Attosecond pump–attosecond probe spectroscopy of Auger decay†
Received
9th February 2021
, Accepted 14th May 2021
First published on 17th May 2021
Abstract
Attosecond pump–attosecond probe spectroscopy is becoming possible due the development of sub-femtosecond free electron laser (FEL) pulses as well as intense high-order harmonic generation-based attosecond sources. Here we investigate theoretically whether these developments can provide access to direct time-resolved measurement of Auger decay through detection of the total yield of an ionic decay product, in analogy to the photodissociation product detection in femtochemistry. We show that the ion yield based measurement is generally possible and in the case of the inner-valence hole decay can be background-free. Extensive first principles calculations are used to optimise the probe photon energies for a variety of prototypical systems.
1 Introduction
Upon extreme ultraviolet or X-ray photo-ionisation, an electron hole can be formed either below or above the double ionisation threshold. In the latter case (inner shell or inner sub-shell ionisation), the hole can decay by electron emission in what is called the Auger effect:1 an outer-shell electron fills the vacancy, and another outer-shell electron is ionised. The Auger decay is mediated by electron–electron repulsion and is a basic manifestation of electron correlation in nature. Since the electron hole can be energetically above several doubly ionised states, the Auger electron spectrum can contain energy separated lines (in atoms) or bands (in molecules due to vibrational degrees of freedom) corresponding to each of the open decay channels. Moreover, a core vacancy can be situated above the triple or quadruple ionisation threshold, in which case recombination of an outer shell electron into the vacancy can result in the simultaneous emission of two or three electrons, in processes known as double or triple Auger decay.2,3
A primary characteristic of the Auger process is its decay lifetime, which for light atoms is typically within a few-femtosecond or sub-femtosecond range. Auger electron spectroscopy does not give access to the lifetime if the Auger electron is coupled to other degrees of freedom exhibiting unbound motion. Indeed, in such cases, the widths of the peaks in its energy spectrum reflect energy partition due to this coupling rather than the lifetime broadening. For example, Auger decay in molecules with dissociative doubly ionised final states results in broad bands reflecting the dissociation dynamics.4 In such cases, time-resolved spectroscopy powered by the availability of attosecond pulses turns out to be a viable alternative to study Auger dynamics. Time-resolved spectroscopy of Auger dynamics is also needed to shed light on some electronic processes that remain poorly understood, for instance the ultra-fast super-Coster–Kronig decays,5 where photoionisation and the Auger decay can occur on the similar time scale, leading one to question the basic two-step picture of the process, or Coster–Kronig decays in the inner valence shell in molecules, where multiple overlapping resonances can lead to rich decay patterns that differ from a simple exponential decay.
The first time-resolved study of Auger decay, employing attosecond pump and IR (streaking) probe was conducted by Drescher and co-workers,6 see ref. 7 for the theoretical analysis. Although applications of the streaking technique to molecular photoionisation have been reported,8 the first attosecond streaking study of molecular Auger decay has been reported only very recently.9 The suggested alternative approaches to resolving Auger dynamics include high-order harmonic generation (HHG) spectroscopy based on the detection of HHG radiation caused by recombination of a photoelectron with the parent ion undergoing Auger decay,10 and molecular Auger interferometry.11 These proposals still await their experimental realisation.
The most straightforward way to observe Auger dynamics in time would be using the attosecond pump–attosecond probe capability that has recently become available.12,13 Moreover, the study of hole migration in molecules shows that the application of the attosecond pump–probe approach based on the single-photon laser enabled Auger decay (spLEAD)14–16 can lead to a background-free measurement based on the detection of a certain molecular charge state that is produced, independently of the energy of the product species. It is therefore natural to ask whether the same kind of capability can be achieved in the realm of Auger decay. Here we answer this question theoretically by considering both core and inner-valence processes in a variety of atomic and molecular species. We show that the background-free measurement of Auger decay based on a charge state detection is possible under some conditions for inner-valence vacancies. We analyse the optimal conditions for attosecond pump–probe Auger measurement both in the inner-valence and in the core and produce specific recommendations for future experimental work.
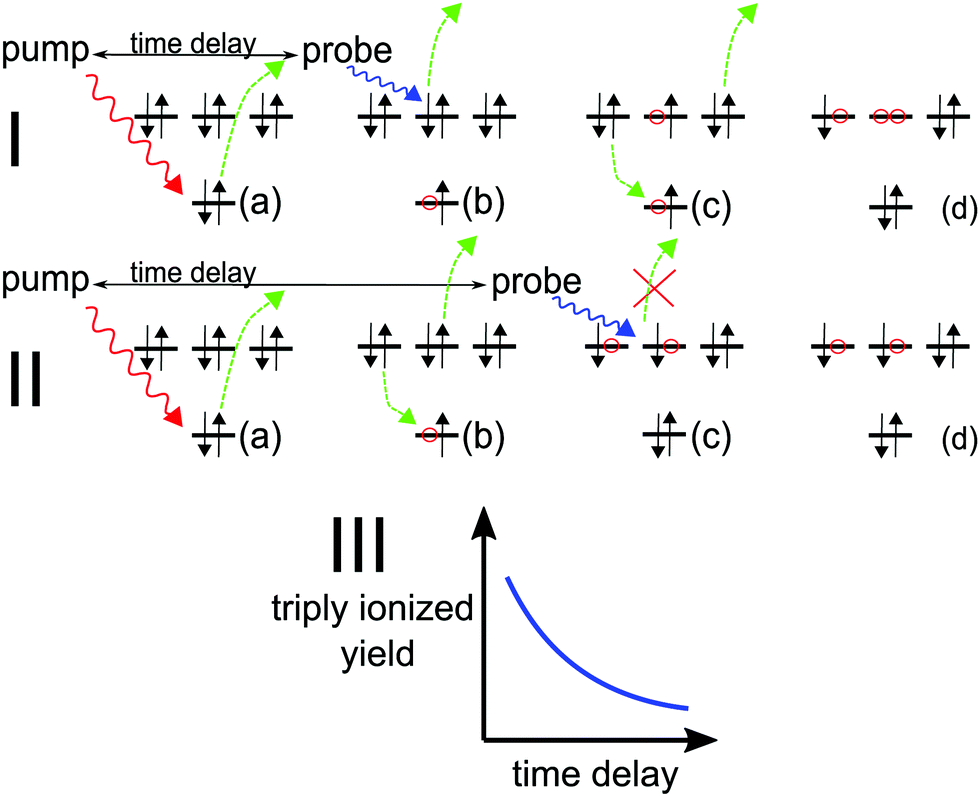
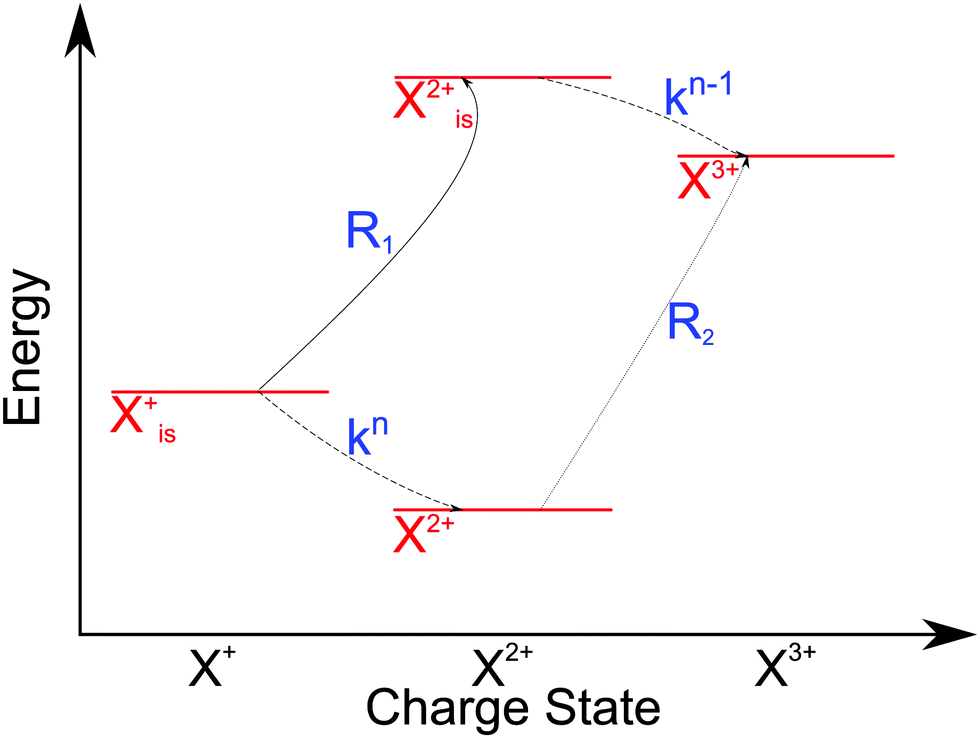
2 Auger pump–probe scheme
The pump–probe scheme to determine the time evolution of a decaying electronic system is graphically depicted in Fig. 1. A short (relative to the Auger lifetime), perturbative pump pulse ionises an electron from an inner (sub)shell (is) and a second pulse probes the decaying system selectively. The scheme thus requires that (i) the probe field photon has sufficient energy (ħω) to ionise the unstable system in the outer shell (os) before Auger decay has occurred (see Fig. 1, Ib),| | | IP(is−1) + ħω ≥ DIP(is−1os−1) | (1) |
(ii) The resulting inner-shell–outer-shell-ionised state is able to undergo further Auger decay to a triply ionised final product, i.e. the probe excites the initial ionic state to the doubly ionised state that lies above TIP (see Fig. 1, Ic),| | | IP(is−1) + ħω ≥ TIP(os−3) | (2) |
(iii) the photon does not have enough energy to ionise the system from the doubly ionised decay product after the Auger decay (see Fig. 1, IIc),| | | ħω ≤ Emin[(os−2) → (os−3)], | (3) |
where the functional Emin returns the lowest-energy dipole-allowed transition from any of the doubly ionised final Auger states to any of the triply ionised states that differ from it by only one hole.
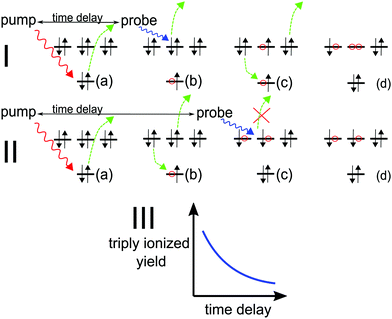 |
| | Fig. 1 (a) Pump pulse ionises the inner shell electron; (I.b–d) probe pulse precedes Auger decay and ionises from the outer valence, subsequent Auger decay produces X3+; (II.b–d) probe pulse follows Auger decay and is not energetic enough to further ionise the molecule, leaving it in the X2+ state. (III) Pump–probe time-delay dependence of the X3+ yield reflects the Auger decay time profile. | |
Provided all the above energy conditions are met, production of the triply ionised species (X3+) by the probe can only be a result of the probe pulse interacting with the system before it decays. Therefore, the change in the X3+ yield as a function of the pump–probe delay grants temporal insight into the Auger decay dynamics. For molecules that dissociate in high charge states, the parent (triply ionised) ion yield can be reconstructed by applying the coincidence17 or covariance mapping18 spectroscopy to the resulting fragments.
For a background-free measurement, the energy window defined by the conditions (i–iii) must be large enough to accommodate the energy bandwidth of the probe pulse, which in its turn should be large enough for the pulse to be short relative to Auger lifetime. A probe pulse duration of at most 0.5 fs and 1 fs (bandwidths of up to ∼4 eV) will be required for temporal resolution of inner-valence- and core-hole systems, respectively.
We assume that the probe pulse electric field has a sech envelope shape with a time bandwidth product of 1.9782:
| | 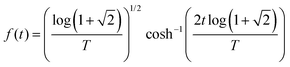 | (4) |
where
T corresponds to the Full Width Half Maximum (FWHM) of the pulse. As this pulse has tails that extend to infinity, we quantify the bandwidth (δ
ω) of the pulse that contains a fixed proportion (
P) of the pulse intensity as follows:
| | 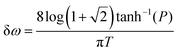 | (5) |
For example, P = 90% of the intensity of a 0.5 fs pulse is contained within the bandwidth of δω = 4.35 eV. If a smaller energy window is required due to the constraints in conditions (i–iii), one can choose a photon energy that is lower than the minimum energy threshold. For instance, the lower integration limit ω′ for the tail on the right that contains 10% of the pulse intensity 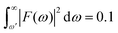 , where F(ω) is the pulse envelope's Fourier transform, is 1.623 eV (from the center of the distribution) and for the 1% tail the same quantity is 3.394 eV; the difference between these two quantities is just 1.771 eV. This would mean that if the system's energy gap was of this size, approximately 10% of the probe-ionised population would be above TIP and just 1% of the decay product in the DI state at the maximum energy threshold could still be ionised to X3+. A larger energy gap can reduce this residual X3+ signal even further.
, where F(ω) is the pulse envelope's Fourier transform, is 1.623 eV (from the center of the distribution) and for the 1% tail the same quantity is 3.394 eV; the difference between these two quantities is just 1.771 eV. This would mean that if the system's energy gap was of this size, approximately 10% of the probe-ionised population would be above TIP and just 1% of the decay product in the DI state at the maximum energy threshold could still be ionised to X3+. A larger energy gap can reduce this residual X3+ signal even further.
In what follows we will assume initial population of a single Auger-active state that decays exponentially and will study the possibility to recover the rate of the exponential decay using the suggested attosecond pump–probe technique. To this end, we will model the pump–probe experiment using rate equations which have proven to be very useful in describing the total yields of the atomic and molecular charge states in X-ray FEL experiments, see for example ref. 19 and 20. Generalisation of the attosecond pump–probe spectroscopy to non-exponential Auger dynamics requires a coherent quantum mechanical treatment and will be reported elsewhere. For considering the possible sources of background, it is important to distinguish between two cases: (i) where the ionised state lies between the double and triple ionisation potential (DIP and TIP) so that it can only undergo single Auger (Coster–Kronig21) decay, as typical for inner-valence holes, e.g. O(2s), in polyatomic molecules; and (ii) core-hole systems that can also undergo double Auger decay.
2.1 Inner valence hole decay
We first formulate the rate equations for the case of a single inner-valence hole state populated by the pump pulse in the most general case where the energy window dictated by eqn (2) and (3) may not be large enough to ensure no probe ionisation of the X2+ final decay product:| | 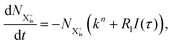 | (6) |
| | 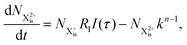 | (7) |
| | 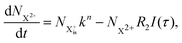 | (8) |
| | 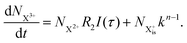 | (9) |
Here, 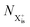 is the population of the initial sub-shell (inner-valence) hole state,
is the population of the initial sub-shell (inner-valence) hole state, 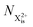 is the population of the DI (inner-outer-valence ionised) state, and NX2+ and NX3+ are the populations of the stable final X2+ and X3+ states; I(τ) is the normalised intensity envelope function of the probe pulse centred at τ = t − t′ for a time delay of t′ [cf.eqn (4)], R1 and R2 are the ionisation rate coefficients from the inner-valence-hole state and the final Auger decay states (X2+), respectively, and kn is the Auger decay rate for n electrons in the valence. In the fully detailed description, every ionisation state consists of a series of individual quantum states, each possessing their own ionisation and/or decay rate. Here we are not interested in such individual state populations, which can only be reliably computed using fully coherent time-dependent Schrödinger equation approach, but rather in the total population of the singly, doubly and triply charged states. Therefore, the decay rates and photoionisation rate coefficients in eqn (6)–(9) should be thought of as averages over the individual quantum states contributing to these ionic populations. Furthermore, we assume that the pump pulse is chosen such that the shake-up and shake-off populations are negligible, as is the effect of the post-collision interaction (PCI). These effects will be considered in Section 4. The scheme of the transitions described by the rate eqn (6)–(9) is given in Fig. 2.
is the population of the DI (inner-outer-valence ionised) state, and NX2+ and NX3+ are the populations of the stable final X2+ and X3+ states; I(τ) is the normalised intensity envelope function of the probe pulse centred at τ = t − t′ for a time delay of t′ [cf.eqn (4)], R1 and R2 are the ionisation rate coefficients from the inner-valence-hole state and the final Auger decay states (X2+), respectively, and kn is the Auger decay rate for n electrons in the valence. In the fully detailed description, every ionisation state consists of a series of individual quantum states, each possessing their own ionisation and/or decay rate. Here we are not interested in such individual state populations, which can only be reliably computed using fully coherent time-dependent Schrödinger equation approach, but rather in the total population of the singly, doubly and triply charged states. Therefore, the decay rates and photoionisation rate coefficients in eqn (6)–(9) should be thought of as averages over the individual quantum states contributing to these ionic populations. Furthermore, we assume that the pump pulse is chosen such that the shake-up and shake-off populations are negligible, as is the effect of the post-collision interaction (PCI). These effects will be considered in Section 4. The scheme of the transitions described by the rate eqn (6)–(9) is given in Fig. 2.
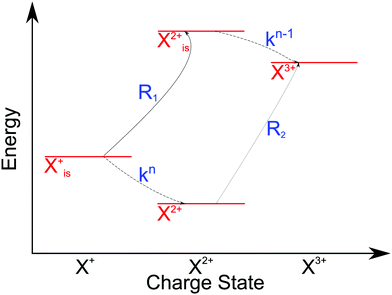 |
| | Fig. 2 Transition scheme of the attosecond pump–attosecond probe experiment for the inner-valence ionisation case, not taking into account shake-up and post-collision interaction effects. The scheme corresponds directly to the rate eqn (6)–(9). | |
Approximating the envelope function of the probe pulse with I(t) = δ(t), we obtain the final populations 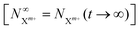 as a functions of the time delay as:
as a functions of the time delay as:
| | 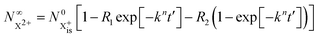 | (10) |
| | 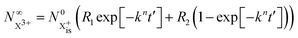 | (11) |
where
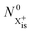
is the inner-valence-hole population at
t = 0. The
R2 rate coefficient is defined as the proportion of final doubly ionised states that can still be ionised by the probe multiplied by the ionisation rate for these states; if the energy gap is wide enough, this parameter will be zero and the X
3+ yield will be background free, if it is not, there will be a residual signal proportional to
R2. Even for the latter case, the X
3+ (and also X
2+) population will depend exponentially on the time delay, allowing one to recover the Auger decay rate from the attosecond pump–probe measurement of the total ion yield.
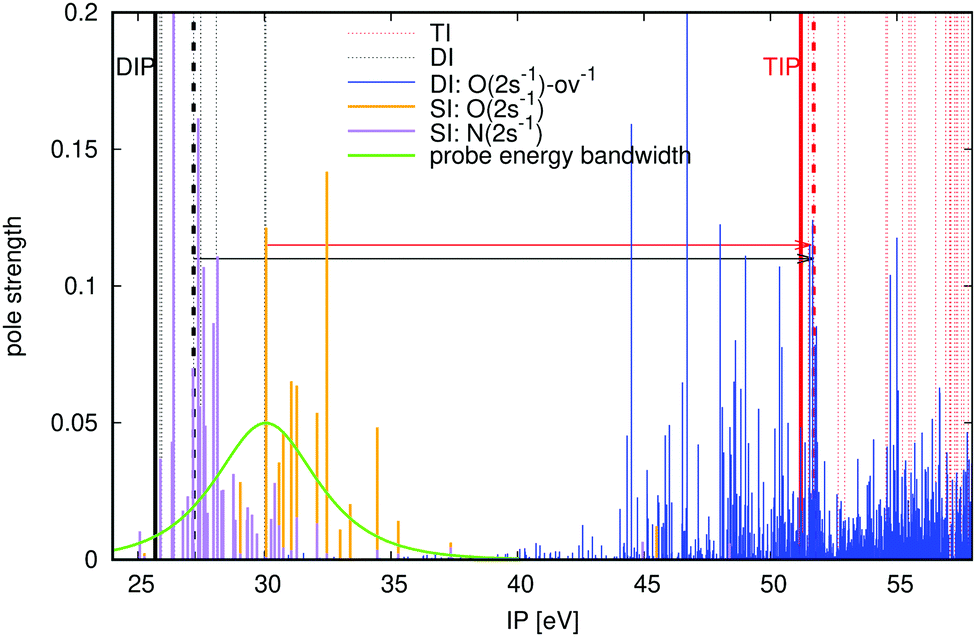
In order to demonstrate the viability of the proposed pump–probe scheme, we consider O 2s Coster–Kronig decay in the urea molecule CO(NH2)2. The relevant energies of urea at equilibrium geometry are shown in Fig. 3 (all ab initio calculations performed in this paper use the extended second-order algebraic diagrammatic construction [ADC(2)x, also known as ADC(2)-E] schemes for the singly, doubly and triply ionised states,22–24 see Section 3 for the geometries and the basis sets). Our pump pulse of ∼30 eV central frequency can also populate the nitrogen Auger active hole (shown in purple), but these initial states are either incapable of resulting in an X3+ final state for the chosen probe photon energy, or have negligible pole strengths (see Fig. 3). The possible intermediate O 2s-ov-ionised states that can then be populated by the probe are shown in Fig. 3 in blue. There is a high density of such states on either side of the TIP at 51.20 eV, so a probe energy that is energetic enough to reach the TIP will reliably populate Auger active intermediate states, satisfying the condition of eqn (2). The probe photon energy must be chosen to be at least 21.57 eV in accordance with eqn (1). The upper limit on the photon energy (solid black arrow) for urea is determined by the lowest-energy one-electron dipole-allowed transition (51.71 − 27.20 eV = 24.51 eV) from the doubly ionised final Auger states to triply ionised states that differ by only one hole. We therefore arrive at the probe photon energy window of 24.51 − 21.57 = 2.94 eV.
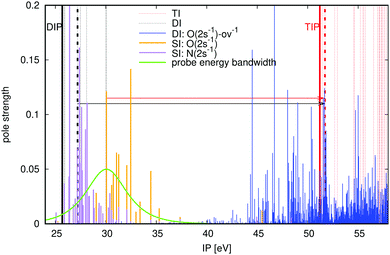 |
| | Fig. 3 ADC(2)x calculated ionisation energies and pole strengths of urea molecule. Electronic states legend: inner-valence-ionised states in the 25–35 eV region are orange lines for O 2s and purple lines for N 2s; inner-outer-valence-ionised states (where inner valence is O 2s) above 45 eV are blue lines; triply ionised states in the outer valence region are red dashed lines; doubly ionised Auger decay products below 30 eV are black dashed lines. The thick dashed black and red lines denote the specific states that define the lowest-energy critical transition for the probe, see eqn (3). The lower and upper limits on the probe photon energy are denoted with solid red and black arrows, respectively. The dashed arrows and associated probe energy bandwidths (0.5 fs pulse duration) show the probe transitions. DIP and TIP are labeled. | |
The probe energy bandwidth for a duration of 0.5 fs is superimposed on Fig. 3 for a photon energy below the minimum, such that 10% of the pulse intensity lies above the threshold of eqn (3). The equivalent percentage of Auger final states that the pulse ionises for this energy gap is just 0.079%, such that the resulting X3+ background is negligible. It should be borne in mind that fast Coster–Kronig decay processes can require shorter probe pulse durations: for a 0.25 fs duration, the residual X3+ signal is almost 10 times higher at 0.62%.
2.2 Single state core hole decay
For the core-hole states, the X3+ yield within the proposed scheme receives contributions from two pathways: single Auger decay (SA) from the core-valence doubly-ionised state produced by the probe and double Auger decay (DA) from the core-ionised state, and is therefore a priori not background free. The probability ratio between single and double Auger decay is between 5–20% for atoms and small molecules.25 A fitted trend, reported in ref. 25, shows that the double Auger decay probability P(DA) has a linear dependence on the number of closest neighboring valence electrons Nve of the form P(DA) = 0.415Nve + 5.46.
Besides the double Auger background, the X3+ yield in core-ionised systems can be further compromised by photo-ionisation of the intermediate states of the sequential double Auger decay. On the other hand, the effect of shake-off or shake-up caused by the pump photo-ionisation and the PCI recapture of the probe photoelectron by the Auger electron can be eliminated by carefully choosing the energy of the pump and probe photons; see Section 4 for detailed discussion.
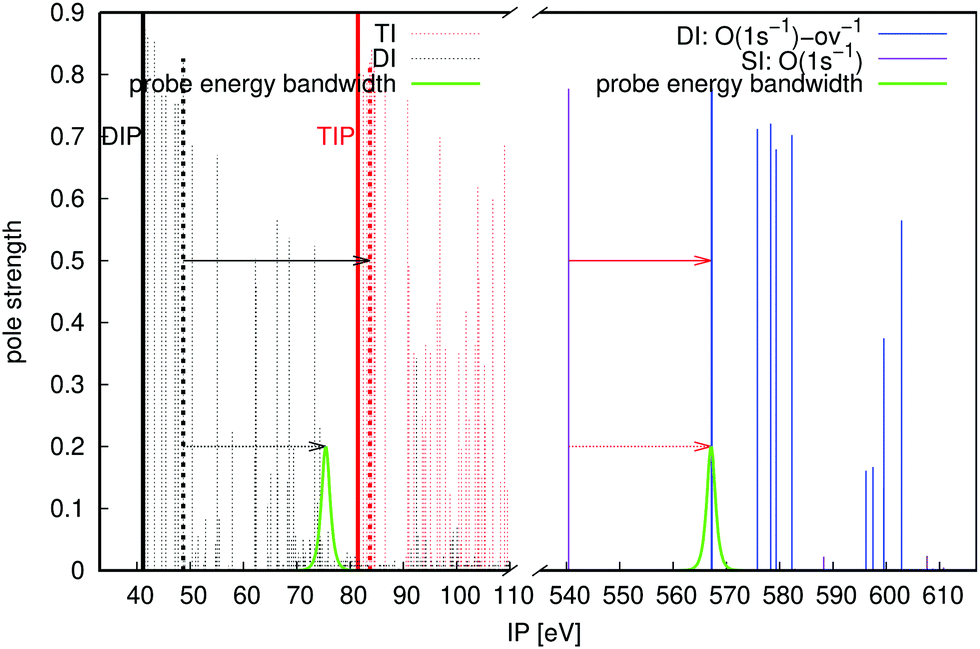
We illustrate the core-hole pump–probe spectroscopy using the O 1s decay in CO, see Fig. 4. The targeted Auger active state is at 540.38 eV above the ground state of the neutral at the equilibrium bond length of the molecule. The nearest intermediate core-valence-ionised state dictates the minimal probe energy of 26.76 eV (solid red arrow). The upper limit on the photon energy (solid black arrow) is 35.04 eV (see eqn (3)). This leaves an ample energy window of 8.24 eV, enough to accommodate the bandwidth of the 1 fs probe pulse, short on the time scale of the Auger decay.
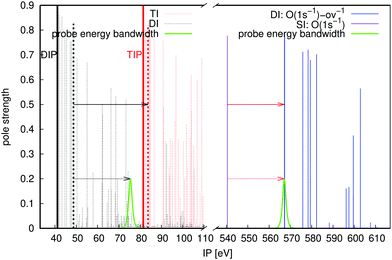 |
| | Fig. 4 Energies and pole strengths of different states for the CO molecule, and energy windows; set out in the same scheme as Fig. 3. The probe energy bandwidths correspond to a 1 fs pulse duration. | |
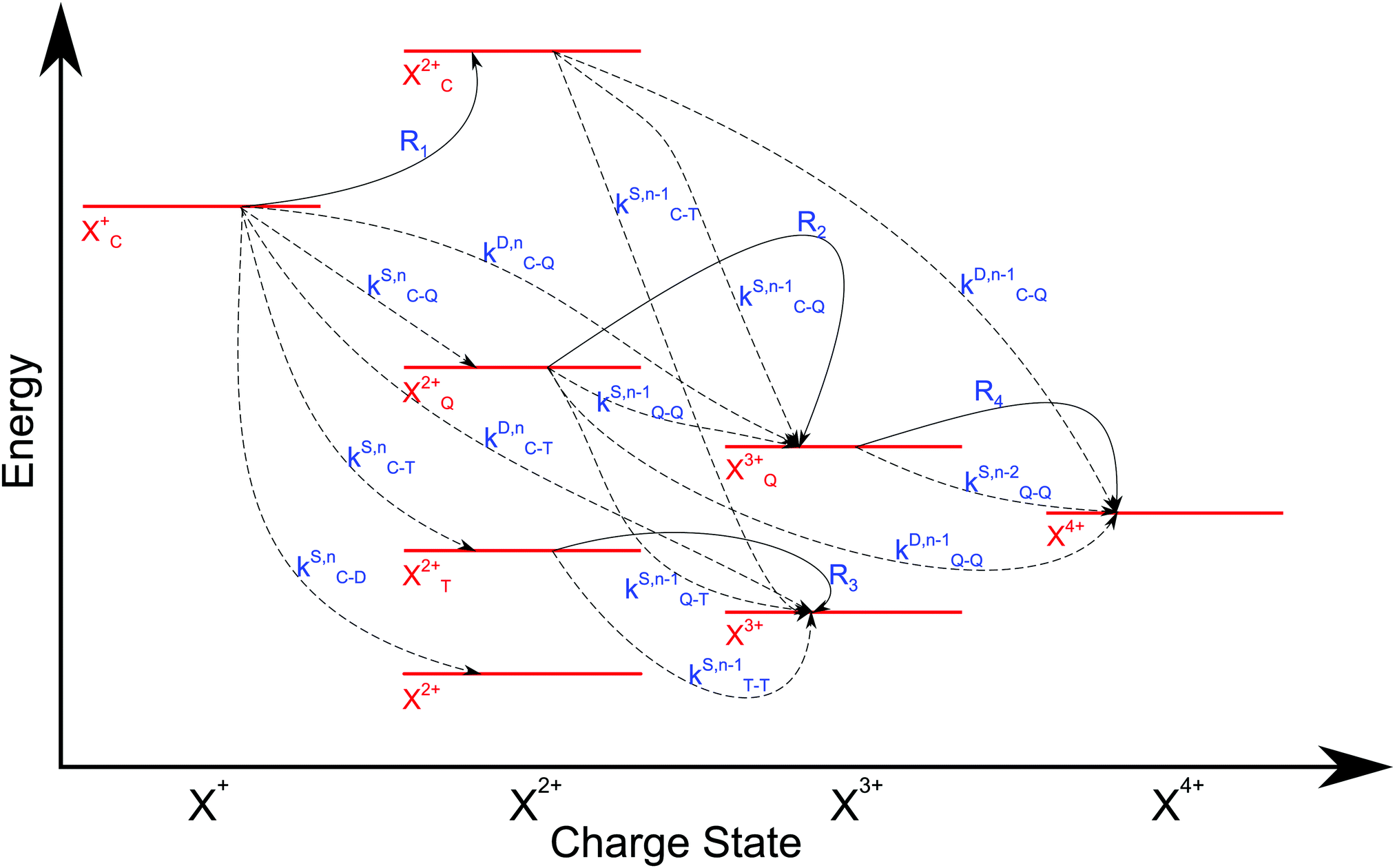
We now formulate the core-hole state's rate equations (assuming the energy of the initial state lies above the QIP), taking all possible decay pathways into consideration. The possibility of sequential double or triple Auger decay depends on the partial widths of the preceding stage(s) of decay: for example, if the normal Auger decay of a core-hole leads to a final state with energy above TIP, then a further decay to X3+ will be energetically allowed. Moreover, if the energy of the excited doubly charged ion after the first stage of decay remains above QIP, then a further two sequential decay processes can occur resulting in the X4+ species, provided that the first of these two decays keeps the ion energy above QIP (otherwise it will reach the X3+ final state).
We subdivide the NX2+ population into the following categories: 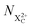 is the population of the X2+ system where the initial Auger active core-hole is still present (denoted by C) and there is an additional hole in the valence caused by the ionising probe pulse;
is the population of the X2+ system where the initial Auger active core-hole is still present (denoted by C) and there is an additional hole in the valence caused by the ionising probe pulse; 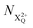 and
and 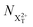 refers to the system where the core-hole has moved to a hole of lower energy, but the ion's overall energy remains above the QIP threshold or between the TIP and QIP threshold, respectively; and NX2+ refers to the system's final state where the two holes are both in the valence and the system is no longer energetically capable of further decay (the total ion energy is therefore below TIP). In a similar vein, we differentiate the X3+ population into the sub-population
refers to the system where the core-hole has moved to a hole of lower energy, but the ion's overall energy remains above the QIP threshold or between the TIP and QIP threshold, respectively; and NX2+ refers to the system's final state where the two holes are both in the valence and the system is no longer energetically capable of further decay (the total ion energy is therefore below TIP). In a similar vein, we differentiate the X3+ population into the sub-population 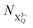 where the total ion energy is above QIP (and therefore still capable of further decay to the X4+ species), and the final decay product of NX3+.
where the total ion energy is above QIP (and therefore still capable of further decay to the X4+ species), and the final decay product of NX3+.
As well as the possible sequential Auger decay processes that can occur between the populations delineated above, there is also the possibility of simultaneous double Auger decay occurring where the two electrons share the available energy with a continuous distribution. This latter event can only happen to the initial state or the 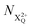 population; the latter's total ionic energy is above the QIP threshold, thus enabling the decay process
population; the latter's total ionic energy is above the QIP threshold, thus enabling the decay process 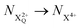 to take place.
to take place.
We define the ionisation rate coefficient from the core-hole as R1, the ionisation rate coefficient from the X2+ species with total energy above TIP 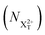 and QIP
and QIP 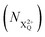 as R2 and R3, respectively, and the ionisation rate coefficient from the X3+ species with total energy above QIP
as R2 and R3, respectively, and the ionisation rate coefficient from the X3+ species with total energy above QIP 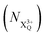 as R4. The rate equations governing the core-hole decay are then given as:
as R4. The rate equations governing the core-hole decay are then given as:
| | 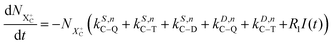 | (12) |
| | 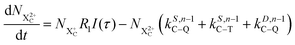 | (13) |
| | 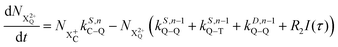 | (14) |
| | 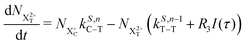 | (15) |
| | 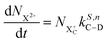 | (16) |
| | 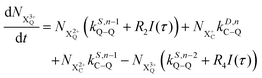 | (17) |
| | 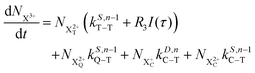 | (18) |
| | 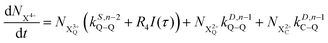 | (19) |
I(
τ) is the probe pulse envelope that is centered on
τ =
t −
t′ for a time delay of
t′. All the relevant Auger decay rates are designated as
k; the superscript specifies the type of Auger decay (single or double) and indicates the number of valence electrons (where
n corresponds to the neutral species), and the subscript denotes the specific transition type: Q, T and D means that the unstable hole is in a position such that the total ion energy is above the specified threshold (QIP, TIP and DIP, respectively) but below the next ionisation threshold, and subscript C denotes populations of the states with the original Auger active core-hole present. So for instance:
kD,n−1C–T refers to the simultaneous double Auger decay rate (
D) from an ion with one valence hole (
n − 1). The specific transition that this rate determines (C–T) is from the initial core-state to a state where the total ion energy is above TIP but below QIP. The possibility of simultaneous triple Auger decay is not considered in this model, as its rate is about two orders of magnitude smaller than the double Auger rate.
3 The scheme of the transitions described by the rate
eqn (12)–(19) is given in
Fig. 5.
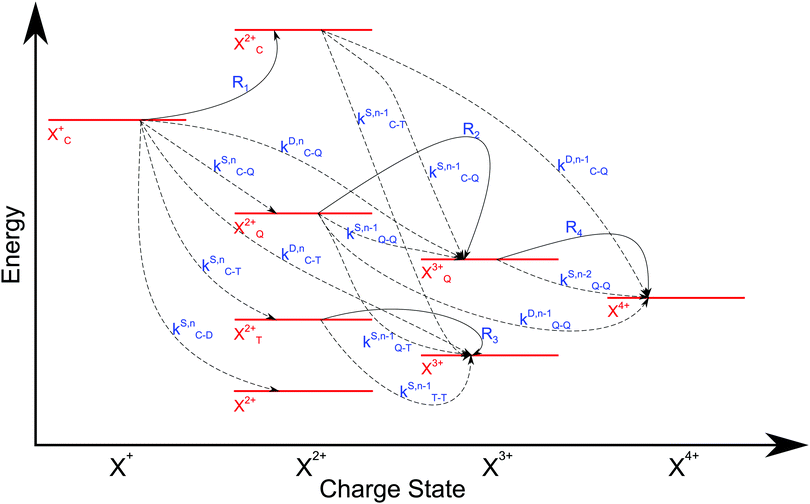 |
| | Fig. 5 Transition scheme of the attosecond pump–attosecond probe experiment for the core ionisation case, not taking into account shake-up and post-collision interaction effects. The scheme corresponds directly to the rate eqn (12)–(19). | |
The equations above assume that the probe cannot affect the final populations of NX4+, NX3+ and NX2+, but will ionise all states that can still decay, e.g. X2+ final Auger states that lie above TIP. There is a broader assumption: the probe has insufficient energy to shift the ion's energy from lying above one potential threshold (TIP) to another (QIP). This assumption can actually be relaxed, which requires the addition of an extra term to eqn (16)
where the quantity
R5 includes the proportion of the final X
2+ decay product that can be probe-ionised above the TIP threshold. This term would also need to be added to
eqn (18) with the opposite sign. A similar adjustment can also be made to the X
3+ population term in
eqn (18)where
R6 includes the proportion of the final X
3+ decay product that can be probe-ionised above the QIP threshold, and this term must likewise be added to
eqn (19) with the opposite sign. However, core-hole systems tend to have longer decay time constants than inner valence systems, so a longer probe pulse duration can be afforded with a smaller energy bandwidth. Additionally, the core-hole energetics are usually more favourable for large energy gaps. For these two reasons, we can in general safely assume that the photon energy constraints are met in their entirety and
R5 and
R6 in the extra terms
eqn (20) and (21) respectively, can be set to zero.
We therefore have nine single Auger decay rates and four double Auger decay rates to be determined in principle. However, we are actually interested in the total decay rate from the initial state, defined as the sum of the partial rates given in eqn (12):
| | | knC–t = kS,nC–Q + kS,nC–T + kS,nC–D + kD,nC–Q + kD,nC–T | (22) |
Approximating the envelope function of the probe pulse as δ(τ), we determine the calculated end population for 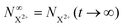 to be
to be
| | 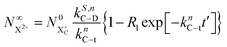 | (23) |
The end population for 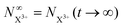 is
is
| | 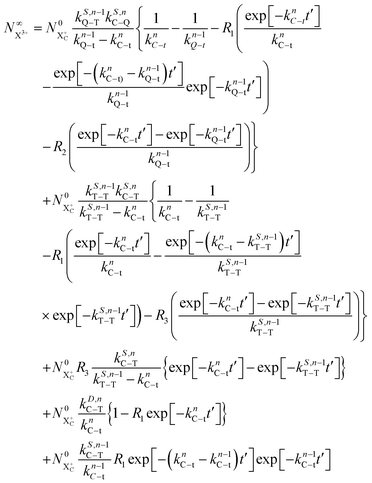 | (24) |
The resulting expression for the 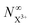 yield does not exhibit a simple exponential decay due to the competing triple ionisation pathways unaccounted for by the simple scheme in Fig. 1. However, there is only one path that contributes to the
yield does not exhibit a simple exponential decay due to the competing triple ionisation pathways unaccounted for by the simple scheme in Fig. 1. However, there is only one path that contributes to the 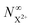 yield, and its final population as a function of the probe time delay has a simple exponential time dependence revealing the system's total decay rate, which we aim to measure.
yield, and its final population as a function of the probe time delay has a simple exponential time dependence revealing the system's total decay rate, which we aim to measure.
As mentioned earlier, the requirement of the upper limit on the photon energy can be relaxed for the core-hole system; as long as there is some subset of final doubly ionised states that cannot be further ionised by the probe, even if some can be ionised to X3+ states, the 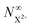 population will still exhibit the same simple exponential dependence. In such a case, and using eqn (20), we modify eqn (23) to
population will still exhibit the same simple exponential dependence. In such a case, and using eqn (20), we modify eqn (23) to
| | 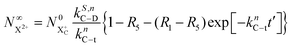 | (25) |
As in
eqn (20), (
R5) is the ionisation rate coefficient from
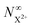
and is defined as the proportion of final DI states that can still be ionised by the probe multiplied by the ionisation rate coefficient for these states. The difference between
R1 and
R5 must, however, be large enough for the measurement of
knC–t to remain feasible.
In Fig. 6, we show the time-delay dependence of the 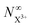 and
and 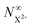 quantities, obtained by solving numerically eqn (12)–(19) for a 0.1 fs (FWHM) duration sech probe pulse and a variety of kntot values within the 0.1–1.0 fs−1 range, as well as the analytical exponential solution of eqn (23). It can be seen that the latter, at time-delays sufficiently large for the whole probe pulse to occur after the pump ionisation event (t = 0), agrees well with the corresponding numerical solution.
quantities, obtained by solving numerically eqn (12)–(19) for a 0.1 fs (FWHM) duration sech probe pulse and a variety of kntot values within the 0.1–1.0 fs−1 range, as well as the analytical exponential solution of eqn (23). It can be seen that the latter, at time-delays sufficiently large for the whole probe pulse to occur after the pump ionisation event (t = 0), agrees well with the corresponding numerical solution.
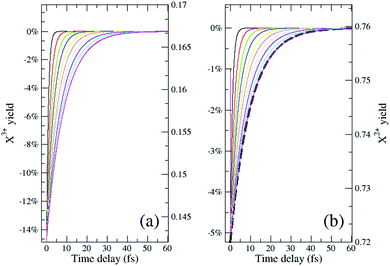 |
| | Fig. 6 The final yield (measured as a percentage of the no-probe yield on the left axis and in absolute terms on the right axis) of the ionised decay products (a) NX3+, and (b) NX2+, as a function of the pump–probe delay. The ionisation rate coefficients in eqn (12)–(19) are assumed to be: R1 = 20%, R2 = 15%, R3 = 10%, R4 = 10% and the probability of (direct and sequential) double and (sequential only) triple Auger are set to 24.5% and 0.5% of the single Auger probability. Different curves correspond to distinct kntot values within the 0.1–1.0 fs−1 range. The numerical results model the probe pulse as a 0.1 fs (FWHM) duration sech pulse. The solution of eqn (23) for knC–t = 0.1 fs−1 is shown in (b) by the dashed line. | |
Although there is a large background for the measurement of 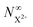 , the extent of variation on this signal is equivalent to the probe's ionisation rate coefficient R1 on the core-hole population (or R1 − R5 for the case described by eqn (25)) as can be seen by the dashed curve in Fig. 6. R1 is dictated by the intensity of the laser pulse and should be chosen as large enough to enable accurate experimental measurement of the variation of the signal over the background, but not too large so as to activate multi-photon transitions. An estimated value for the ionisation rate coefficient as low as 1% should suffice to accurately measure the variability of the yield based on the sensitivity of current experimental methods and their high collection efficiency.
, the extent of variation on this signal is equivalent to the probe's ionisation rate coefficient R1 on the core-hole population (or R1 − R5 for the case described by eqn (25)) as can be seen by the dashed curve in Fig. 6. R1 is dictated by the intensity of the laser pulse and should be chosen as large enough to enable accurate experimental measurement of the variation of the signal over the background, but not too large so as to activate multi-photon transitions. An estimated value for the ionisation rate coefficient as low as 1% should suffice to accurately measure the variability of the yield based on the sensitivity of current experimental methods and their high collection efficiency.
3
Ab initio calculations of the energy window for the probe pulse
The idea of the pump–probe scheme introduced in this study is general and non-system-specific. Comparison of the energetic constraints (1) and (3) shows that the available photon energy window is up to the order of Coulombic repulsion between the charge clouds of two outer-valence holes, or 1/R, where R is of the order of the chemical bond length:| | 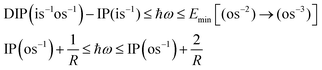 | (26) |
where we assume that the coulombic repulsion between the outer valence holes in the triply ionised state is roughly three times larger than in the doubly ionised state. This very crude electrostatic model leads us to expect the energy windows of up to 10 eV, with the probe frequencies in the range of ∼20–30 eV for molecular systems. One could argue that in very large molecules, the energy window can close, since it would always be possible to ionise a large molecule at a location very distant from the localisation of the outer-valence holes of the final Auger state. For such very large systems, however, measurement of the Auger lifetime will be problematic already because of the difficulty in resolving the initial core vacancy, unless it belongs, for example, to a uniquely present hetero-atom. Instead, one should be able to infer the Auger lifetime in such or another chemical environment using much smaller “model” compounds for which our proposed pump–probe technique would be suitable. In this case, the additional inter-atomic contributions can be taken into account analytically.26
In order to estimate the energy gap for the probe photon for a representative series of inner-valence- and core-ionised systems, we apply the extended second order ADC(2)x methods for the singly, doubly and triply ionised closed-shell systems.22–24 The second-order ADC schemes are the highest-order ones in the ADC hierarchy which have been developed for single, double and triple ionisation and therefore represent the obvious consistent choice of the approximation level within the ADC hierarchy. While the absolute accuracy of the second-order ADC schemes is only moderate, in analogy with the related MP2 method for the ground state, the differences between ADC(2)x energies are expected to be far more precise due to error cancellation. For example, benzene Auger spectrum calculation involving a large number of doubly ionised final states shows that the second-order ADC energy differences are within 0.3 eV or less from the true (experimental) values.27 In the present work we are interested in the energy gaps for the probe photon energies, therefore in energy differences. The ADC(2)x energy gaps calculated at the equilibrium geometries of the neutral systems are given in Table 1. All of the predicted energy gaps in are well within the expected accuracy of the ADC(2)x energy differences and, with the exception of CIF+ (F 2s−1) and CH3F+ (F 2s−1), can comfortably contain the energy bandwidth of a pulse with duration on the order of 0.5–1 fs. The technical details of the calculations are provided in Table 1.
Table 1 A sample of common inner-valence- and core-hole excited atomic and molecular systems with the available energy window for the probe pulse where the lower limit is given in eqn (1) and (2) and the upper limit is given in eqn (3). The results for CO and urea are determined for the scenarios detailed in Fig. 3 and 4, where specific poles are targeted. All energies are in units of eV and data is obtained from ADC(2)x calculations. * without h-functions; ** without g-functions
| System |
Geometry |
ADC(2)x basis set |
Lower limit |
Upper limit |
Gap |
| Core-hole systems |
| Xe+ (4d−1) |
N/A |
cc-pwCV5Z-PP |
22.70 |
29.13 |
6.43 |
| Ar+ (2s−1) |
N/A |
cc-pwCV5Z |
30.84 |
39.01 |
8.17 |
| Ne+ (1s−1) |
N/A |
cc-pwCV5Z |
50.04 |
62.13 |
12.09 |
| CO+ (C 1s−1) |
1.1283 Å |
cc-pVQZ |
28.86 |
35.04 |
6.18 |
| CO+ (O 1s−1) |
1.1283 Å |
cc-pVQZ |
26.80 |
35.04 |
8.24 |
| C6H5NO+ (N 1s−1) |
6-31G** |
cc-pVTZ |
14.86 |
17.28 |
2.42 |
|
|
| Inner-valence-hole systems |
| CIF+ (F 2s−1) |
1.6283 Å |
Aug-cc-pCV5Z |
33.14 |
33.58 |
0.44 |
| CH3F+ (F 2s−1) |
28
|
Aug-cc-pCV5Z |
32.36 |
32.98 |
0.61 |
| C2H5F+ (F 2s−1) |
29
|
Aug-cc-pVTZ |
24.18 |
26.40 |
2.22 |
| HCOO (O 2s−1) |
CCD/6-31G(2df,p) |
Aug-cc-pCV5Z* |
13.18 |
20.36 |
7.18 |
| Glycine+ (O 2s−1) |
GlyI conformer30 |
Aug-cc-pCVQZ** |
15.52 |
20.31 |
4.79 |
| Glycine+ (N 2s−1) |
GlyI conformer30 |
Aug-cc-pCVQZ** |
23.15 |
25.34 |
2.19 |
| Urea+ (O 2s−1) |
C
2v, CCSD(T)/aug-cc-pVQZ |
cc-pVTZ |
21.56 |
24.51 |
2.95 |
| Furan+ (O 2s−1) |
31
|
cc-pVTZ |
17.42 |
21.91 |
4.49 |
4 Post-collision interaction and shake-off/shake-up effects
We now consider the influence of shake-off/shake-up effects and of the PCI on the proposed attosecond pump–attosecond probe experiment. Various excitation processes can occur during atomic inner-shell photo-ionisation, where the final state comprises a removed core electron and the excitation of further electrons to either excited bound states (shake-up) or to the continuum (shake-off). These processes leave their mark as satellites in the Auger electron and photoelectron spectra.32
The photoelectron deriving from an inner-shell photo-ionisation process can be recaptured after the subsequent Auger decay of the system (PCI) due to energy exchange with the fast Auger electron and interaction with the Coulombic field of the dication.33,34 This can be understood by separating the interactions into delineated stages as in a classical model; we have a system that is impacted with a projectile (photon or electron) that ejects an electron from the system with excess energy. The system then relaxes with the additional energy carried away by the Auger electron. If the first electron has lower energy than the latter, then it initially sees a singly-charged ion as the Auger electron is screening the dication. When it is overtaken by the Auger electron it slows in the stronger potential of the dication; simultaneously the Auger electron is now shielded from the dication and subsequently gains in energy. This effect is detectable in the Auger spectrum. It is possible for the overtaken electron to be recaptured by the dication.35,36 An approximate formula for the recapture probability P(Eex), where Eex is the excess energy of the slow photoelectron, is found37 to be
| | | P(Eex) = 1 − exp[−Γ/Eex] | (27) |
where
Γ is the linewidth of the Auger transition.
In our model, we take the possibility of shake-off into account by assuming that the populations of the charge states are initialised (by the pump probe) with a factor G for the inner-shell-hole state and (1 − G) for the inner-shell-valence-hole state, and we consider the shake-off initial population 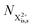 separately from the
separately from the 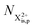 population (that derives from the probed inner-shell-hole
population (that derives from the probed inner-shell-hole 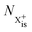 ).
).
Furthermore, the possibility of the probe photoelectrons to be recaptured due to PCI is included through the parameter Fp, which denotes the proportion of probe photoelectrons that are not recaptured by the Auger electron. We assume that the pump photon can be chosen to ensure that the pump photoelectron has sufficient excess energy to preclude its recapture. We introduce a further quantity Fs which denotes the proportion of shake-off photoelectrons that are not recaptured by the Auger electron (this will be in general different to Fp). We can assume that the shake-off electron does not measurably interact with the probe photoelectron, and the pump photoelectron can always be chosen to have greater excess energy than the probe photoelectron to ensure that these do not interact either.
Post-collision recapture from an initial (is–os) state takes place in two stages: (a) decay from 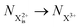 and (b) recapture NX3+ → NX2+. We simplify this in the model by assuming that both stages happen simultaneously, as we are not concerned with the time dynamics of this process. The shake-off state
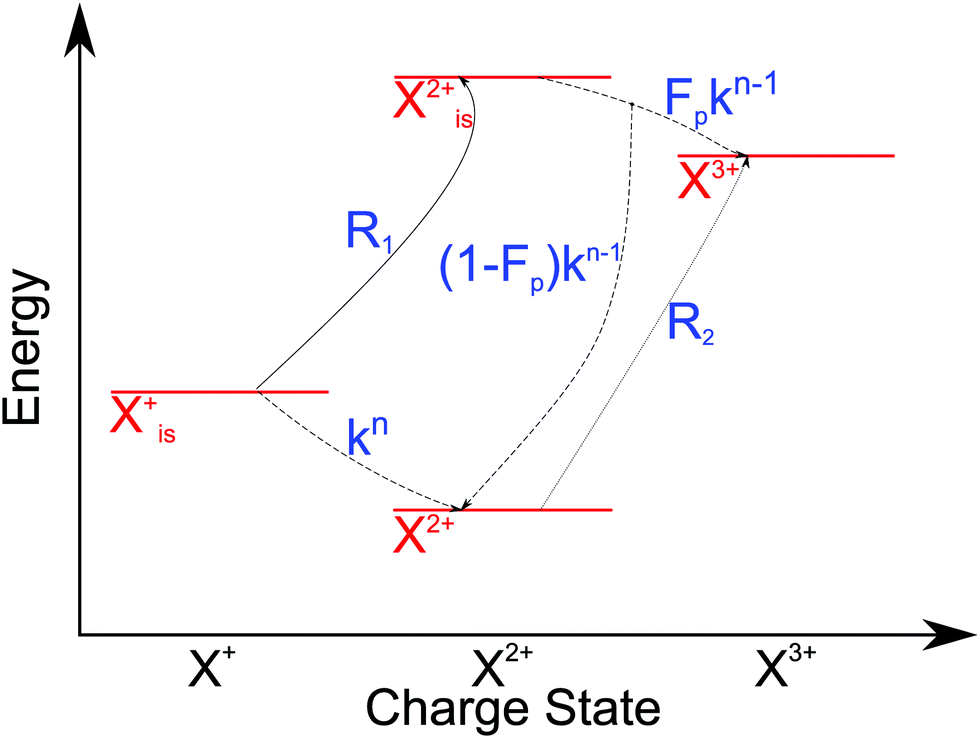
and (b) recapture NX3+ → NX2+. We simplify this in the model by assuming that both stages happen simultaneously, as we are not concerned with the time dynamics of this process. The shake-off state 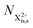 can be further ionised by the probe in proportion R3 to the final state NX3+, which can no longer decay as it is below QIP for inner-valence systems. The updated rate equations for the inner-valence system are therefore as follows:
can be further ionised by the probe in proportion R3 to the final state NX3+, which can no longer decay as it is below QIP for inner-valence systems. The updated rate equations for the inner-valence system are therefore as follows:
| | 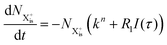 | (28) |
| | 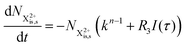 | (29) |
| | 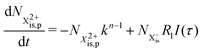 | (30) |
| | 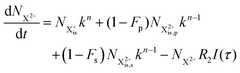 | (31) |
| | 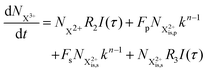 | (32) |
The modified transition diagram including the PCI effect is given in
Fig. 7.
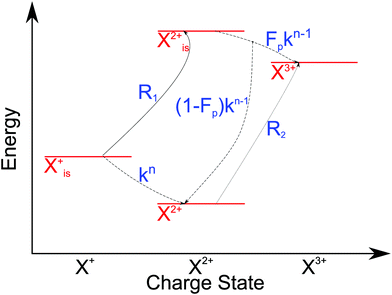 |
| | Fig. 7 Transition scheme of the attosecond pump–attosecond probe experiment for the inner-valence ionisation case including the effect of the electron recapture due to post-collision interaction. The scheme corresponds directly to the rate eqn (28)–(32). | |
We solve the rate equations by approximating the pulse with I(τ) = δ(τ) to obtain the final population of the X2+ charge state,
| | 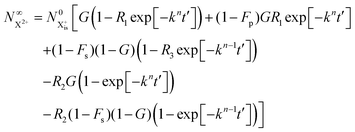 | (33) |
and the X
3+ charge state,
| | 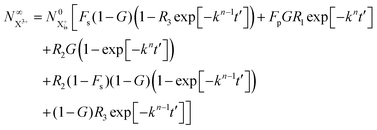 | (34) |
We now have two exponents in the final population expressions: all terms proportional to exp[−kn−1t′] derive from the 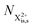 shake-off population which decays at the rate kn−1 before the probe interaction time of t′. Hence, we retrieve the original single exponent dependence by ensuring that the shake-off population produced by the pump photon is negligible (G = 1).
shake-off population which decays at the rate kn−1 before the probe interaction time of t′. Hence, we retrieve the original single exponent dependence by ensuring that the shake-off population produced by the pump photon is negligible (G = 1).
The PCI and shake-off modification for the X2+ final population given in eqn (33) also applies to the expression in eqn (25) for the X2+ final population of the core-hole state, as in both cases this population is only coupled to the initial state and the probe-ionised initial state. We can therefore similarly retain the single exponent dependence by ensuring that there is no shake-off for core-hole systems. As an example, for the case of argon 1s ionisation, a value of the excess (above threshold) energy of the pump-photon lower than ∼30 eV is sufficient to ensure that there is no Auger satellite-group intensity corresponding to shake-off, even from the outer valence 3p orbital.32
Although shake-up does not directly affect the charge state populations, it is nevertheless problematic for two reasons: (i) it can reduce the size of the probe photon energy window by lowering the energy threshold for ionising the final X2+ state [see eqn (2)] as this species may have an electron occupying a Rydberg state, and (ii) the measured decay time may correspond to the excited shake-up state, which could be different to the true decay time of the ground inner-shell-hole state.
For the argon 1s example, the energy threshold for 1s ionisation accompanied by 3p → 4p shake-up is ∼15 eV. It is therefore simple to ensure that the pump photon is chosen to keep the excess energy of the photoelectron below this limit, thereby removing the possibility of shake-up and shake-off (and the consequent possibility of recapture of the shake-off electron). For inner-valence systems such as urea (see Fig. 3), the allowed excess energy of the pump photoelectron should be kept within ∼20 eV.
As mentioned above, the pump photoelectron should also have enough excess energy to preclude its recapture by the ion after Auger decay. As indicated in the empirical formula given in eqn (27), this condition can in general be met with an excess of just ∼5 eV (for Auger decay times on the order of ∼10 fs) regardless of the precise energy of the Auger electron (see ref. 36 and 38). This is because the two electrons have energies of different orders of magnitude, so their interaction is minimal. The dicationic field after the decay is insufficient to capture the photoelectron provided the latter has this threshold energy value. For faster Auger decay times, higher excess energy for the pump photoelectron will be required to prevent recapture from taking place.
The probe photoelectron poses a more serious problem with regards to recapture, as the energy window imposes narrow constraints on the choice of probe photon energy. For the core-hole system there is in general an energy window of sufficient size to allow for an excess probe photoelectron energy of 5 eV. As an example, the CO molecule discussed above has an energy gap of 8.24 eV. Conversely, for inner-valence systems we can be forced to use probe photon energies at the ionisation threshold, making the very slow probe photoelectron vulnerable to recapture. However, the Auger electron produced by the decay of inner-valence systems typically has very low energy as well – equivalent to the difference between the targeted inner-outer-valence-ionised state and the TIP threshold that lies directly below (see ionisation energies and pole strengths of urea molecule in Fig. 3). The PCI effect in this regime will be influenced by the electron–electron interaction and the probability of recapture may (depending on the system) be non-negligible.
Thus, by careful choice of the pump photon (which will be in general system-dependent), we can ensure that G and by extension Fs are set to unity. The final state populations consequently differ from the original rate equations by the term on the second line in eqn (33) for the 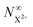 population and by the term on the second line in eqn (34) for the
population and by the term on the second line in eqn (34) for the 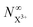 population. This distinction simply modifies the signal that we propose to measure in the inner-valence case by a factor of Fp, which can be ascertained for a given system and chosen probe energy. The equations for the core-hole case can remain unchanged from the original model, provided we ensure that the probe photon gives the resulting photoelectron enough excess energy (but less than that of the pump photoelectron) to preclude recapture by the dication.
population. This distinction simply modifies the signal that we propose to measure in the inner-valence case by a factor of Fp, which can be ascertained for a given system and chosen probe energy. The equations for the core-hole case can remain unchanged from the original model, provided we ensure that the probe photon gives the resulting photoelectron enough excess energy (but less than that of the pump photoelectron) to preclude recapture by the dication.
5 Conclusion
In this work, we have introduced and explored the efficacy of the ion-yield-based attosecond pump–attosecond probe scheme for the investigation of the Auger decay dynamics in atoms and molecules. The proposed scheme has been demonstrated to be applicable to the determination of the lifetimes of isolated resonance states decaying due to the Auger effect. However, the constraints arising for such a measurement are not related to the exponential characters of the time evolution, and any type of decay, exponential or non-exponential, should be amenable to the suggested pump–probe scheme, provided it is not too fast for being interrogated using a probe pulse with the central frequency within the energy window dictated by the constraints (1)–(3).
We have shown that in inner-valence-hole systems it is in principle possible to map these dynamics onto the background-free total yield of the X3+ ions that is modulated only by the PCI effect. For core-hole systems, on the other hand, triply charged state measurement is not background-free because of double Auger decay. Moreover, a detailed analysis shows that the pump–probe delay variation of the X2+ (rather than X3+) yield provides the best diagnostics for the temporal evolution of the core-hole. Therefore, appropriate choice of the pump and the probe pulse frequencies eliminates the need in an energy-resolved measurement. However, coincident detection or covariance mapping spectroscopy of the fragment ions may be needed to detect the yields of the unstable X2+ and X3+ systems.
The generality of the proposed attosecond pump–attosecond probe spectroscopy has been confirmed by extensive ab initio calculations on representative set of atoms and molecules. Our theoretical results underscore the usefulness of attosecond pulse sources in the extreme ultraviolet (∼20–60 eV) region,39 in particular if synchronised with attosecond soft X-ray pulses.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This work was funded by EPSRC/DSTL MURI-MIR grant EP/I08680/1. P. K. acknowledges financial support by the Czech Science Foundation (Project GAČR No. 17-10866S).
References
-
B. Crasemann, Atomic inner-shell physics, Springer Science & Business Media, 2013 Search PubMed.
- T. A. Carlson and M. O. Krause, Phys. Rev. Lett., 1965, 14, 390 CrossRef CAS.
- A. Mueller, A. Borovik Jr, T. Buhr, J. Hellhund, K. Holste, A. Kilcoyne, S. Klumpp, M. Martins, S. Ricz and J. Viefhaus,
et al.
, Phys. Rev. Lett., 2015, 114, 013002 CrossRef CAS PubMed.
- S. Svensson, L. Karlsson, P. Baltzer, M. Keane and B. Wannberg, Phys. Rev. A: At., Mol., Opt. Phys., 1989, 40, 4369 CrossRef CAS PubMed.
- R. Bruhn, B. Sonntag and H. Wolff, Phys. Lett. A, 1978, 69, 9–11 CrossRef.
- M. Drescher, M. Hentschel, R. Kienberger, M. Uiberacker, V. Yakovlev, A. Scrinzi, T. Westerwalbesloh, U. Kleineberg, U. Heinzmann and F. Krausz, Nature, 2002, 419, 803–807 CrossRef CAS PubMed.
- O. Smirnova, V. S. Yakovlev and A. Scrinzi, Phys. Rev. Lett., 2003, 91, 253001 CrossRef PubMed.
- M. Huppert, I. Jordan, D. Baykusheva, A. Von Conta and H. J. Wörner, Phys. Rev. Lett., 2016, 117, 093001 CrossRef PubMed.
-
https://arxiv.org/abs/2105.08854
.
- J. Leeuwenburgh, B. Cooper, V. Averbukh, J. P. Marangos and M. Ivanov, Phys. Rev. Lett., 2013, 111, 123002 CrossRef PubMed.
- M. Khokhlova, B. Cooper, K. Ueda, K. Prince, P. Kolorenč, M. Y. Ivanov and V. Averbukh, Phys. Rev. Lett., 2019, 122, 233001 CrossRef CAS PubMed.
- T. Barillot, P. Matia-Hernando, D. Greening, D. Walke, T. Witting, L. Frasinski, J. Marangos and J. Tisch, Chem. Phys. Lett., 2017, 683, 38–42 CrossRef CAS.
- J. Duris, S. Li, T. Driver, E. G. Champenois, J. P. MacArthur, A. A. Lutman, Z. Zhang, P. Rosenberger, J. W. Aldrich and R. Coffee,
et al.
, Nat. Photonics, 2020, 14, 30–36 CrossRef CAS.
- B. Cooper and V. Averbukh, Phys. Rev. Lett., 2013, 111, 083004 CrossRef PubMed.
- D. Iablonskyi, K. Ueda, K. L. Ishikawa, A. Kheifets, P. Carpeggiani, M. Reduzzi, H. Ahmadi, A. Comby, G. Sansone and T. Csizmadia,
et al.
, Phys. Rev. Lett., 2017, 119, 073203 CrossRef CAS PubMed.
- D. You, K. Ueda, M. Ruberti, K. L. Ishikawa, P. A. Carpeggiani, T. Csizmadia, L. G. Oldal, N. Harshitha, G. Sansone and P. K. Maroju,
et al.
, New J. Phys., 2019, 21, 113036 CrossRef CAS.
- G. Dujardin, S. Leach, O. Dutuit, P.-M. Guyon and M. Richard-Viard, Chem. Phys., 1984, 88, 339–353 CrossRef CAS.
- L. Frasinski, K. Codling and P. Hatherly, Science, 1989, 246, 1029–1031 CrossRef CAS PubMed.
- L. Young, E. P. Kanter, B. Krässig, Y. Li, A. March, S. Pratt, R. Santra, S. Southworth, N. Rohringer and L. DiMauro,
et al.
, Nature, 2010, 466, 56–61 CrossRef CAS PubMed.
- A. Rudenko, L. Inhester, K. Hanasaki, X. Li, S. Robatjazi, B. Erk, R. Boll, K. Toyota, Y. Hao and O. Vendrell,
et al.
, Nature, 2017, 546, 129–132 CrossRef CAS PubMed.
- D. Coster and R. d. L. Kronig, Physica, 1935, 2, 13–24 CrossRef CAS.
- J. Schirmer and A. Barth, Z. Phys. A: At. Nucl., 1984, 317, 267–279 CrossRef CAS.
- A. Tarantelli and L. Cederbaum, Phys. Rev. A: At., Mol., Opt. Phys., 1992, 46, 81 CrossRef PubMed.
- J. Schirmer, A. Trofimov and G. Stelter, J. Chem. Phys., 1998, 109, 4734–4744 CrossRef CAS.
- A. H. Roos, J. Eland, J. Andersson, R. Squibb, D. Koulentianos, O. Talaee and R. Feifel, Sci. Rep., 2018, 8, 16405 CrossRef PubMed.
- N. V. Kryzhevoi, V. Averbukh and L. S. Cederbaum, Phys. Rev. B: Condens. Matter Mater. Phys., 2007, 76, 094513 CrossRef.
- S. A. Tarantelli, J. Chem. Phys., 1987, 86, 2201 CrossRef.
- J. Demaison, J. Breidung, W. Thiel and D. Papousek, Struct. Chem., 1999, 10, 129–133 CrossRef CAS.
-
K.-H. Hellwege and A. M. Hellwege, Structure data of free polyatomic molecules, Springer Verlag, 1976 Search PubMed.
- A. G. Csaszar, J. Am. Chem. Soc., 1992, 114, 9568–9575 CrossRef CAS.
-
G. Herzberg, Molecular Spectra and Molecular Structure: Electric spectra and electronic structure of polyatomic molecules, Van Nostrand, 1950, vol. 3 Search PubMed.
- G. B. Armen, T. Åberg, K. R. Karim, J. C. Levin, B. Crasemann, G. S. Brown, M. H. Chen and G. E. Ice, Phys. Rev. Lett., 1985, 54, 182 CrossRef CAS PubMed.
- A. Niehaus, J. Phys. B: At. Mol. Phys., 1977, 10, 1845 CrossRef CAS.
- A. Russek and W. Mehlhorn, J. Phys. B: At. Mol. Phys., 1986, 19, 911 CrossRef CAS.
- M. Van der Wiel, G. Wight and R. Tol, J. Phys. B: At. Mol. Phys., 1976, 9, L5 CrossRef CAS.
- W. Eberhardt, S. Bernstorff, H. Jochims, S. Whitfield and B. Crasemann, Phys. Rev. A: At., Mol., Opt. Phys., 1988, 38, 3808 CrossRef CAS PubMed.
- J. A. Samson, W. C. Stolte, Z. He, J. N. Cutler and D. Hansen, Phys. Rev. A: At., Mol., Opt. Phys., 1996, 54, 2099 CrossRef CAS PubMed.
- R. Guillemin, S. Sheinerman, R. Püttner, T. Marchenko, G. Goldsztejn, L. Journel, R. Kushawaha, D. Céolin, M. N. Piancastelli and M. Simon, Phys. Rev. A: At., Mol., Opt. Phys., 2015, 92, 012503 CrossRef.
- D. Fabris, T. Witting, W. Okell, D. Walke, P. Matia-Hernando, J. Henkel, T. Barillot, M. Lein, J. Marangos and J. Tisch, Nat. Photonics, 2015, 9, 383–387 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1cp00623a |
|
| This journal is © the Owner Societies 2021 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *a,
Přemysl
Kolorenč
*a,
Přemysl
Kolorenč
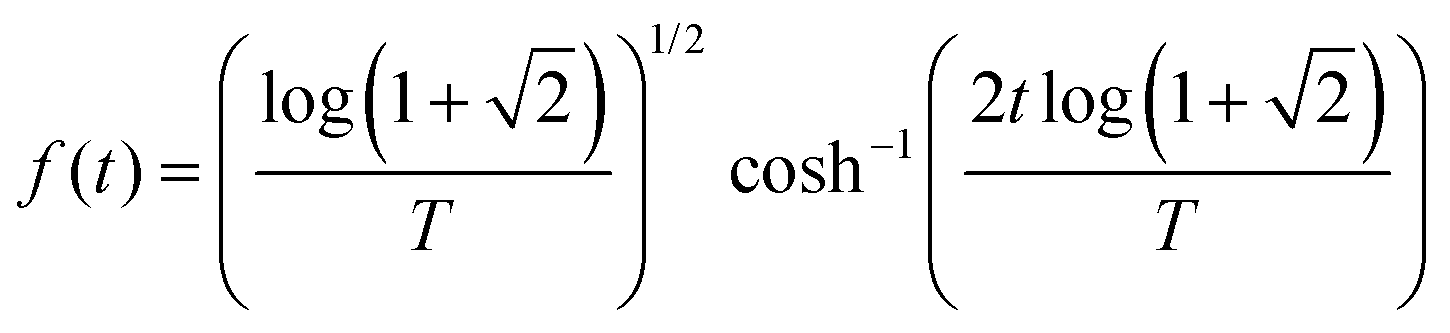
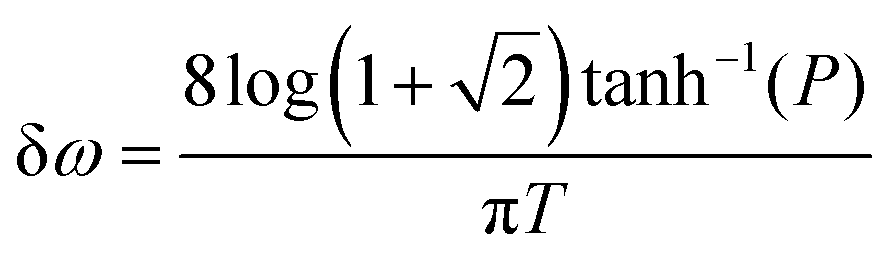
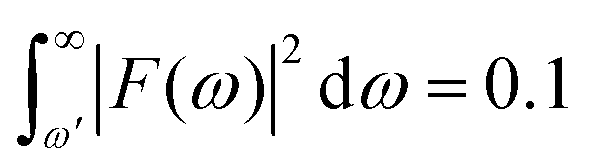 , where F(ω) is the pulse envelope's Fourier transform, is 1.623 eV (from the center of the distribution) and for the 1% tail the same quantity is 3.394 eV; the difference between these two quantities is just 1.771 eV. This would mean that if the system's energy gap was of this size, approximately 10% of the probe-ionised population would be above TIP and just 1% of the decay product in the DI state at the maximum energy threshold could still be ionised to X3+. A larger energy gap can reduce this residual X3+ signal even further.
, where F(ω) is the pulse envelope's Fourier transform, is 1.623 eV (from the center of the distribution) and for the 1% tail the same quantity is 3.394 eV; the difference between these two quantities is just 1.771 eV. This would mean that if the system's energy gap was of this size, approximately 10% of the probe-ionised population would be above TIP and just 1% of the decay product in the DI state at the maximum energy threshold could still be ionised to X3+. A larger energy gap can reduce this residual X3+ signal even further.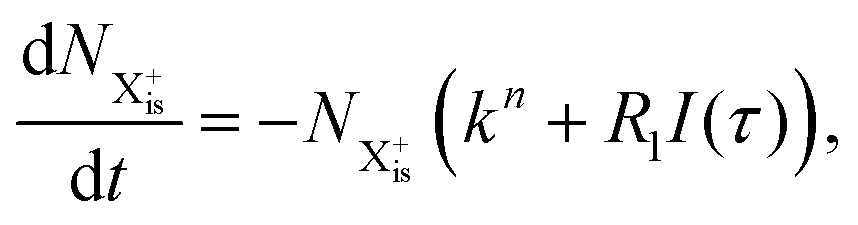
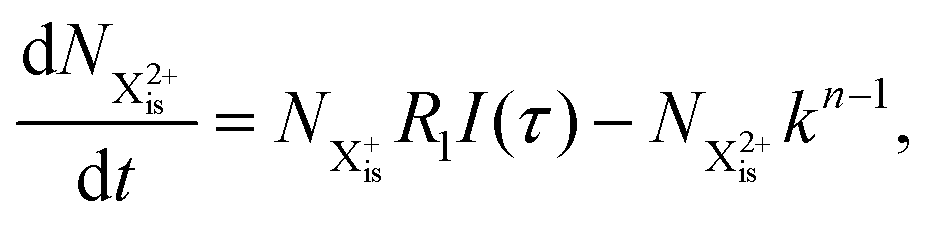
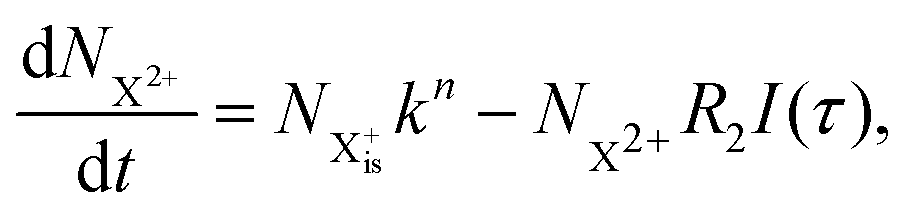
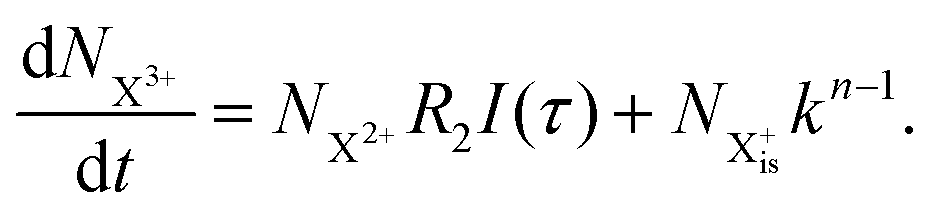
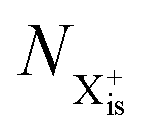 is the population of the initial sub-shell (inner-valence) hole state,
is the population of the initial sub-shell (inner-valence) hole state, 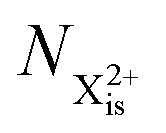 is the population of the DI (inner-outer-valence ionised) state, and NX2+ and NX3+ are the populations of the stable final X2+ and X3+ states; I(τ) is the normalised intensity envelope function of the probe pulse centred at τ = t − t′ for a time delay of t′ [cf.eqn (4)], R1 and R2 are the ionisation rate coefficients from the inner-valence-hole state and the final Auger decay states (X2+), respectively, and kn is the Auger decay rate for n electrons in the valence. In the fully detailed description, every ionisation state consists of a series of individual quantum states, each possessing their own ionisation and/or decay rate. Here we are not interested in such individual state populations, which can only be reliably computed using fully coherent time-dependent Schrödinger equation approach, but rather in the total population of the singly, doubly and triply charged states. Therefore, the decay rates and photoionisation rate coefficients in eqn (6)–(9) should be thought of as averages over the individual quantum states contributing to these ionic populations. Furthermore, we assume that the pump pulse is chosen such that the shake-up and shake-off populations are negligible, as is the effect of the post-collision interaction (PCI). These effects will be considered in Section 4. The scheme of the transitions described by the rate eqn (6)–(9) is given in Fig. 2.
is the population of the DI (inner-outer-valence ionised) state, and NX2+ and NX3+ are the populations of the stable final X2+ and X3+ states; I(τ) is the normalised intensity envelope function of the probe pulse centred at τ = t − t′ for a time delay of t′ [cf.eqn (4)], R1 and R2 are the ionisation rate coefficients from the inner-valence-hole state and the final Auger decay states (X2+), respectively, and kn is the Auger decay rate for n electrons in the valence. In the fully detailed description, every ionisation state consists of a series of individual quantum states, each possessing their own ionisation and/or decay rate. Here we are not interested in such individual state populations, which can only be reliably computed using fully coherent time-dependent Schrödinger equation approach, but rather in the total population of the singly, doubly and triply charged states. Therefore, the decay rates and photoionisation rate coefficients in eqn (6)–(9) should be thought of as averages over the individual quantum states contributing to these ionic populations. Furthermore, we assume that the pump pulse is chosen such that the shake-up and shake-off populations are negligible, as is the effect of the post-collision interaction (PCI). These effects will be considered in Section 4. The scheme of the transitions described by the rate eqn (6)–(9) is given in Fig. 2.
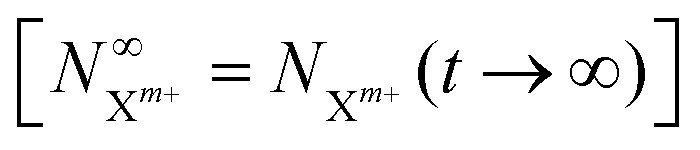 as a functions of the time delay as:
as a functions of the time delay as: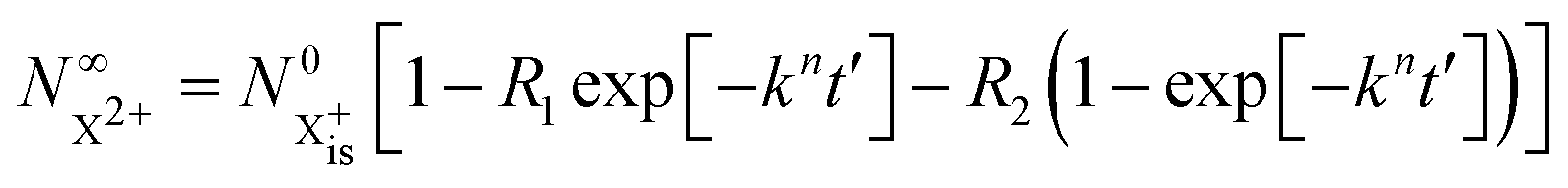
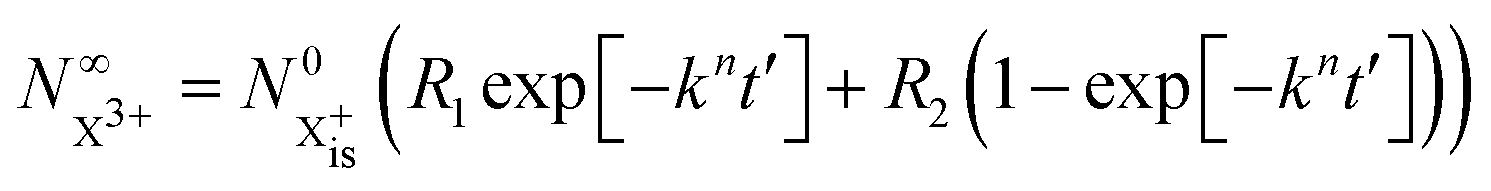
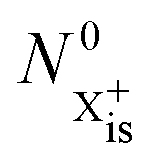 is the inner-valence-hole population at t = 0. The R2 rate coefficient is defined as the proportion of final doubly ionised states that can still be ionised by the probe multiplied by the ionisation rate for these states; if the energy gap is wide enough, this parameter will be zero and the X3+ yield will be background free, if it is not, there will be a residual signal proportional to R2. Even for the latter case, the X3+ (and also X2+) population will depend exponentially on the time delay, allowing one to recover the Auger decay rate from the attosecond pump–probe measurement of the total ion yield.
is the inner-valence-hole population at t = 0. The R2 rate coefficient is defined as the proportion of final doubly ionised states that can still be ionised by the probe multiplied by the ionisation rate for these states; if the energy gap is wide enough, this parameter will be zero and the X3+ yield will be background free, if it is not, there will be a residual signal proportional to R2. Even for the latter case, the X3+ (and also X2+) population will depend exponentially on the time delay, allowing one to recover the Auger decay rate from the attosecond pump–probe measurement of the total ion yield.
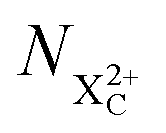 is the population of the X2+ system where the initial Auger active core-hole is still present (denoted by C) and there is an additional hole in the valence caused by the ionising probe pulse;
is the population of the X2+ system where the initial Auger active core-hole is still present (denoted by C) and there is an additional hole in the valence caused by the ionising probe pulse; 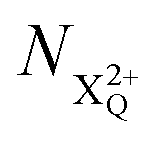 and
and 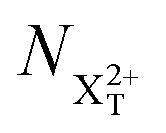 refers to the system where the core-hole has moved to a hole of lower energy, but the ion's overall energy remains above the QIP threshold or between the TIP and QIP threshold, respectively; and NX2+ refers to the system's final state where the two holes are both in the valence and the system is no longer energetically capable of further decay (the total ion energy is therefore below TIP). In a similar vein, we differentiate the X3+ population into the sub-population
refers to the system where the core-hole has moved to a hole of lower energy, but the ion's overall energy remains above the QIP threshold or between the TIP and QIP threshold, respectively; and NX2+ refers to the system's final state where the two holes are both in the valence and the system is no longer energetically capable of further decay (the total ion energy is therefore below TIP). In a similar vein, we differentiate the X3+ population into the sub-population 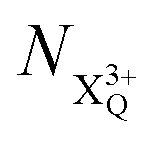 where the total ion energy is above QIP (and therefore still capable of further decay to the X4+ species), and the final decay product of NX3+.
where the total ion energy is above QIP (and therefore still capable of further decay to the X4+ species), and the final decay product of NX3+.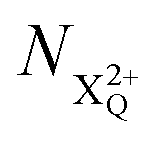 population; the latter's total ionic energy is above the QIP threshold, thus enabling the decay process
population; the latter's total ionic energy is above the QIP threshold, thus enabling the decay process 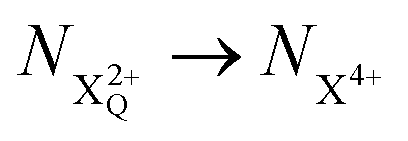 to take place.
to take place.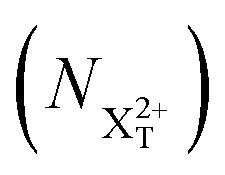 and QIP
and QIP 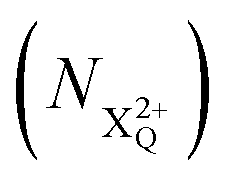 as R2 and R3, respectively, and the ionisation rate coefficient from the X3+ species with total energy above QIP
as R2 and R3, respectively, and the ionisation rate coefficient from the X3+ species with total energy above QIP 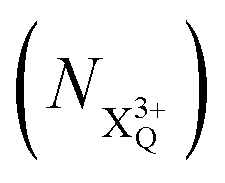 as R4. The rate equations governing the core-hole decay are then given as:
as R4. The rate equations governing the core-hole decay are then given as: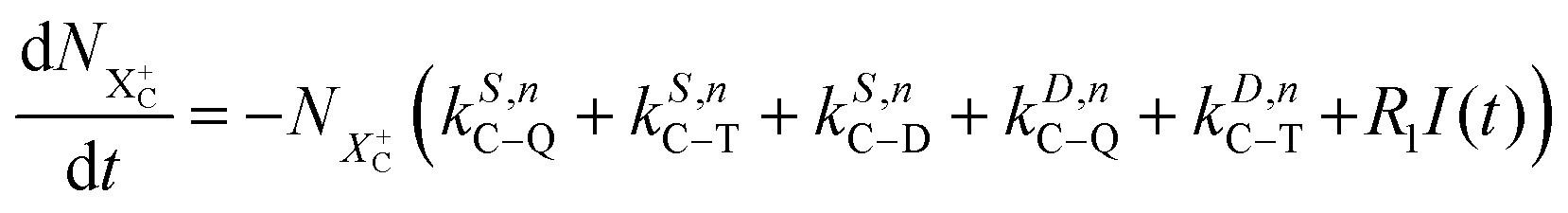
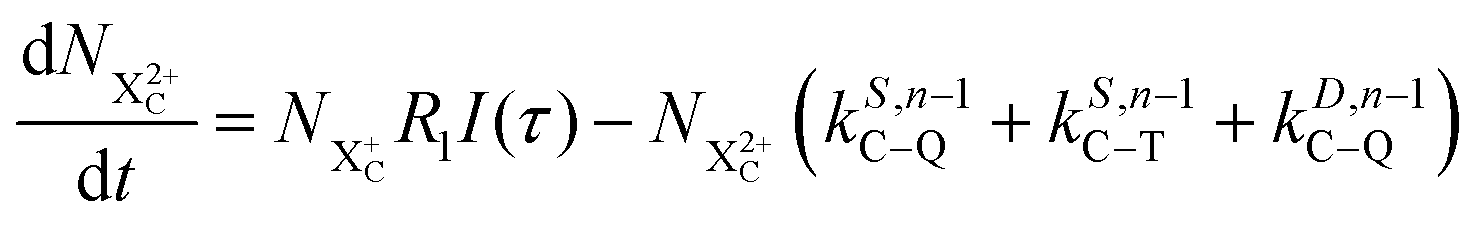
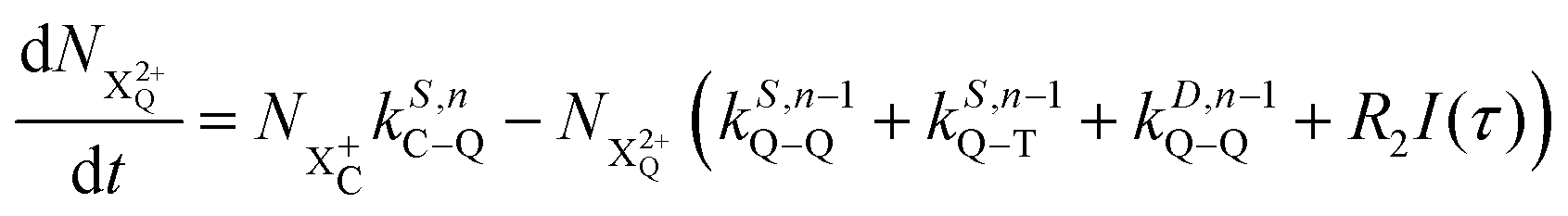
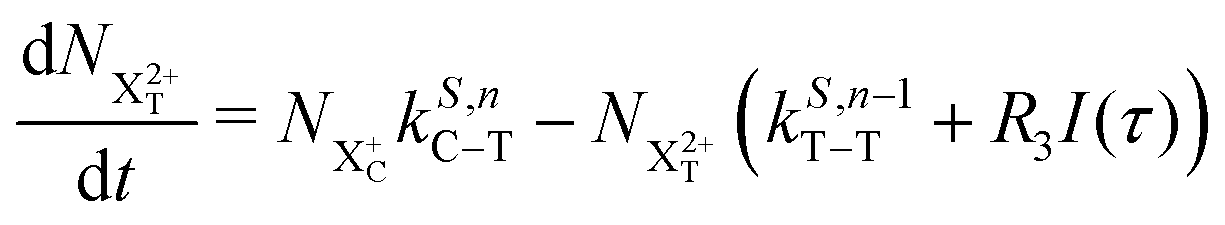
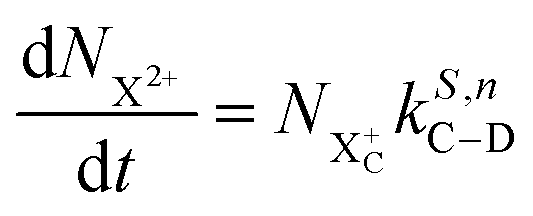
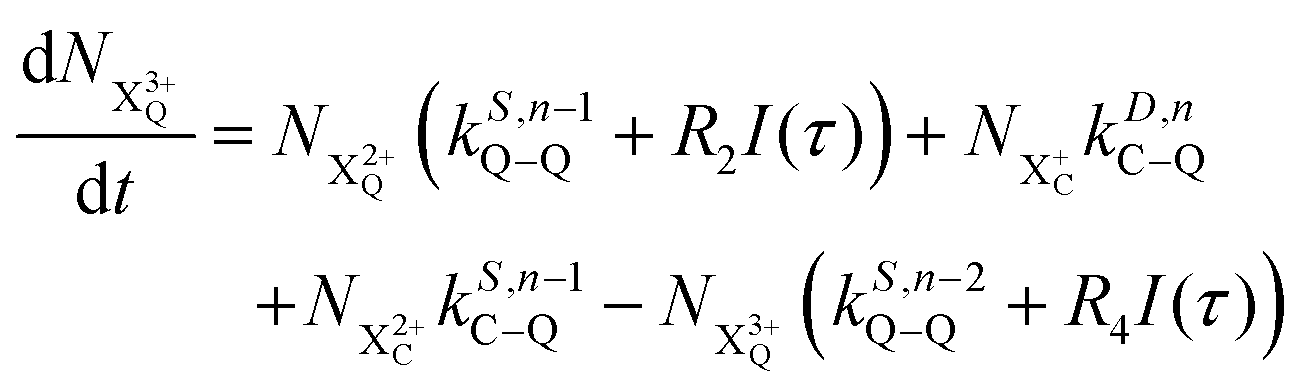
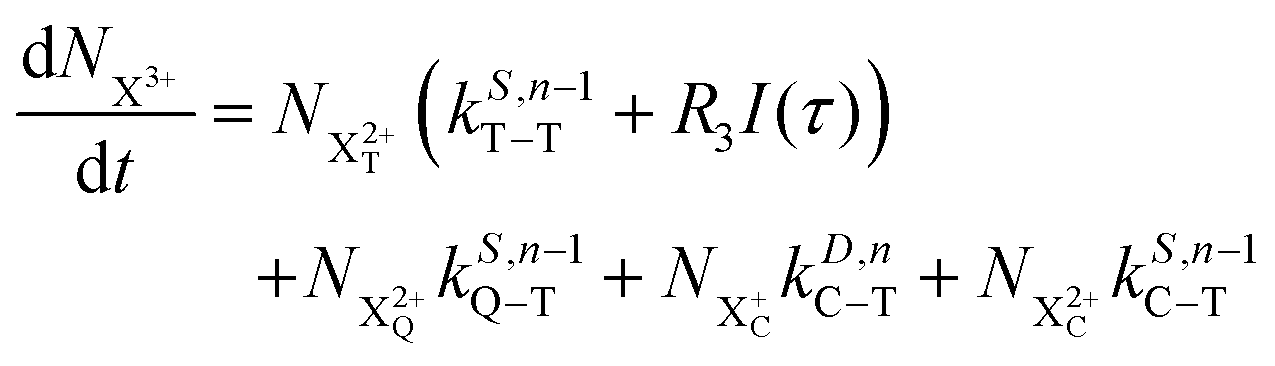
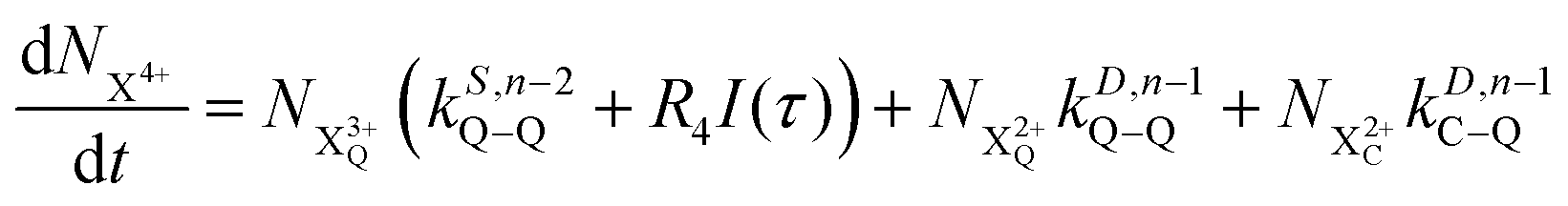
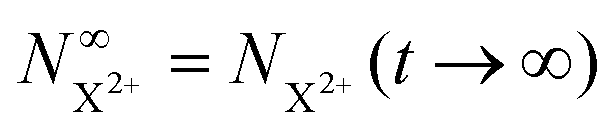 to be
to be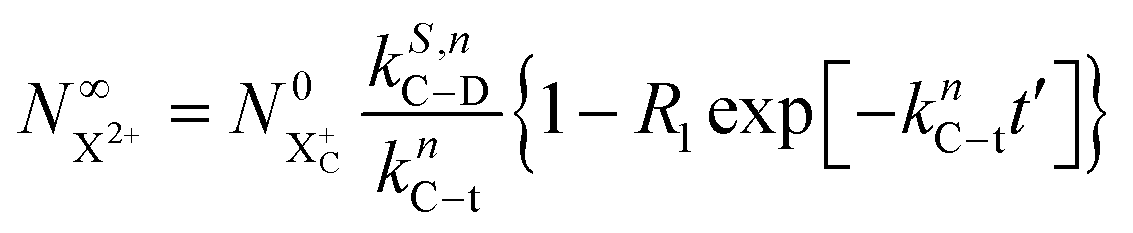
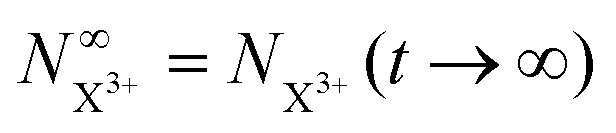 is
is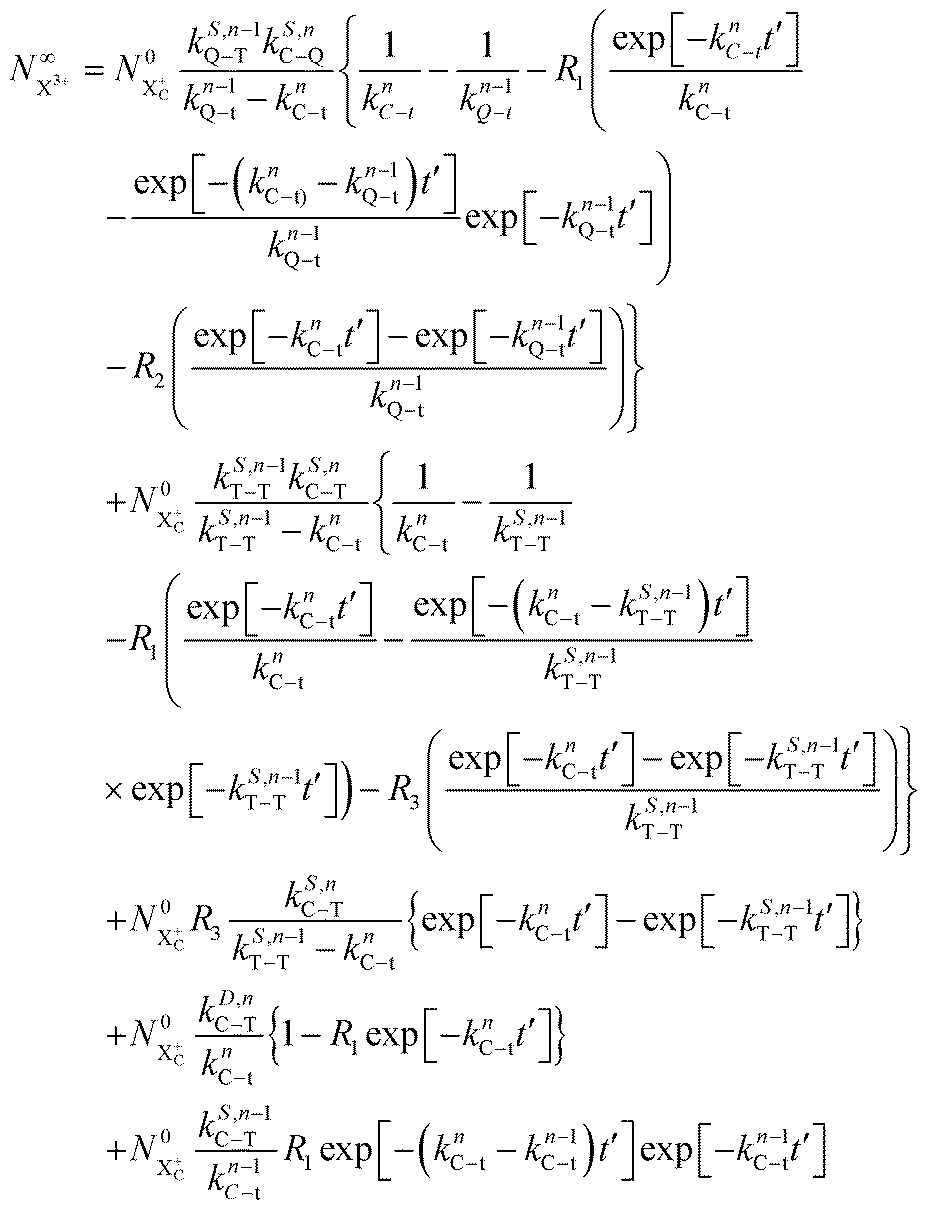
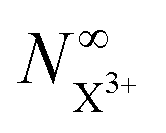 yield does not exhibit a simple exponential decay due to the competing triple ionisation pathways unaccounted for by the simple scheme in Fig. 1. However, there is only one path that contributes to the
yield does not exhibit a simple exponential decay due to the competing triple ionisation pathways unaccounted for by the simple scheme in Fig. 1. However, there is only one path that contributes to the 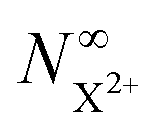 yield, and its final population as a function of the probe time delay has a simple exponential time dependence revealing the system's total decay rate, which we aim to measure.
yield, and its final population as a function of the probe time delay has a simple exponential time dependence revealing the system's total decay rate, which we aim to measure.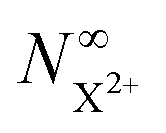 population will still exhibit the same simple exponential dependence. In such a case, and using eqn (20), we modify eqn (23) to
population will still exhibit the same simple exponential dependence. In such a case, and using eqn (20), we modify eqn (23) to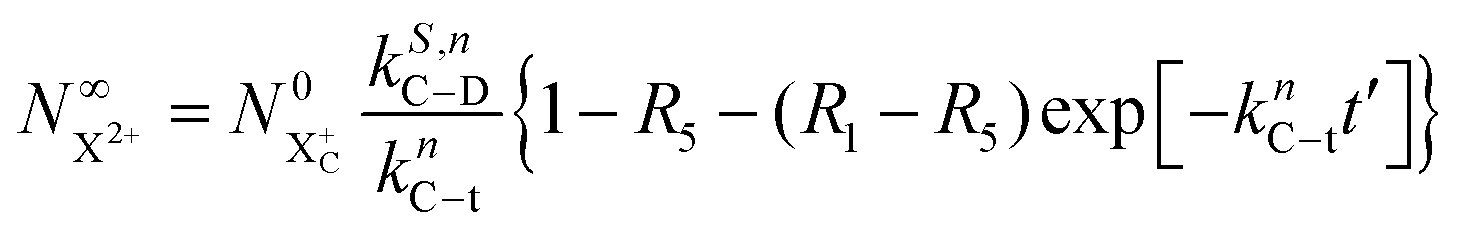
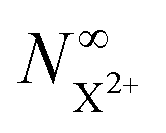 and is defined as the proportion of final DI states that can still be ionised by the probe multiplied by the ionisation rate coefficient for these states. The difference between R1 and R5 must, however, be large enough for the measurement of knC–t to remain feasible.
and is defined as the proportion of final DI states that can still be ionised by the probe multiplied by the ionisation rate coefficient for these states. The difference between R1 and R5 must, however, be large enough for the measurement of knC–t to remain feasible.
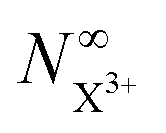 and
and 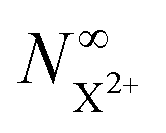 quantities, obtained by solving numerically eqn (12)–(19) for a 0.1 fs (FWHM) duration sech probe pulse and a variety of kntot values within the 0.1–1.0 fs−1 range, as well as the analytical exponential solution of eqn (23). It can be seen that the latter, at time-delays sufficiently large for the whole probe pulse to occur after the pump ionisation event (t = 0), agrees well with the corresponding numerical solution.
quantities, obtained by solving numerically eqn (12)–(19) for a 0.1 fs (FWHM) duration sech probe pulse and a variety of kntot values within the 0.1–1.0 fs−1 range, as well as the analytical exponential solution of eqn (23). It can be seen that the latter, at time-delays sufficiently large for the whole probe pulse to occur after the pump ionisation event (t = 0), agrees well with the corresponding numerical solution.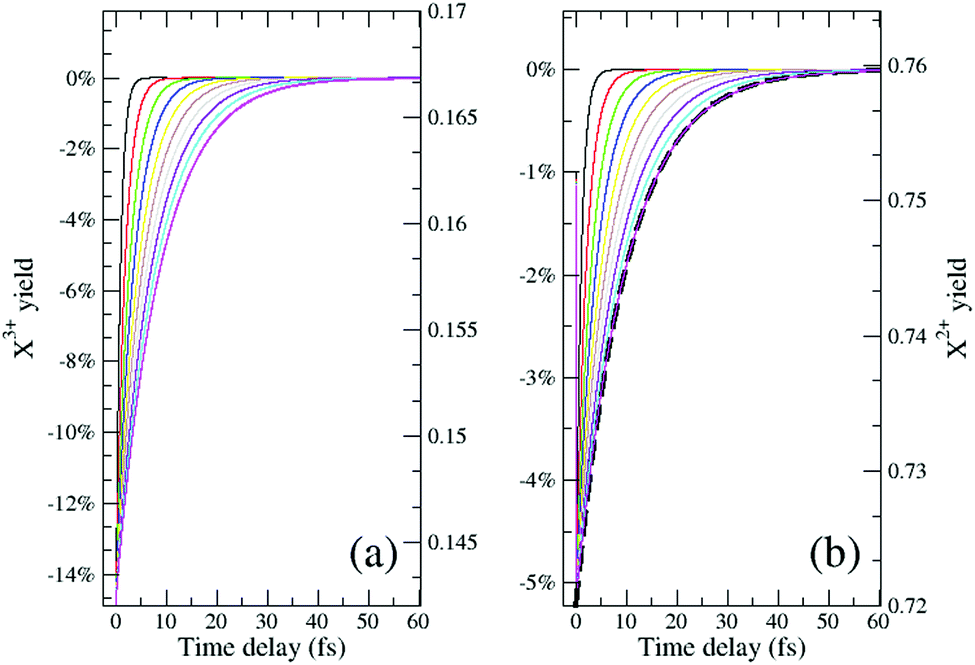
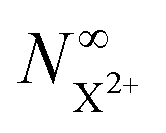 , the extent of variation on this signal is equivalent to the probe's ionisation rate coefficient R1 on the core-hole population (or R1 − R5 for the case described by eqn (25)) as can be seen by the dashed curve in Fig. 6. R1 is dictated by the intensity of the laser pulse and should be chosen as large enough to enable accurate experimental measurement of the variation of the signal over the background, but not too large so as to activate multi-photon transitions. An estimated value for the ionisation rate coefficient as low as 1% should suffice to accurately measure the variability of the yield based on the sensitivity of current experimental methods and their high collection efficiency.
, the extent of variation on this signal is equivalent to the probe's ionisation rate coefficient R1 on the core-hole population (or R1 − R5 for the case described by eqn (25)) as can be seen by the dashed curve in Fig. 6. R1 is dictated by the intensity of the laser pulse and should be chosen as large enough to enable accurate experimental measurement of the variation of the signal over the background, but not too large so as to activate multi-photon transitions. An estimated value for the ionisation rate coefficient as low as 1% should suffice to accurately measure the variability of the yield based on the sensitivity of current experimental methods and their high collection efficiency.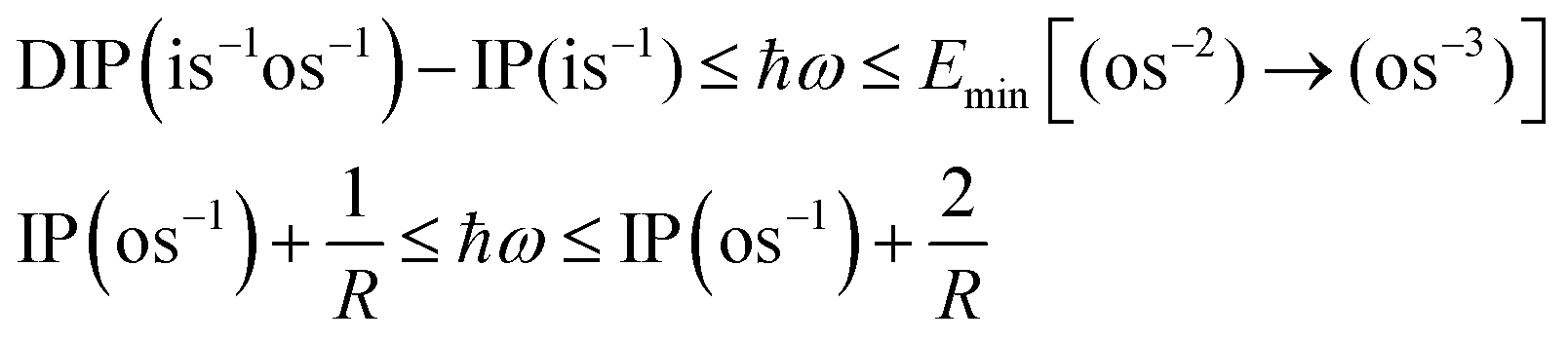
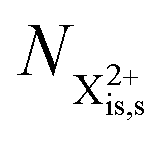 separately from the
separately from the 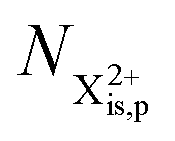 population (that derives from the probed inner-shell-hole
population (that derives from the probed inner-shell-hole 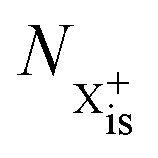 ).
).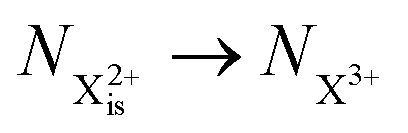 and (b) recapture NX3+ → NX2+. We simplify this in the model by assuming that both stages happen simultaneously, as we are not concerned with the time dynamics of this process. The shake-off state
and (b) recapture NX3+ → NX2+. We simplify this in the model by assuming that both stages happen simultaneously, as we are not concerned with the time dynamics of this process. The shake-off state 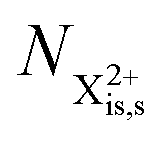 can be further ionised by the probe in proportion R3 to the final state NX3+, which can no longer decay as it is below QIP for inner-valence systems. The updated rate equations for the inner-valence system are therefore as follows:
can be further ionised by the probe in proportion R3 to the final state NX3+, which can no longer decay as it is below QIP for inner-valence systems. The updated rate equations for the inner-valence system are therefore as follows: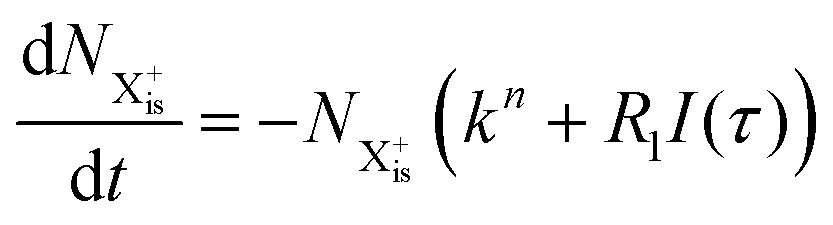
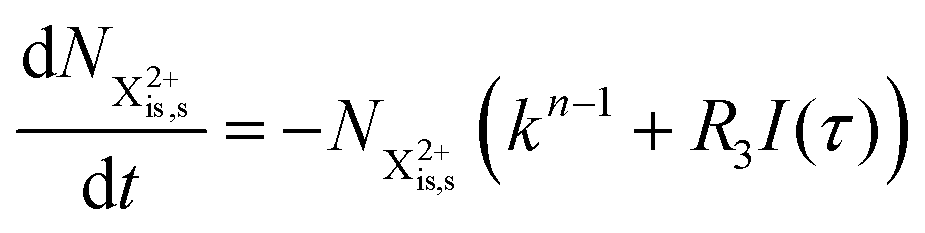
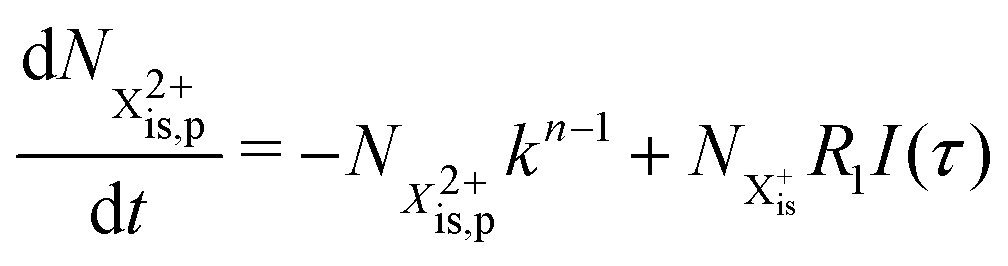
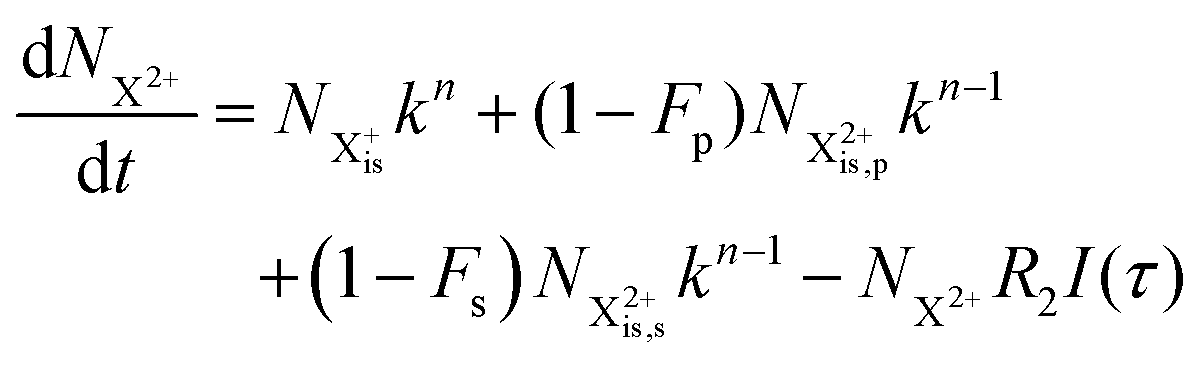
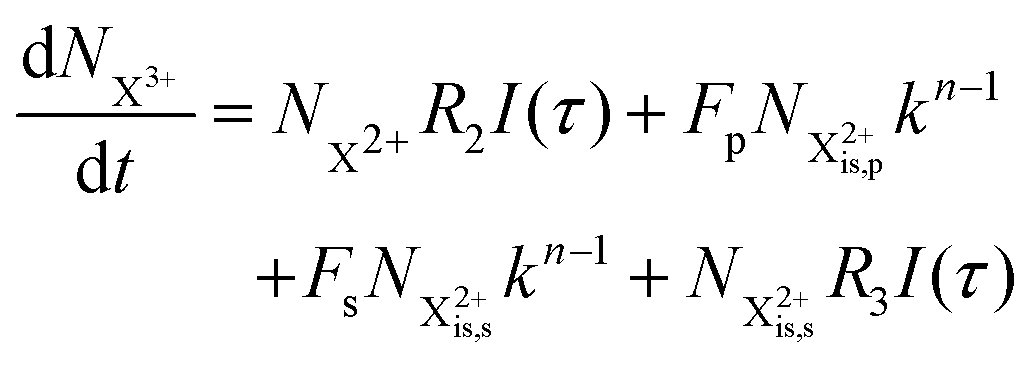
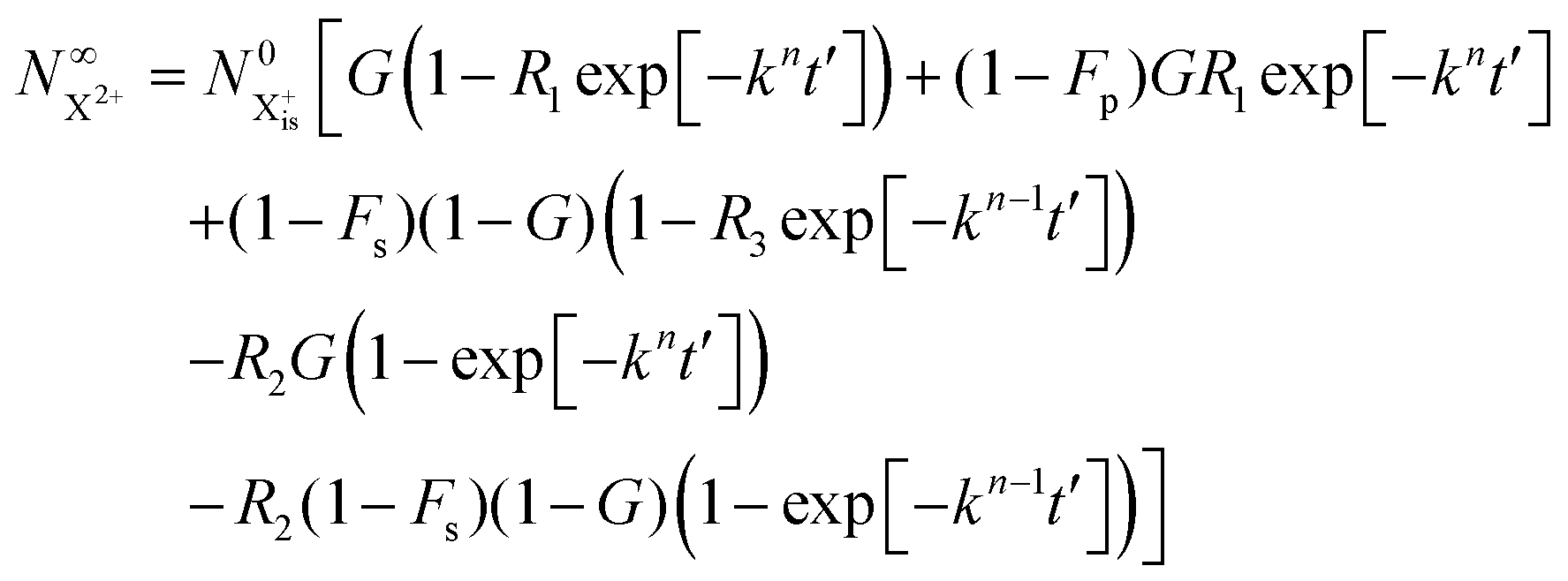
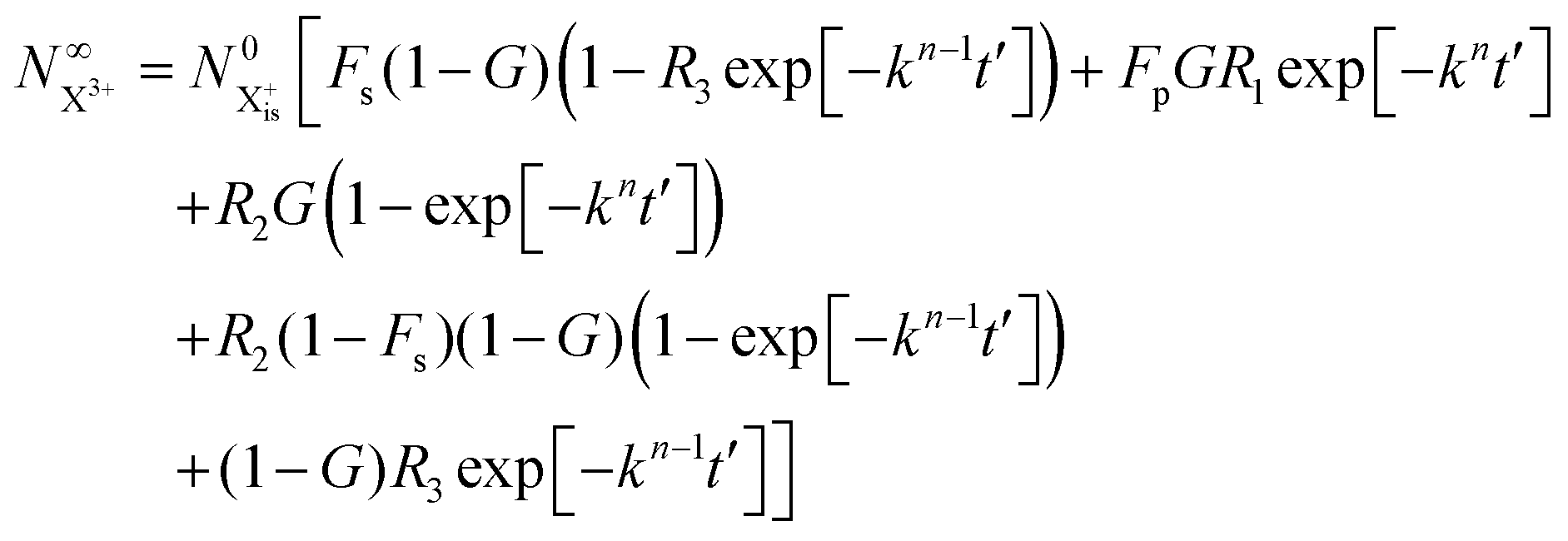
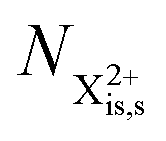 shake-off population which decays at the rate kn−1 before the probe interaction time of t′. Hence, we retrieve the original single exponent dependence by ensuring that the shake-off population produced by the pump photon is negligible (G = 1).
shake-off population which decays at the rate kn−1 before the probe interaction time of t′. Hence, we retrieve the original single exponent dependence by ensuring that the shake-off population produced by the pump photon is negligible (G = 1).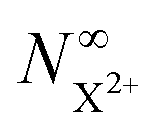 population and by the term on the second line in eqn (34) for the
population and by the term on the second line in eqn (34) for the 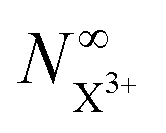 population. This distinction simply modifies the signal that we propose to measure in the inner-valence case by a factor of Fp, which can be ascertained for a given system and chosen probe energy. The equations for the core-hole case can remain unchanged from the original model, provided we ensure that the probe photon gives the resulting photoelectron enough excess energy (but less than that of the pump photoelectron) to preclude recapture by the dication.
population. This distinction simply modifies the signal that we propose to measure in the inner-valence case by a factor of Fp, which can be ascertained for a given system and chosen probe energy. The equations for the core-hole case can remain unchanged from the original model, provided we ensure that the probe photon gives the resulting photoelectron enough excess energy (but less than that of the pump photoelectron) to preclude recapture by the dication.