectrosc. Relat. Phenom., 1988, 47, 197–214 Search PubMed.
- J. B. Varley, J. R. Weber, A. Janotti and C. G. Van de Walle, Appl. Phys. Lett., 2010, 97, 142106 Search PubMed.
- N. Ueda, H. Hosono, R. Waseda and H. Kawazoe, Appl. Phys. Lett., 1997, 70, 3561–3563 Search PubMed.
- L. Nagarajan, R. A. De Souza, D. Samuelis, I. Valov, A. Börger, J. Janek, K.-D. Becker, P. C. Schmidt and M. Martin, Nat. Mater., 2008, 7, 391–398 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1cp01074c |
|
| This journal is © the Owner Societies 2021 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence a,
Pin-Cheng
Lin
a,
Pin-Cheng
Lin