
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDesign of robust 2,2′-bipyridine ligand linkers for the stable immobilization of molecular catalysts on silicon(111) surfaces†
Samantha I.
Johnson
 ab,
James D.
Blakemore
c,
Bruce S.
Brunschwig
d,
Nathan S.
Lewis
de,
Harry B.
Gray
e,
William A.
Goddard
III
*ae and
Petter
Persson
*b
ab,
James D.
Blakemore
c,
Bruce S.
Brunschwig
d,
Nathan S.
Lewis
de,
Harry B.
Gray
e,
William A.
Goddard
III
*ae and
Petter
Persson
*b
aMaterials Research Center, California Institute of Technology, Pasadena, CA 91125, USA. E-mail: wag@wag.caltech.edu
bTheoretical Chemistry Division, Chemistry Department, Lund University, Box 124, SE-221 00 Lund, Sweden. E-mail: Petter.Persson@teokem.lu.se
cDepartment of Chemistry, University of Kansas, Lawrence, Kansas 66045, USA
dMolecular Materials Resource Center, Beckman Institute, California Institute of Technology, Pasadena, California 91125, USA
eDivision of Chemistry and Chemical Engineering, California Institute of Technology, Pasadena, CA 91125, USA
First published on 6th April 2021
Abstract
The attachment of the 2,2′-bipyridine (bpy) moieties to the surface of planar silicon(111) (photo)electrodes was investigated using ab initio simulations performed on a new cluster model for methyl-terminated silicon. Density functional theory (B3LYP) with implicit solvation techniques indicated that adventitious chlorine atoms, when present in the organic linker backbone, led to instability at very negative potentials of the surface-modified electrode. In prior experimental work, chlorine atoms were present as a trace surface impurity due to required surface processing chemistry, and thus could plausibly result in the observed surface instability of the linker. Free energy calculations for the Cl-atom release process with model silyl-linker constructs revealed a modest barrier (14.9 kcal mol−1) that decreased as the electrode potential became more negative. A small library of new bpy-derived structures has additionally been explored computationally to identify strategies that could minimize chlorine-induced linker instability. Structures with fluorine substituents are predicted to be more stable than their chlorine analogues, whereas fully non-halogenated structures are predicted to exhibit the highest stability. The behavior of a hydrogen-evolving molecular catalyst Cp*Rh(bpy) (Cp* = pentamethylcyclopentadienyl) immobilized on a silicon(111) cluster was explored theoretically to evaluate differences between the homogeneous and surface-attached behavior of this species in a tautomerization reaction observed under reductive conditions for catalytic H2 evolution. The calculated free energy difference between the tautomers is small, hence the results suggest that use of reductively stable linkers can enable robust attachment of catalysts while maintaining chemical behavior on the electrode similar to that exhibited in homogeneous solution.
Introduction
In artificial photosynthesis, solar photons with the aid of suitable light absorbers and electrocatalysts drive reactions that split water, evolving dioxygen (O2).1 The protons and electrons produced in this process can form chemical fuels, either as H2 or as other energy-rich products arising, for example, from reduction of carbon dioxide (CO2). Many technical challenges remain in successful implementation of artificial photosynthesis, including facile approaches to interface catalysts with light absorbers and other device components. Molecular electrocatalysts can be selective, are readily tuned, and have been successfully demonstrated to produce solar fuels at reasonable rates.2–5 Heterogeneous catalytic systems6,7 offer the advantages of simplified product separation and longevity. Hence, immobilization of a homogeneous, molecular catalyst on the surface of an electrode can, in principle, be beneficial,8–14 while potentially avoiding pitfalls such as catalyst deactivation by dimerization or aggregation,15 or insolubility in the chosen solvent/electrolyte.16Extensive effort has focused on preparation of molecular catalysts with reactive functionalities that enable polymerization of catalysts onto, for example, conducting carbon electrodes.17–19 Noncovalent interactions such as π–π interactions have also been exploited to promote association between polycyclic aromatic groups appended to a catalyst and the surfaces of graphitic electrodes. For example, a pyrene-appended Re(CO)3(bpy)Cl catalyst exhibited selective conversion of CO2 to CO when the complex was immobilized on a graphitic carbon electrode. However, loss of catalytic activity attributed to loss of the catalyst from the electrode was observed on a timescale of hours.20 Similarly, a pyrene-appended iridium POCOP (POCOP = κ3-C6H3-2,6-(OPtBu2)2) catalyst was immobilized onto a carbon nanotube gas diffusion electrode and used for formate production from CO2, yielding a turnover number (TON) of >54![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 with little loss in activity.21
000 with little loss in activity.21
The performance of the surface-attached Ir POCOP system can be attributed, in part, to insolubility of the organometallic iridium species in the aqueous electrolyte used for electrocatalysis. Covalent attachment of electrocatalysts to (photo)electrode surfaces might therefore be expected to result in more stable systems than those that only exploit noncovalent interactions. Covalent attachment of catalysts to metallic or semiconducting electrode surfaces exploits a variety of ligands and attachment strategies.22 Stable systems include attachment of a Co porphyrin to a conductive diamond electrode.23 In this system, a relatively long (>9 CH2 units) sp3-hybridized alkane group with a terminal azide was coupled using a CuI-catalyzed click reaction (CuAAC) to a conductive diamond surface that contained a functionalized alkyne. The catalyst was stable for at least 1000 electrochemical cycles and showed a turnover frequency (TOF) of 0.8 s−1 for reduction of CO2 to CO.23 In related work, Fe4N clusters have been attached to glassy carbon electrodes using similar click reactions, yielding stability for >100 h and improved turnover numbers relative to the homogeneous system.24 Other carbon-bound catalyst systems include Co and Ni25,26 catalysts grafted onto glassy carbon surfaces, as well as Rh and Co27,28 catalysts bound to graphite. Ni(P2N2)2 hydrogen-evolution catalysts have also been bound to glassy carbon electrodes, although due to Ni–P bond cleavage the catalysts decompose in acidic acetonitrile.29 H2 evolution catalysts have been attached to semiconducting GaP electrodes,30,31 and Re catalysts have been bound to SiO232 and TiO2.15,33
To obtain Si surfaces modified with vinyl ferrocene, a chlorinated Si(111) surface was first reacted with vinyl-tagged ferrocene. The remaining surface sites were then terminated with methyl groups, using a methyl Grignard reagent.34,35 These covalently bound ferrocene systems exhibited stable redox cycling on both planar silicon(111)31,36 and Si microwire arrays.37 Highlighting the challenge of understanding chemical reactivity occurring on a semiconductor surface, only small amounts of chlorine were detected by X-ray photoelectron (XP) spectroscopy, and an infrared vibrational stretch associated with formation of a C–C double bond in the linker group was not observed.38 Silicon(111) surfaces have also been modified using UV-induced attachment of 4-vinyl-2,2′-bipyridyl. Following attachment of this organic group, the bipyridine on the surface was readily metalated to form surface-attached [Cp*Rh], [Cp*Ir], and [Ru(acac)2] complexes. X-ray photoelectron as well as X-ray absorption spectroscopic data on the immobilized [Cp*Rh] complexes are consistent with formation of the desired metal complexes. However, when the surface-modified electrode was biased to the negative potentials required to reduce Rh(III) to Rh(I) (centered at ∼−1 V vs. ferrocenium/ferrocene), the complexes, and substituted bipyridyl ligand, were lost from the electrode surface within the timescale of three complete cyclic voltammetric cycles.36
Computational approaches have been explored to understand the stabilities and activities of a variety of surface-immobilized redox-active compounds on semiconductor surfaces. Extensive work has been performed on photoanodes in dye-sensitized solar cells (DSSC). Computation has also revealed important phenomena and reactivity in molecular catalysis in particulate photocatalytic systems33,39 as well as for immobilized molecular catalysts deposited on Au clusters (for comparison of theoretical structures to experimental data).40 Typically, molecular calculations are completed with a discrete treatment using Gaussian basis sets,41 while surfaces are approached using a periodic formalism employing plane wave basis sets.42,43 Surface attached molecular entities are challenging because they require treatment at both length scales. One way to bridge between these length scales is to use a molecular cluster, a large molecule of units approximating the surface.44 The active molecule can then be appended onto this cluster. This approach allows us to maximize the accuracy of the properties of the molecular entities and has been used in catalytic33,40 and in DSSC literature.45–47
We report herein a computational investigation of the 4-vinyl-2,2′-bipyridine linker system attached to Si(111) via a photoinitiated olefin immobilization. The results, which provide a plausible failure mechanism supported by the available spectroscopic data, inform a predictive paradigm of alternative linker and catalyst structures that could afford reductively stable surface-immobilized catalysts on Si(111). The results indicate that chlorine used as part of the surface-attachment process is expected theoretically to deleteriously affect catalyst stability when the electrode is biased to very negative electrode potentials. We have additionally investigated theoretically the effect of surface attachment on the behavior of surface-attached Rh-based H2 evolution catalysts. This work is relevant to the important role played by metal-bpy groups in redox catalysis in conjunction with the beneficial use of such systems when attached to electrode surfaces.48–51
Methods
Geometry optimizations were performed using the B3LYP functional52,53 with the Los Alamos small core potential on transition metals.54 Double-zeta basis sets were used on transition metals and the 6-311G** basis set was used on organic atoms.41,55 A Poisson–Boltzmann polarizable continuum acetonitrile solvent model was also used in geometry optimizations. Iso values of 0.005 were evaluated for spin density and orbital plots. Silicon clusters were initially cut along the (111) plane using Crystal,56 and were then trimmed to minimize the number of doubly H-terminated Si atoms. A similar method has been used previously for oxide clusters.57,58 The clusters were large enough to enable a full ring of CH3–Si bonds around the primary bond, minimizing the outward bending of neighboring CH3–Si units. This bending has been observed in smaller clusters and affects the electronic behavior of the system.59 Due to their large size, the energies for large clusters consisted only of electronic and solvation components.(CH3)3Si)3Si-Moieties were used for mechanistic assessment, including transition state calculations for dissociation, because frequency calculations were computationally intractable on larger clusters. All (CH3)3Si)3Si-species were optimized using the methods described above; however, full free energies were calculated from single-point electronic energies, entropies, and enthalpies, in accordance with previously used methods.60,61 Single-point energies were calculated as follows:
| G = EM06 + Gsolv + EZPE + Hvib + HTR − T(Svib + Selec) |
Zero-point energies, EZPE, vibrational enthalpies, Hvib, and vibrational and electronic entropies, Svib and Selec respectively, were obtained from frequency calculations. Translational and rotational enthalpies were taken as 12/2kBT. Single-point electronic energies were calculated with the M06 functional62 and the 6-311G**++ basis set.63,64 Frequency calculations provided the entropic and enthalpic terms. Transition states were verified using these frequency calculations and intrinsic reaction coordinate calculations. All calculations were performed in Jaguar,59 except where otherwise noted.
Results and discussion
Experimental and computational work on H-terminated Si surfaces has shown that photoexcitation of Si–H bond electrons leads to H-atom loss, producing a dangling Si radical.65 This radical then reacts with vinyl-tagged molecules, affording a radical on the β-carbon of the vinyl linker.65,66 For a Cl-terminated Si(111) surface, the analogous attachment mechanism would yield a Cl on the β-carbon of the linker. For ease of reference, the carbon of the linker furthest from the bpy moiety is referred to as the α-carbon and the one closer to bpy is referred to as the β-carbon, while the subscript Si is used to indicate attachment to the Si cluster. Support for this process is evident by comparing the calculated free energies of the various bpy conformations attached to Si(111) clusters with chlorine (Fig. 1). In 1Si, which provides the lowest energy conformation, the Cl is on the β-carbon. The energy of 2Si, with Cl on the α-carbon, is higher by 11.9 kcal mol−1, largely due to an unfavorable steric interaction with neighboring methyl groups bonded to surficial Si atoms. The sp2 analog of the linker shown in Fig. 1 as 3Si, in which HCl is lost in the linking process, is higher than the energy of 1Si by 7.1 kcal mol−1 (due to the lack of steric repulsion in 1Si).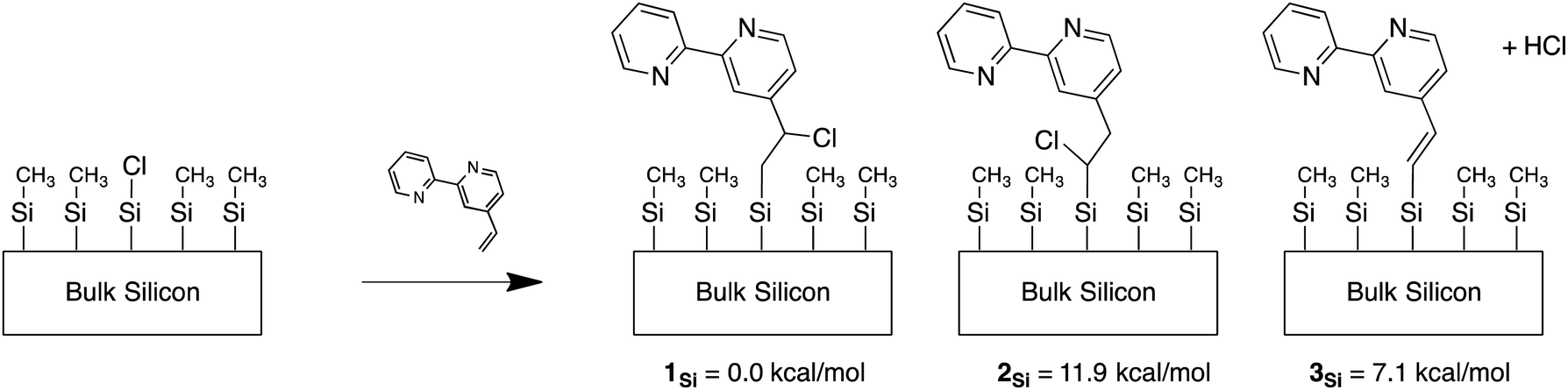 | ||
| Fig. 1 Relative energies (compared to 1Si) for various Cl binding motifs. | ||
To investigate the effect of chlorination on the behavior of the surface-bound complex, large basis set calculations were performed on bipyridine with Cl substituted on the linker (Fig. 2). The molecules were singly reduced, in accord with expectations for the surface chemistry under catalytic conditions. The computations suggest that the reduced complexes 2 and 3 should remain intact, whereas the reduced complex 1 would release a chloride ion under reaction conditions, yielding bpy with a two-carbon radical linker. Spin density is useful in qualitatively determining the “location” of radical population upon reduction. The radical doublet is primarily centered on the β-carbon of the linker, as indicated by atomic charges and spin populations (see Table 1). As shown in Fig. 3, the spin density of the reduced complex extends from a pyridine ring into the linker. The bpy ligand can be reversibly reduced in solution,67 so it should be able to host an extra electron without participation of the linker. However, the linker is theoretically expected to decompose upon reduction when Cl is initially on the β carbon atom (complex 1).
 | ||
| Fig. 2 Molecular complexes with chlorinated linkers. | ||
| Atom | Atomic charges | Spin |
|---|---|---|
| Cl | −0.889 | 0.020 |
| β-Carbon | −0.365 | 0.693 |
| α-Carbon | −0.105 | −0.056 |
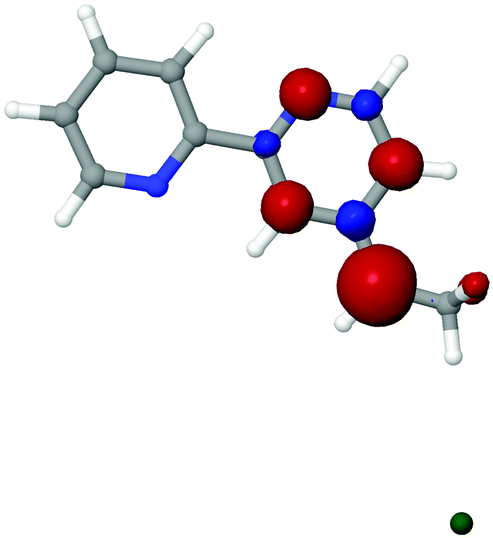 | ||
| Fig. 3 Spin density of the reduced chlorinated bpy complex 1. The chloride ion is released from the β-carbon. | ||
When the reduction process was evaluated computationally with complex 1 attached to a Si cluster, the same decomposition process was observed, and chlorine dissociated. The spin density plot of the attached complex (Fig. 4) was similar to that of the molecular species (Fig. 3).
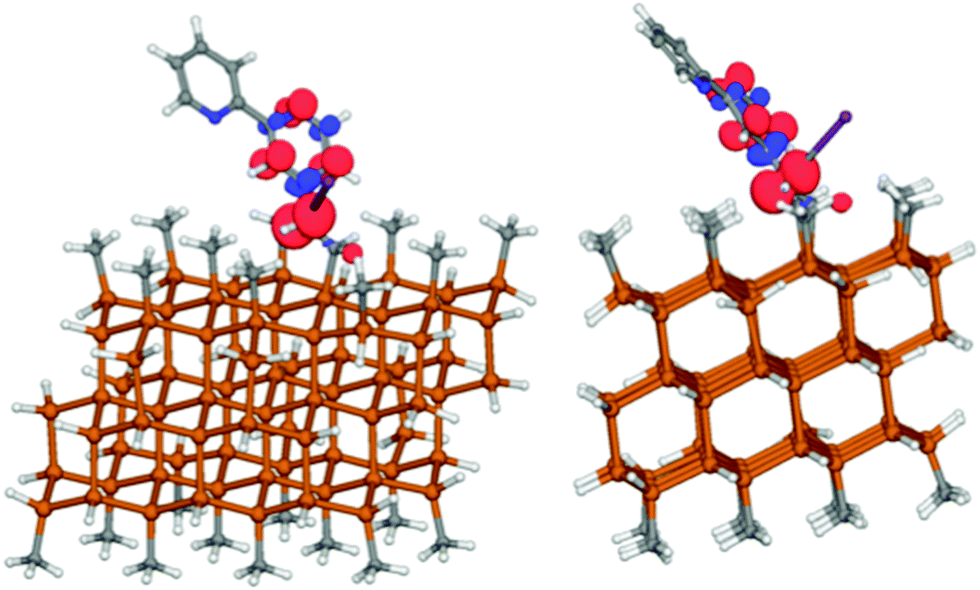 | ||
| Fig. 4 Spin density from two views of the reduced chlorinated complex (1) attached to a silicon cluster. The C–Cl distance is 3.62 Å, suggesting that chlorine has migrated from the linker. | ||
The molecular and cluster calculations collectively suggest a decomposition pathway reliant on the presence of chlorine in the system. During the attachment process, a chlorine radical, formed by photoexcitation that cleaves the Si–Cl bond, is ejected. However, it can recombine with the radical on the β-carbon formed when the vinyl-bpy (v-bpy) ligand attaches to the bare Si atom on the surface, which in turn creates a chlorinated linkage between the bpy complex and the Si surface (Fig. 1, 1Si). This is in agreement with experiments investigating the analogous process in H-terminated Si.65 When the chlorinated bpy linkage is reduced, the chlorine dissociates (as chloride), leaving a neutral doublet radical on the β-carbon. The reaction essentially reverses the attachment process, and thus allows the v-bpy ligand to dissociate from the surface.
To further assess the plausibility of this hypothesis, a (CH3)3Si)3Si-moiety was evaluated computationally as an analogue for the Si(111) surface so that a transition state search can be carried out. Scheme 1 presents the proposed mechanism, using the bpy-bound molecule, 4, as the ground state. H-terminated silanes have been shown previously to undergo an analogous photoactivation process, leading to the formation of a silyl radical that reacts with alkenes.68
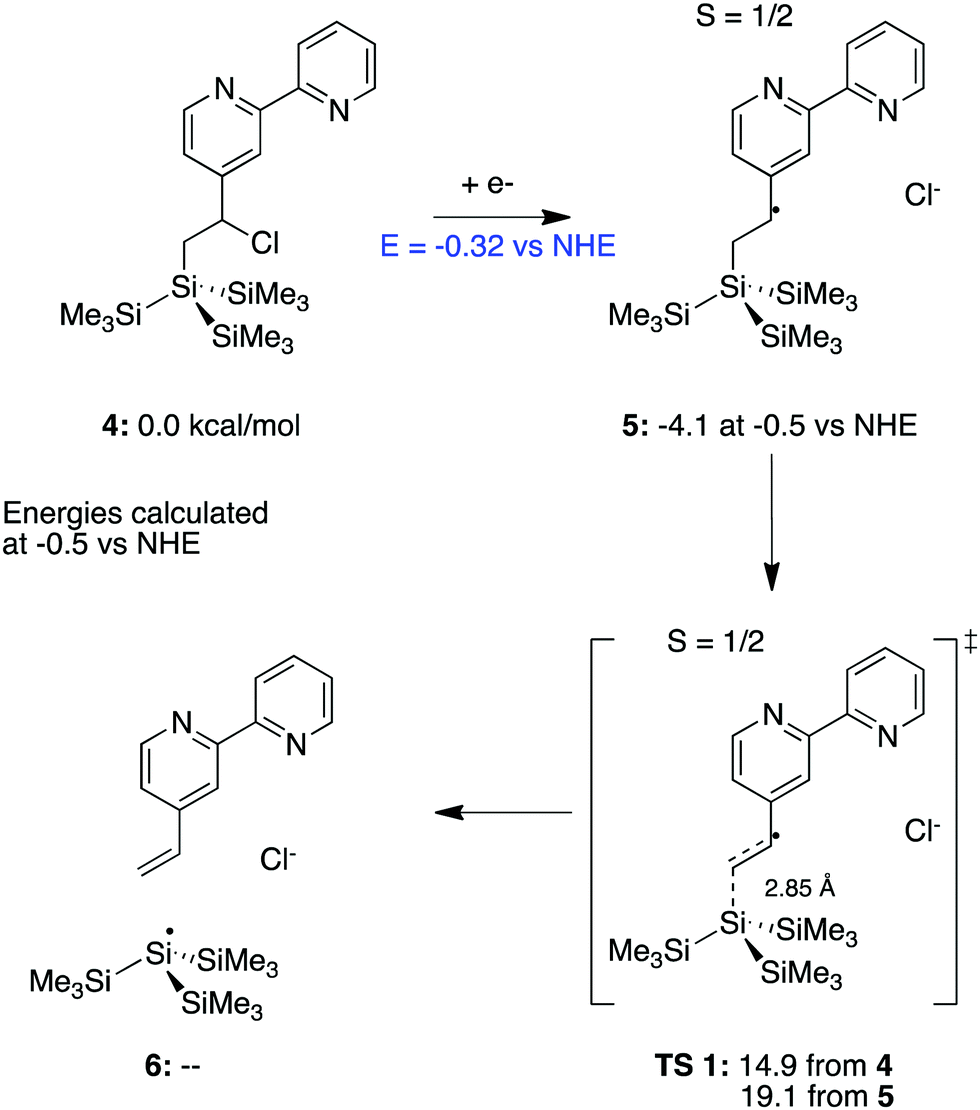 | ||
| Scheme 1 The transition state for dissociation using a tristrimethylsilane small molecule model system is shown in TS 1. While the overall barrier is dependent on the operating potential, the barrier reported from 5 is constant at 19.1 kcal mol−1. All free energies are given in kcal mol−1, except where otherwise specified. Where relevant, energies are calculated at −0.5 V vs. NHE. | ||
The reduction of the chlorinated bpy system initiates loss of the chloride ion and is calculated to occur at −0.32 V vs. NHE. The overall barrier for the separation of the de-chlorinated bpy complex from (CH3)3Si)3Si- is calculated as 14.9 kcal mol−1, using the free energy of an electron at −0.5 V vs. NHE (chosen as a midpoint in the experimentally used range of potentials) from 4. The barrier for dissociation referenced to the immediately preceding 5 (Scheme 1) is independent of the energy of the electron and thus, the electrode potential. This barrier was calculated to be 19.1 kcal mol−1. After dissociation, the unbound bpy moiety would be free to dissociate into solution, leaving the silane behind, shown in 6.
However, the overall barrier depends on the potential at which the reduction occurs (Fig. 5). As the electrode potential is made more negative, loss of the bpy complex occurs more rapidly, and when the electrode potential is −1.1 V vs. NHE, no barrier was computed for the loss of chloride. Studies have shown that computationally obtained bond strengths scaled with the number of Si groups attached to the silane of interest,69 so on the bulk Si crystal surface the reported molecular value represents an upper bound to the actual barrier and dissociation may occur more readily than calculated here. Effectively, the chlorine substituent weakens the attachment of the linker to the surface.
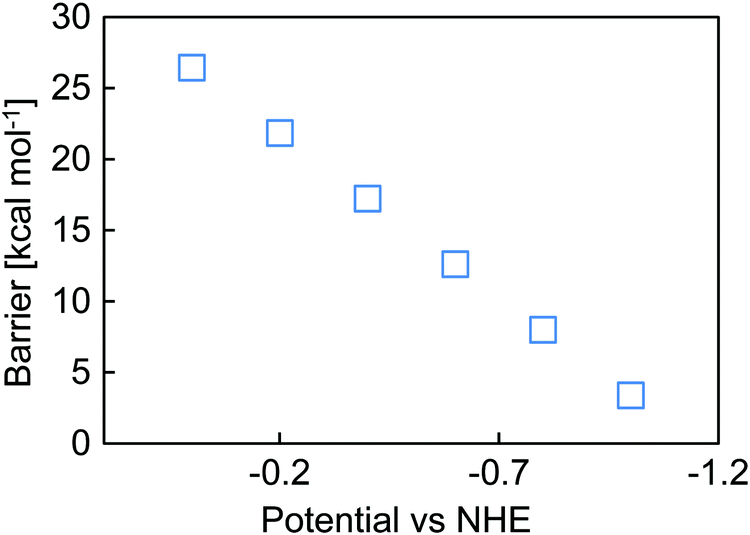 | ||
| Fig. 5 Overall dissociation barrier from (CH3)3Si)3Si as a function of applied potential (V). | ||
Mutually analogous behavior was observed for the molecular and cluster calculations, so the former can be used to evaluate how changes to the complex affect the robustness of its attachment to the surface. One modification involves replacement of the chlorine with other halogens. Structures for the reduced bpy in solution with a fluorine and bromine attached to either the α- or β-carbons of the linker are shown in Fig. 6, complexes 7 and 8.
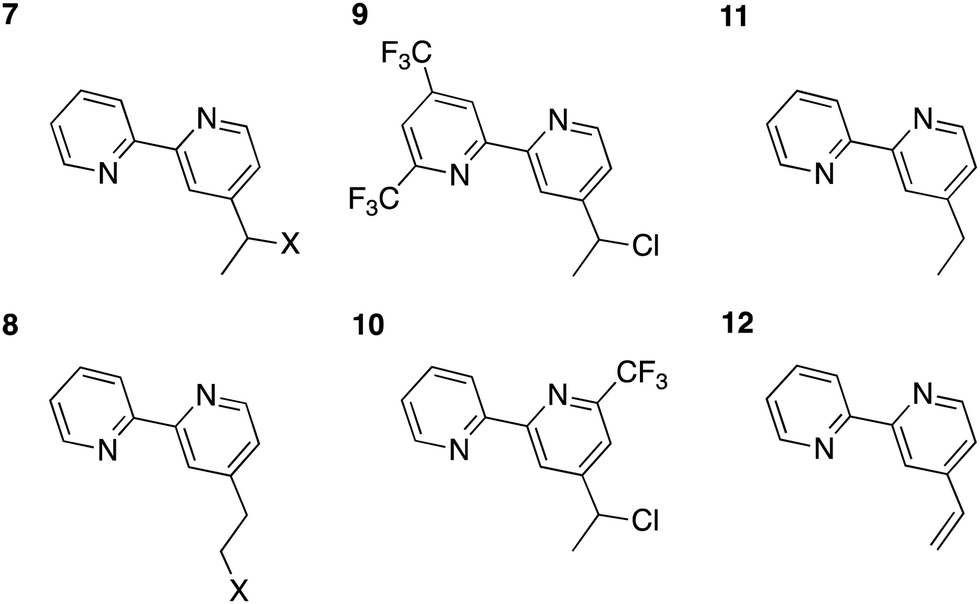 | ||
| Fig. 6 Molecular test analogues for exploitation of bpy redox non-innocence. In the halogenated species 7 and 8, X= F or Br. | ||
Bromine is normally a better leaving group than chlorine, hence it is reasonable that upon reduction, the bromine atom is computed to be readily lost from either position. A fluorine at either position, however, is computed to be stable upon reduction. The spin density in the brominated ligand is quite similar to the chlorinated analogue and extends out on the linker (Fig. 7). However, the fluorinated analogue exhibits much different behavior, with the bulk of spin density localized on the bpy. Consequently, the computations indicate that the fluorine does not dissociate.
 | ||
| Fig. 7 Spin density plots for modified bpy ligands. The density in the fluorinated (7F), bis-CF3 (9) and sp3-hybridized (11) linkers, localized on the bpy, behavior expected for a non-innocent ligand. | ||
The geometry and spin density distribution of the attached ligand is shown in Fig. 8. In the reduced molecular complex, the C–F bond distance in the linker is 1.42 Å, whereas in the bound complex, the distance is 1.44 Å. This very slight lengthening found theoretically is negligible. Similar to the molecular complex, the spin density is largely localized on the bpy ligand, with only a small amount on the fluorine. The spin density on the surface-attached fluorine is 0.006 (which is negligible), whereas the molecular species had no spin density on the fluorine. The difference is not substantial relative to the amount of spin density on the C and N atoms of the bipyridyl group, which ranged from 0.15 to 0.25 on both the molecular and surface-attached complexes. The relatively small difference in fluorine spin densities may be attributed to differences in the basis set used in each case and/or to differences in the chemical environment. In all cases, notably, the spin density was negligible on the β-carbon of the linker and suggest that controlling electron movement within the linker will be key to stability.
 | ||
| Fig. 8 Spin density plot for the surface-attached fluorinated complex. Density is localized on the bpy ligand. | ||
Upon reduction, localization of the added electron on bpy is computationally assessed to leave the linker intact, so approaches to prevent electron movement to the linker can be investigated to increase the robustness of linkers. One logical approach would thus involve addition of electron-withdrawing groups to the bpy moiety. Calculations for both reduced 4′-(1-chloroethyl)-4,6-bis(trifluoromethyl)-2,2′-bipyridine (9) and 4-(1-chloroethyl)-6-(trifluoromethyl)-2,2′-bipyridine (10) were therefore also performed. Trifluoromethyl (CF3) groups are both strongly electron-withdrawing70 and meta-directing, so they were strategically placed to maximize the electron density on the bpy. Fig. 7 shows the spin density plots of 9 and 10. Although 9 showed improvement relative to the original complex 2, some residual spin density was still calculated to be present on the β-carbon of the linker. With one CF3 group in the meta position to the vinyl linkage, as in complex 10, the spin density still extended onto the linker, thus these modifications seem not to be effective enough.
A preferable approach may involve elimination of halogen atoms on the linkers altogether, through either a sp2 or sp3 hybridized framework. Such complexes (11 and 12), and their spin densities, are shown in Fig. 6 and 7. The conjugation between the bpy and the vinyl linkage is clear, as the spin density in the reduced molecule is delocalized over the entire complex. In the sp3 ethyl linkage, the electron is confined to the bpy complex. Berry's work23 with Co porphyrin catalysts utilized a long (greater than 9 CH2 units, largely sp3 hybridized) linkage that exhibited substantial stability. While the length of the linker precludes rapid electron tunneling via the linker, the catalyst can potentially approach the surface, allowing for efficient electron transport via direct contact between the catalyst and the electrode. Berry reported that the cyclic voltammetry of the system stabilized after the first 300 cycles, suggesting that a stable conformation had been reached.
An acetylide sp-hybridized linker was also explored computationally, based on studies involving ethynyl and propynyl functionalization of Si71 and the modification of the Si band structure with fluorinated phenyl groups.72 Such sp-hybridized groups have been used in rigid-rod linker constructions for dye-sensitized solar cells (DSSCs).73–75 To assess these groups as linkers for redox catalytic applications, 4-ethynyl-2,2′-bipyridine moieties were reduced both molecularly and on the cluster (Fig. 9).
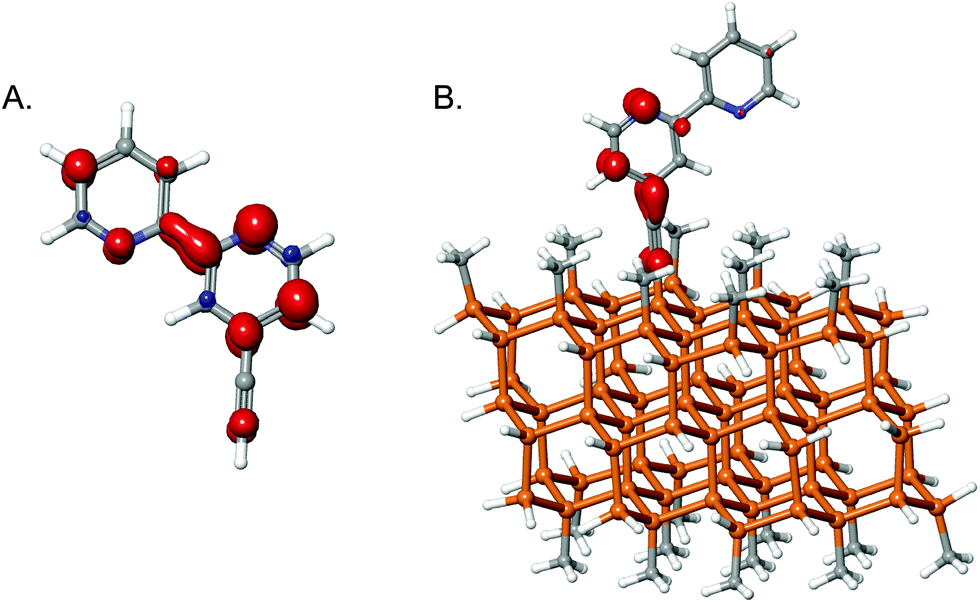 | ||
| Fig. 9 Spin density on reduced 4-ethynyl-2,2′-bipyridine (A) in molecular form and (B) on the cluster surface. | ||
As shown in Fig. 9A, some spin density was calculated for the α-carbon, though none was present on the β-carbon. Once bound to the cluster (Fig. 9B), some electron density extended onto the β-carbon, but most of the spin density was found on the first pyridine ring and on the α-carbon. This behavior contrasts with unbound analogues, but is corroborated by the change in bond length calculated upon reduction. In 2 bound to the cluster (see Fig. 4), the C–C bond length in the linker was calculated to decrease from 1.54 to 1.48 Å due to the loss of the Cl. In contrast, in the sp-hybridized system, the linker extended negligibly, from 1.23 Å to 1.24 Å. By acquiring some electron density, the linker likely will not be as stable as in a fully sp3-hybridized system, but it appears to be more robust than chlorinated linkers, and may facilitate more rapid rates of electron transfer via improved coupling. Using DSSCs as models, Li et al. calculated that fully sp3-hybridized linkers (three carbons in length) reduced the rate of electron injection from a dye into a TiO2 cluster by a factor of ∼7 relative to use of alkene linkers.46 The times for electron injection were in the tens of femtoseconds range. While it is true that TiO2 is different than Si, the fact that orbital overlap needs to occur is still true and the qualitative trend will likely still exist. Even in very active or selective catalysts, the turnover frequencies are usually 0.1 to 10 s−1.2,20,76 Often electron transfer is not the rate-limiting step, but rather binding of the substrate, protonation, or hydride transfer. Hence even with very slow electron-transfer rates, the linker will not likely be limiting for the catalysis.
As an extension of the theoretical methods described above, the Cp*Rh(bpy) H2 evolution catalyst was attached to a Si cluster using a sp hybridized linker. In the molecular system, this catalyst undergoes a two-electron reduction followed by protonation, and both experimental and theoretical work have shown a preference for protonation on the Cp* ring.77,78 The highest occupied molecular orbital (HOMO) electron density is distributed throughout the catalyst (bpy ligand, the Cp* ligand, and the Rh metal center) (Fig. 10, panel A). Interestingly, some density is present computationally on the first carbon of the linker, in contrast to Fig. 9B, in which density is observed on both carbons. Hence the metal and the linker influence one another to determine the robustness of the surface-attached species.
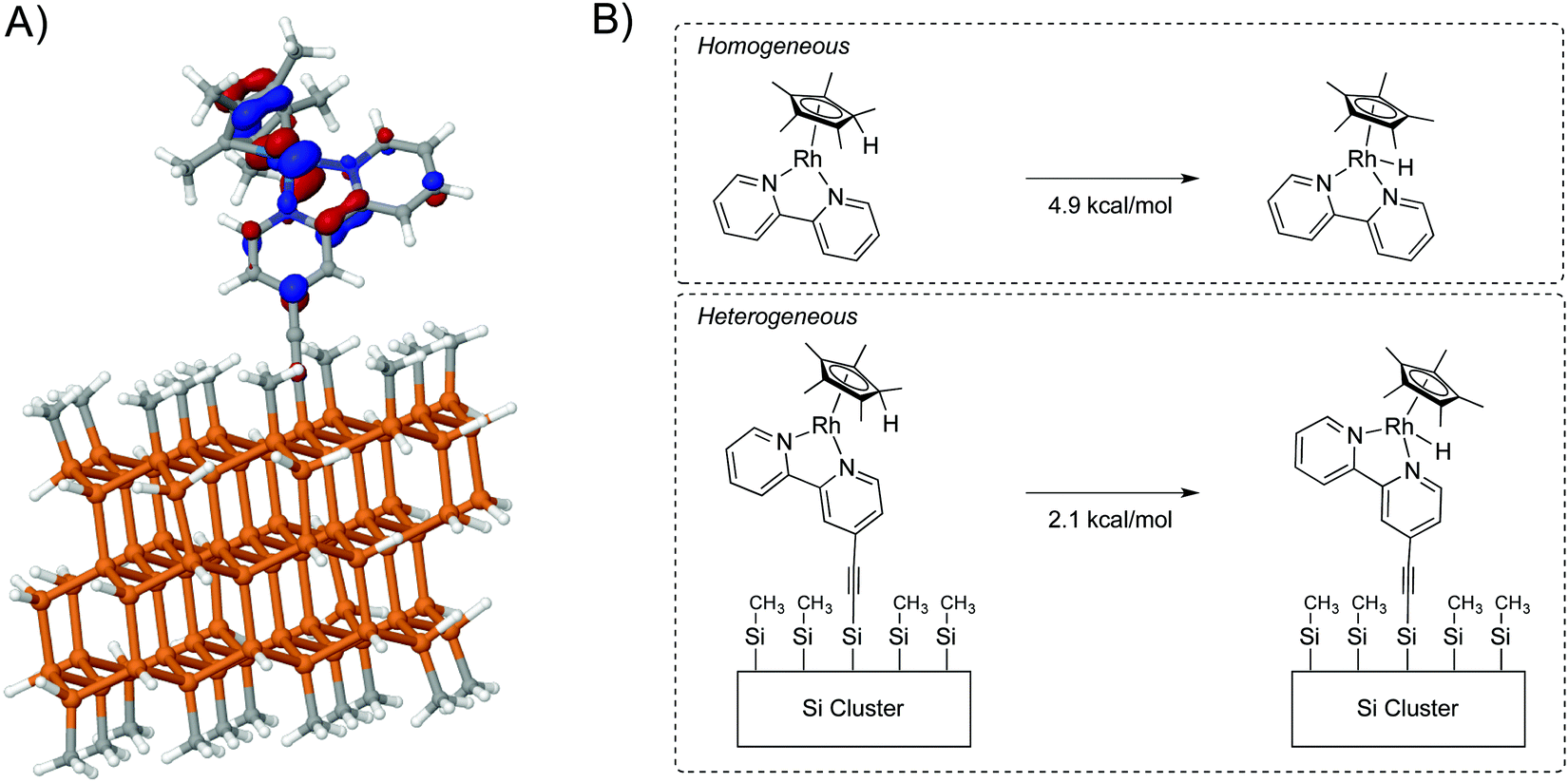 | ||
| Fig. 10 (A) The HOMO of the reduced Rh catalyst on the Si cluster (B) Electronic and solvation energies of homogeneous and heterogenized Rh clusters. While there is a difference in energies, the tautomer preference is preserved. | ||
Panel B of Fig. 10 compares tautomers of homogeneous and surface-attached systems. The large size of the cluster limits the level of theory that can be used, so the difference in energy (E + ΔGsolv) between metal hydride and protonated ligand analogues was calculated to be 4.9 kcal mol−1, with preference for the protonated ligand. The difference in energy between attached Cp*Rh(H) and (Cp*H)Rh complexes was calculated as 2.1 kcal mol−1, again with a preference for the protonated ligand. The overall chemistry was not changed, but a shift in the relative energies is observed computationally.
Comparison of the tautomerization energy to the previous Hammett parameter analysis78 implies that the Si cluster is more electron-withdrawing than modified bipyridine. The calculations notably suggest that surface attachment itself constitutes another approach to tune the behavior of a catalyst, which can be tuned further by applying a voltage.79 This expectation is consistent with the change in mechanism for CO2 reduction to formate reported by Berben and coworkers for Fe4N clusters covalently bound to graphitic carbon. In homogeneous catalysis, the hydride is generated by stepwise electron transfer and coupled proton transfer steps, whereas for the linker-tuned system catalysis occurs via a concerted electron/proton transfer step.24 An interesting extension of this work would be to examine how the bound (Cp*H)Rh moiety affects the band structure of the electrode, as has been recently investigated by Rose and coworkers.80
In summary, the detachment of bipyridine-based moieties from Si(111) was assessed with the goal of identifying covalently-bound systems with improved robustness under operating conditions. Studies of cluster and molecular model revealed the use of chlorine in the attachment process as a potential source of linker weakness in electrochemical processes at very negative electrode potentials. Linkers having sp-hybridization, previously used for band-edge modification of Si, were also explored computationally. Attachment, reduction, and protonation of Cp*Rh(bpy) were evaluated computationally using a sp hybridized linker, with findings similar to those established for the homogeneous catalyst. Based on the combination of ab initio calculations and experimental data, the apparent robustness of sp-hybridized linker systems appears to make them promising candidates for construction of covalently modified Si-based catalytic electrodes.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
Research at Caltech was supported by the National Science Foundation Center for Chemical Innovation Solar Fuels (NSF CHE-1305124). S. I. J. was supported by a National Science Foundation Graduate Research Fellowship (Grant No. DGE-1144469), a National Science Foundation Graduate Research Opportunities Worldwide Grant, and by the generous support of the Resnick Sustainability Institute. S. I. J. and P. P. were supported by the Swedish Research Council (VR). W. A. G. was partially supported by the Liquid Sunlight Alliance, which is supported by the U.S. Department of Energy, Office of Science, Office of Basic Energy Sciences, Fuels from Sunlight Hub under Award Number DE-SC0021266. P. P. acknowledges the Swedish Energy Agency (STEM) as well as the Swedish supercomputing facilities LUNARC and NSC for support. S. I. J. thanks Drs. Robert J. Nielsen and Noah Plymale for helpful discussions. S. I. J. and P. P. thank Drs. Lisa A. Fredin and Marta Galynska for helpful discussions.References
- N. S. Lewis and D. G. Nocera, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 15729–15735 CrossRef CAS.
- P. Kang, C. Cheng, Z. Chen, C. K. Schauer, T. J. Meyer and M. Brookhart, J. Am. Chem. Soc., 2012, 134, 5500–5503 CrossRef CAS PubMed.
- E. E. Benson, C. P. Kubiak, A. J. Sathrum and J. M. Smieja, Chem. Soc. Rev., 2009, 38, 89–99 RSC.
- C. Costentin, M. Robert and J.-M. Saveant, Chem. Soc. Rev., 2013, 42, 2423–2436 RSC.
- P. G. Jessop, F. Joó and C.-C. Tai, Coord. Chem. Rev., 2004, 248, 2425–2442 CrossRef CAS.
- B. D. Matson, C. T. Carver, A. Von Ruden, J. Y. Yang, S. Raugei and J. M. Mayer, Chem. Commun., 2012, 48, 11100–11102 RSC.
- K. P. Kuhl, E. R. Cave, D. N. Abram and T. F. Jaramillo, Energy Environ. Sci., 2012, 5, 7050–7059 RSC.
- H. D. Abruña, Coord. Chem. Rev., 1988, 86, 135–189 CrossRef.
- R. W. Murray, Acc. Chem. Rev., 1980, 13, 135–141 CrossRef CAS.
- R. L. McCreery, Chem. Rev., 2008, 108, 2646–2687 CrossRef CAS PubMed.
- M. S. Wrighton, Science, 1986, 231, 32–37 CrossRef CAS PubMed.
- J. L. Inglis, B. J. MacLean, M. T. Pryce and J. G. Vos, Coord. Chem. Rev., 2012, 256, 2571–2600 CrossRef CAS.
- F. Franco, C. Rettenmaier, H. S. Jeon and B. Roldan Cuenya, Chem. Soc. Rev., 2020, 49, 6884–6946 RSC.
- D.-H. Nam, P. De Luna, A. Rosas-Hernández, A. Thevenon, F. Li, T. Agapie, J. C. Peters, O. Shekhah, M. Eddaoudi and E. H. Sargent, Nat. Mater., 2020, 19, 266–276 CrossRef CAS PubMed.
- C. D. Windle, E. Pastor, A. Reynal, A. C. Whitwood, Y. Vaynzof, J. R. Durrant, R. N. Perutz and E. Reisner, Chem. – Eur. J., 2015, 21, 3746–3754 CrossRef CAS PubMed.
- C. M. Hanna, C. D. Sanborn, S. Ardo and J. Y. Yang, ACS Appl. Mater. Interfaces, 2018, 10, 13211–13217 CrossRef CAS PubMed.
- S. Chardon-Noblat, A. Deronzier, R. Ziessel and D. Zsoldos, J. Electroanal. Chem., 1998, 444, 253–260 CrossRef CAS.
- G. J. Samuels and T. J. Meyer, J. Am. Chem. Soc., 1981, 103, 307–312 CrossRef CAS.
- X. Zhang, Z. Wu, X. Zhang, L. Li, Y. Li, H. Xu, X. Li, X. Yu, Z. Zhang, Y. Liang and H. Wang, Nat. Commun., 2017, 8, 14675 CrossRef PubMed.
- J. D. Blakemore, A. Gupta, J. J. Warren, B. S. Brunschwig and H. B. Gray, J. Am. Chem. Soc., 2013, 135, 18288–18291 CrossRef CAS PubMed.
- P. Kang, S. Zhang, T. J. Meyer and M. Brookhart, Angew. Chem., 2014, 126, 8853–8857 CrossRef.
- C. Sun, R. Gobetto and C. Nervi, New J. Chem., 2016, 40, 5656–5661 RSC.
- S. A. Yao, R. E. Ruther, L. Zhang, R. A. Franking, R. J. Hamers and J. F. Berry, J. Am. Chem. Soc., 2012, 134, 15632–15635 CrossRef CAS PubMed.
- D. B. Cluff, A. Arnold, J. C. Fettinger and L. A. Berben, Organometallics, 2019, 38, 1230–1235 CrossRef CAS.
- N. Elgrishi, S. Griveau, M. B. Chambers, F. Bedioui and M. Fontecave, Chem. Commun., 2015, 51, 2995–2998 RSC.
- A. Zhanaidarova, C. E. Moore, M. Gembicky and C. P. Kubiak, Chem. Commun., 2018, 54, 4116–4119 RSC.
- S. Oh, J. R. Gallagher, J. T. Miller and Y. Surendranath, J. Am. Chem. Soc., 2016, 138, 1820–1823 CrossRef CAS PubMed.
- C. J. Kaminsky, J. Wright and Y. Surendranath, ACS Catal., 2019, 9, 3667–3671 CrossRef CAS.
- A. K. Das, M. H. Engelhard, R. M. Bullock and J. A. S. Roberts, Inorg. Chem., 2014, 53, 6875–6885 CrossRef CAS PubMed.
- D. Khusnutdinova, A. M. Beiler, B. L. Wadsworth, S. I. Jacob and G. F. Moore, Chem. Sci., 2017, 8, 253–259 RSC.
- G. F. Moore and I. D. Sharp, J. Phys. Chem. Lett., 2013, 4, 568–572 CrossRef CAS PubMed.
- K. D. Dubois, H. He, C. Liu, A. S. Vorushilov and G. Li, J. Mol. Catal. Chem., 2012, 363–364, 208–213 CrossRef CAS.
- C. L. Anfuso, R. C. Snoeberger, A. M. Ricks, W. Liu, D. Xiao, V. S. Batista and T. Lian, J. Am. Chem. Soc., 2011, 133, 6922–6925 CrossRef CAS PubMed.
- A. Bansal, X. Li, I. Lauermann, N. S. Lewis, S. I. Yi and W. H. Weinberg, J. Am. Chem. Soc., 1996, 118, 7225–7226 CrossRef CAS.
- E. J. Nemanick, P. T. Hurley, B. S. Brunschwig and N. S. Lewis, J. Phys. Chem. B, 2006, 110, 14800–14808 CrossRef CAS PubMed.
- J. R. C. Lattimer, J. D. Blakemore, W. Sattler, S. Gul, R. Chatterjee, V. K. Yachandra, J. Yano, B. S. Brunschwig, N. S. Lewis and H. B. Gray, Dalton Trans., 2014, 43, 15004–15012 RSC.
- O. S. Kang, J. P. Bruce, D. E. Herbert and M. S. Freund, ACS Appl. Mater. Interfaces, 2015, 7, 26959–26967 CrossRef CAS PubMed.
- J. R. C. Lattimer, B. S. Brunschwig, N. S. Lewis and H. B. Gray, J. Phys. Chem. C, 2013, 117, 27012–27022 CrossRef CAS.
- A. C. Durrell, G. Li, M. Koepf, K. J. Young, C. F. A. Negre, L. J. Allen, W. R. McNamara, H.-E. Song, V. S. Batista, R. H. Crabtree and G. W. Brudvig, J. Catal., 2014, 310, 37–44 CrossRef CAS.
- M. L. Clark, B. Rudshteyn, A. Ge, S. A. Chabolla, C. W. Machan, B. T. Psciuk, J. Song, G. Canzi, T. Lian, V. S. Batista and C. P. Kubiak, J. Phys. Chem. C, 2016, 120, 1657–1665 CrossRef CAS.
- W. J. Hehre, R. Ditchfie and J. A. Pople, J. Chem. Phys., 1972, 56, 2257–2261 CrossRef CAS.
- G. Makov and M. C. Payne, Phys. Rev. B, 1995, 51, 4014–4022 CrossRef CAS PubMed.
- C. J. Cramer, Essentials of Computational Chemistry, Wiley, 2004 Search PubMed.
- G. A. Ferguson and K. Raghavachari, J. Chem. Phys., 2006, 125, 154708 CrossRef PubMed.
- P. Persson, M. J. Lundqvist, R. Ernstorfer, W. A. Goddard and F. Willig, J. Chem. Theory Comput., 2006, 2, 441–451 CrossRef CAS PubMed.
- J. Li, H. Wang, P. Persson and M. Thoss, J. Chem. Phys., 2012, 137, 22A529 CrossRef.
- P. Persson, in Solar Energy Conversion: Dynamics of Interfacial Electron and Excitation Transfer, ed. P. Piotrowiak, RSC, 2013, ch. 3, pp. 77–110 Search PubMed.
- L. M. A. Quintana, S. I. Johnson, S. L. Corona, W. Villatoro, W. A. Goddard, M. K. Takase, D. G. VanderVelde, J. R. Winkler, H. B. Gray and J. D. Blakemore, Proc. Natl. Acad. Sci. U. S. A., 2016, 113, 6409 CrossRef PubMed.
- C. M. Bolinger, N. Story, B. P. Sullivan and T. J. Meyer, Inorg. Chem., 1988, 27, 4582–4587 CrossRef CAS.
- M. K. Brennaman, R. J. Dillon, L. Alibabaei, M. K. Gish, C. J. Dares, D. L. Ashford, R. L. House, G. J. Meyer, J. M. Papanikolas and T. J. Meyer, J. Am. Chem. Soc., 2016, 138, 13085–13102 CrossRef CAS PubMed.
- J. D. Blakemore, N. D. Schley, D. Balcells, J. F. Hull, G. W. Olack, C. D. Incarvito, O. Eisenstein, G. W. Brudvig and R. H. Crabtree, J. Am. Chem. Soc., 2010, 132, 16017–16029 CrossRef CAS.
- A. D. Becke, J. Chem. Phys., 1993, 98, 5648–5652 CrossRef CAS.
- C. T. Lee, W. T. Yang and R. G. Parr, Phys. Rev. B, 1988, 37, 785–789 CrossRef CAS PubMed.
- P. J. Hay and W. R. Wadt, J. Chem. Phys., 1985, 82, 299–310 CrossRef CAS.
- M. M. Francl, W. J. Pietro, W. J. Hehre, J. S. Binkley, M. S. Gordon, D. J. Defrees and J. A. Pople, J. Chem. Phys., 1982, 77, 3654–3665 CrossRef CAS.
- R. Dovesi, R. Orlando, B. Civalleri, C. Roetti, V. R. Saunders and C. M. Zicovich-Wilson, Z. Kristallogr., 2005, 220, 571–573 CAS.
- M. Galynska and P. Persson, in Advances in Quantum Chemistry, ed. J. R. Sabin, 2014, vol. 69, pp. 303–332 Search PubMed.
- M. Galynska and P. Persson, Int. J. Quantum Chem., 2013, 113, 2611–2620 CrossRef CAS.
- A. D. Bochevarov, E. Harder, T. F. Hughes, J. R. Greenwood, D. A. Braden, D. M. Philipp, D. Rinaldo, M. D. Halls, J. Zhang and R. A. Friesner, Int. J. Quantum Chem., 2013, 113, 2110–2142 CrossRef CAS.
- S. I. Johnson, R. J. Nielsen and W. A. Goddard, ACS Catal., 2016, 6362–6371 CrossRef CAS.
- M. Zhou, S. I. Johnson, Y. Gao, T. J. Emge, R. J. Nielsen, W. A. Goddard and A. S. Goldman, Organometallics, 2015, 34, 2879–2888 CrossRef CAS.
- Y. Zhao and D. G. Truhlar, Theor. Chem. Acc., 2008, 120, 215–241 Search PubMed.
- R. Krishnan, J. S. Binkley, R. Seeger and J. A. Pople, J. Chem. Phys., 1980, 72, 650–654 CrossRef CAS.
- T. Clark, J. Chandrasekhar, G. W. Spitznagel and P. V. Schleyer, J. Comput. Chem., 1983, 4, 294–301 CrossRef CAS.
- R. L. Cicero, M. R. Linford and C. E. D. Chidsey, Langmuir, 2000, 16, 5688–5695 CrossRef CAS.
- J. M. Buriak, Chem. Mater., 2014, 26, 763–772 CrossRef CAS.
- T. Saji and S. Aoyagui, J. Electroanal. Chem. Interfacial Electrochem., 1975, 58, 401–410 CrossRef CAS.
- C. Chatgilialoglu, Chem. Rev., 1995, 95, 1229–1251 CrossRef CAS.
- Y.-D. Wu and C.-L. Wong, J. Org. Chem., 1995, 60, 821–828 CrossRef CAS.
- C. Hansch, A. Leo and R. W. Taft, Chem. Rev., 1991, 91, 165–195 CrossRef CAS.
- N. T. Plymale, Y.-G. Kim, M. P. Soriaga, B. S. Brunschwig and N. S. Lewis, J. Phys. Chem. C, 2015, 119, 19847–19862 CrossRef CAS.
- N. T. Plymale, A. A. Ramachandran, A. Lim, B. S. Brunschwig and N. S. Lewis, J. Phys. Chem. C, 2016, 120, 14157–14169 CrossRef CAS.
- K. Chitre, A. Batarseh, A. Kopecky, H. Fan, H. Tang, R. Lalancette, R. A. Bartynski and E. Galoppini, J. Phys. Chem. B, 2015, 119, 7522–7530 CrossRef CAS.
- E. Galoppini, Coord. Chem. Rev., 2004, 248, 1283–1297 CrossRef CAS.
- D. Wang, R. Mendelsohn, E. Galoppini, P. G. Hoertz, R. A. Carlisle and G. J. Meyer, J. Phys. Chem. B, 2004, 108, 16642–16653 CrossRef CAS.
- S. Cosnier, A. Deronzier and N. Vlachopoulos, Chem. Commun., 1989, 1259–1261 RSC.
- L. M. A. Quintana, S. I. Johnson, S. L. Corona, W. Villatoro, W. A. Goddard, M. K. Takase, D. G. VanderVelde, J. R. Winkler, H. B. Gray and J. D. Blakemore, Proc. Natl. Acad. Sci. U. S. A., 2016, 113, 6409 CrossRef PubMed.
- S. I. Johnson, H. B. Gray, J. D. Blakemore and W. A. Goddard, Inorg. Chem., 2017, 56, 11375–11386 CrossRef CAS PubMed.
- J. Heo, H. Ahn, J. Won, J. G. Son, H. K. Shon, T. G. Lee, S. W. Han and M.-H. Baik, Science, 2020, 370, 214 CrossRef CAS PubMed.
- D. G. Boucher, K. Kearney, E. Ertekin and M. J. Rose, J. Am. Chem. Soc., 2021, 143(6), 2567–2580 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1cp00545f |
| This journal is © the Owner Societies 2021 |