
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceFrom absolute potentials to a generalized computational standard hydrogen electrode for aqueous and non-aqueous solvents†
Michael
Busch
 *a,
Elisabet
Ahlberg
b and
Kari
Laasonen
a
*a,
Elisabet
Ahlberg
b and
Kari
Laasonen
a
aDepartment of Chemistry and Material Science, School of Chemical Engineering, Aalto University Kemistintie 1, 02150 Espoo, Finland. E-mail: michael.busch@aalto.fi
bDepartment of Chemistry and Molecular Biology, University of Gothenburg, Kemigården 4, 41296 Gothenburg, Sweden
First published on 4th May 2021
Abstract
We describe a simple and efficient procedure to compute a conversion factor for the absolute potential of the standard hydrogen electrode in water to any other solvent. In contrast to earlier methods our procedure only requires the pKa of an arbitrary acid in water and few simple quantum chemical calculations as input. Thus, it is not affected adversely by experimental shortcomings related to measurements in non-aqueous solvents. By combining this conversion factor with the absolute potential in water, the absolute potential in the solvent of interest is obtained. Based on this procedure a new generalized computational standard hydrogen electrode for the computation of electron transfer and proton-coupled electron transfer potentials in non-aqueous solvents and ionic liquids is developed. This enables for the first time the reliable prediction of redox potentials in any solvent. The method is tested through calculation of absolute potentials in 36 solvents. Using the Kamlet–Taft linear solvation energy model we find that the relative absolute potentials consistently increase with decreasing polarisability and decreasing hydrogen bonding ability. For protic solvents good agreement with literature is observed while significant deviations are found for aprotic solvents. The obtained conversion factors are independent of the quantum chemical method, while minor differences are observed between solvation models. This does, however, not affect the global trends.
1 Introduction
The prediction of redox potentials using quantum-chemical calculations is central for understanding mechanisms and developing materials for electro-synthesis,1,2 energy storage3–8 and energy conversion.9–16 Experimentally, these reactions are typically performed in water but non-aqueous solvents and ionic liquids are also far from uncommon.1,17–20 Owing to the importance of these applications, several computational protocols for the prediction of redox potentials have been developed over the last decades. Proton coupled electron transfer (PCET) reactions, for example, are evaluated using the computational standard hydrogen electrode which uses gas phase H2 formed from H+ and e− as a reference reaction.21,22 This intrinsically connects the PCET step to the standard hydrogen electrode (SHE) scale. Naturally, also any other reference may be used,15,23 provided its redox potential is known with sufficient accuracy.ET reactions on the other hand are significantly more difficult to model. From an electrochemical perspective certainly the most natural approach is, similar to the computational SHE, the use of a suitable reference reaction. Owing to the large amount of very detailed experimental studies on ferrocene and its use as an internal ref. 24, this system could be considered ideal for this purpose. Indeed, metallocenes have been considered as reference systems in several computational studies.25,26 But owing to the complex electronic structure of transition metal complexes, significant errors must be expected for these compounds.27–29 These complications could be avoided by instead relying on simple organic molecules such as benzene or toluene. However, cyclic voltammetry (CV) measurements commonly used to determine the experimental redox potentials for organic molecules are, due to the potential coupling of the ET step with a subsequent fast chemical reaction, often unreliable.30,31 Thus, selecting a suitable reference reaction is far from trivial and requires significant experience with experimental electrochemical techniques.
An alternative procedure to computationally determine ET potentials relies on the absolute potential of the hydrogen evolution reaction (HER). Generally, constant values of 4.44 V32,33 or 4.28 V34,35 are used to offset the electrochemical step to the SHE scale in water. The difference between both values is a result of the inclusion of the surface potential in the former (the outer or Volta potential is reported) whereas this contribution is not included in the latter value suggested by Kelly et al. (the inner or Galvani potential is reported). The surface potential is a result of electrostatic interactions at the liquid–gas or liquid–electrode interface.36,37 Recently, it was argued that these contributions should be absent in ideal solutions owing to the absence of ionic interactions.38 However, following the original argument of Lange et al.37 dipole interactions resulting for example from differences in the solvation shell of the molecule between the end pointing towards the liquid and gas phase are sufficient for the surface potential to form. Whether this contribution must be included in a computation strongly depends on the construction of the implicit solvation model and is thus, not obvious.39 Furthermore, information on the detailed determination of the semi-empirical parameters used in the construction of implicit solvation models required for this decision is scarce in the literature.40,41 Additionally, it must be cautioned that besides these “commonly accepted” absolute potentials38 also alternative values ranging from 4.05 V to 4.70 V have been proposed.42–48
A possibility to avoid these complications offers the recently developed “effective absolute potential method” in which the most appropriate absolute potential for a given setup is estimated from the acid dissociation constant of a reference system.42 Thus, ambiguities resulting from the choice of the most suitable absolute potential can be avoided. Additionally, this procedure increases the overall accuracy of the predicted ET potentials by removing systematic errors.42
A major shortcoming of the absolute potential based methods for predicting ET potentials is, however, the difficulty to reference the reaction to the SHE in solvents other than water. When using the constant absolute potential approach, values have only been reported for very few non-aqueous solvents such as acetone or acetonitrile.33,38,49,50 The “effective absolute potential method” on the other hand is limited by the need to rely on accurately measured acid dissociation constants in the solvent of interest. This limitation could in principle be avoided by using experimentally determined conversion factors between different solvents. However, these conversion factors are often unreliable or missing.24,35 Thus, this procedure is effectively limited to water and other protic solvents for which pKa values have been determined with sufficient accuracy.51,52 This is opposed to the computational standard hydrogen electrode used to predict PCET redox potentials. Owing to the use of gas phase hydrogen as reference, the potential is always offset with respect to the SHE in the solvent of interest. Accordingly, PCET and ET steps are related to different reference systems and need to be converted to a common SHE scale if both reactions types are present.
In what follows we will describe a method to compute the required conversion factors based on the pKa of an arbitrary acid in a freely chosen solvent. This procedure will enable the efficient prediction of absolute potentials in any solvent and thus, allow for the efficient computation of redox potentials in non-aqueous media. Additionally, the absolute potential can be used to predict the solvation energy of protons which in turn enables the easy prediction of acid dissociation constants in non-aqueous solvents. The knowledge of acid–base properties in non-aqueous solvents is crucial for problems in many areas of chemistry such as organic synthesis.53,54 The validity of this generalized computational standard hydrogen electrode is tested by computing the absolute potentials of the SHE for 36 organic solvents and ionic liquids. The global trends in the relative stability of the dissolved proton are evaluated taking advantage of linear solvation energy relations.55
2 Computational details
All density functional theory (DFT) calculations using either B3LYP56–58 or M06-2X59 and coupled cluster CCSD(T) computations were performed within Gaussian 16 Rev B.01.60 For B3LYP, Grimme D3 corrections were added in order to properly treat dispersion interactions.61 An ultra-fine grid was used in all cases. For the DFT methods a 6-311++G** basis set, which includes polarization and diffuse functions on all atoms, was used while the aug-cc-pvqz basis set was employed for CCSD(T) calculations. The latter was found to allow for well converged ab initio results.42 Solvation effects were included using the implicit SMD solvation model.62 The SMD parameters for the considered ionic liquids were taken from the work of Bernales et al.63 The influence of the solvation model was tested by varying the cavity scaling (α) in the SMD model between 1.0 and 1.2 and through additional calculations using the Poisson–Boltzmann Finite elements (PBF) solvation model64–66 as implemented into Jaguar.67 Here, the M06-2X functional59 in combination with a 6-311++G** basis set was used. If not explicitly stated otherwise, SMD in combination with the default cavity scaling of α = 1.0 was used. We opted against the use of an explicit solvation model since developing reliable solvation shells would require ab initio molecular dynamics simulations.68 Performing these simulations would be computationally unfeasible owing to the large number of considered solvents and the partially large size of the solvent molecules. Considering the errors associated with the SMD solvation model,62 the use on an implicit solvation model is expected to be only slightly less accurate than using a computationally much more expensive explicit solvation shell.All structures were optimized for the DFT methods in all solvents and only considered converged if no imaginary frequencies were present. This was followed by single-point CCSD(T) computations at the respective converged B3LYP-D3 structures. Zero-point energy and entropy contributions were computed explicitly at the DFT level of theory whereas the corresponding corrections obtained with B3LYP-D3 were used to convert the total energies obtained with CCSD(T) to Gibbs free energies. The dissociation of formic acid into formate and a solvated proton in water was used as reference system for the effective absolute potential method. The pKa of this reaction is 3.77 in water.51
3 Results and discussion
3.1 Predicting absolute potentials
Let us start by assuming an arbitrary ET reaction in a freely chosen solvent (Rxn 1).| A + e− → A− | (1) |
| E(ET) = −nF[G(A−) − G(A)] + Eabs | (2) |
| Eabs = −nFG(e−) | (3) |
A possibility to compute the absolute potential of the SHE offers the recently developed “effective absolute potential method”.42 The procedure relies on the connection between the experimental pKa value of an arbitrary acid and the absolute potential to obtain a computational setup dependent value.42 Following this work, the acid dissociation can be rewritten by combining it with the standard hydrogen electrode (Rxn 4)
| H+ + e− → 0.5H2 E° = 0 V | (4) |
| A–H + e− → A− + 0.5H2 | (5) |
| Eabs(SHE) = −nF[0.5G(H2) + G(A−) − G(A–H) − ΔG(Diss)] | (6) |
| ΔG(Diss) = RT(pKa)ln(10) | (7) |
Note that the IUPAC definition of the SHE as zero level does not make any reference to a specific solvent. Accordingly, it is independent from the solvent the proton is dissolved in or whether the required conditions for the SHE could practically be reached.69 Thus, the HER from a hypothetically 1 M proton solution in Toluene can be considered with the same right as zero level as the equivalent reaction in water. Naturally, conversion factors are therefore needed when converting between the SHE in different solvents. To avoid confusion, we explicitly highlight the solvent which the SHE refers to whenever necessary. Similarly also the definition of the acid dissociation constant is without any reference to a specific solvent and can therefore be used universally.69 Thus, eqn (6) is valid in all solvents.
Assuming a situation where the experimental pKa has been determined in water while the ET reaction of interest takes place in a second solvent X, the electrochemical potential can be converted to the SHE scale in solvent X by adding a constant conversion factor (ESHE; SolvXconversion). E.g. the absolute potential in the new solvent (ESolvXabs(SHE)) is given by:
| ESolvXabs(SHE) = EWaterabs(SHE) + ESHE; SolvXconversion | (8) |
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) SolvXconversion can be obtained by rewriting the acid dissociation reaction as a PCET oxidation step (upper left branch in Fig. 1) followed by an ET reduction (upper right branch in Fig. 1). In the special case, where the solvent of the acid base reaction and the ET reaction are identical (e.g. both take place in water), the Gibbs free energy of the acid dissociation (ΔG(Diss)) simply corresponds to the sum of the free energies of the PCET (ΔG(PCET)) and ET (ΔG(ET)) steps (eqn (9)). Note that the energetics of the ET and PCET steps are not defined as reduction potentials as it is commonly done in electrochemistry but instead assume the reaction directions shown in Fig. 1. I.e. the PCET reaction is defined as an oxidation whereas the ET step is defined as a reduction.
SolvXconversion can be obtained by rewriting the acid dissociation reaction as a PCET oxidation step (upper left branch in Fig. 1) followed by an ET reduction (upper right branch in Fig. 1). In the special case, where the solvent of the acid base reaction and the ET reaction are identical (e.g. both take place in water), the Gibbs free energy of the acid dissociation (ΔG(Diss)) simply corresponds to the sum of the free energies of the PCET (ΔG(PCET)) and ET (ΔG(ET)) steps (eqn (9)). Note that the energetics of the ET and PCET steps are not defined as reduction potentials as it is commonly done in electrochemistry but instead assume the reaction directions shown in Fig. 1. I.e. the PCET reaction is defined as an oxidation whereas the ET step is defined as a reduction.| ΔG(Diss) = ΔG(PCET) + ΔG(ET) | (9) |
 | ||
| Fig. 1 The thermochemical cycle connecting the pKa with proton coupled electron transfer and electron transfer reactions is shown. | ||
Under these circumstances, eqn (9) directly leads to reaction (5) and eqn (6). Assuming on the other hand a situation where the acid dissociation and the electrochemical reactions take place in different solvents, this expression no longer holds. This is a direct result of the methodologies used to compute PCET and ET steps. In the case of a PCET reaction computed through the computational SHE, the electrochemical reaction is offset to the SHE by using gas phase H2 as a reference. Thus, computing the Gibbs free energies of the oxidized and reduced species (e.g. G(A˙) and G(A–H)) in the new solvent X using for example an implicit solvation model will automatically reference the PCET reaction to the HER in solvent X. Calculations of ET reactions on the other hand rely on the absolute potential. This offsets the reaction to the SHE in the the solvent the absolute potential has been determined for and is completely independent from the solvent the redox reaction takes place in. Thus, the PCET and ET steps refer to the SHE in different solvents as zero level. It is insufficient to simply use a suitable solvation model to describe the intermediates in the new solvent but additionally, also the absolute potential of the SHE in the new solvent X is needed to ensure that the PCET and ET steps refer to the same reference. Unfortunately, this value is only known in a very limited number of non-aqueous solvents33,38 and can not be estimated using the “effective absolute potential method”42 due to a lack of reliable pKa values in non-aqueous solvents. Using instead the absolute potential of the SHE in water results in a situation where the ET step is referenced to the SHE in water while the PCET reaction is referenced to the SHE in solvent X. Thus, eqn (9) is no longer valid and a correction to convert both reaction steps to the same SHE reference needs to be introduced. This conversion factor corresponds to the energy difference of the HER in the 2 solvents and is therefore equivalent to the correction for converting the absolute potential of the SHE in water to the SHE in solvent X (ESHE; SolvXconversion). Owing to it being a pure conversion factor, ESHE; SolvXconversion is expected to be method independent provided the computations are able to sufficiently accurately describe the electronic structure of the intermediates of the Born–Haber cycle. Accordingly, eqn (9) can be rewritten to
 | (10) |
| ESHE; SolvXconversion = nF[ΔG(PCET; SolvX) + ΔG(ET;H2O)] | (11) |
ΔG(PCET; SolvX) and ΔG(ET; H2O) can be obtained in the usual manner through eqn (12) and (13).
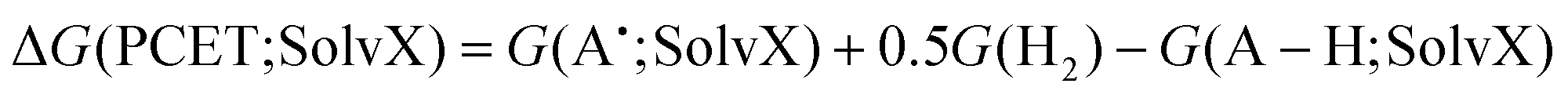 | (12) |
 | (13) |
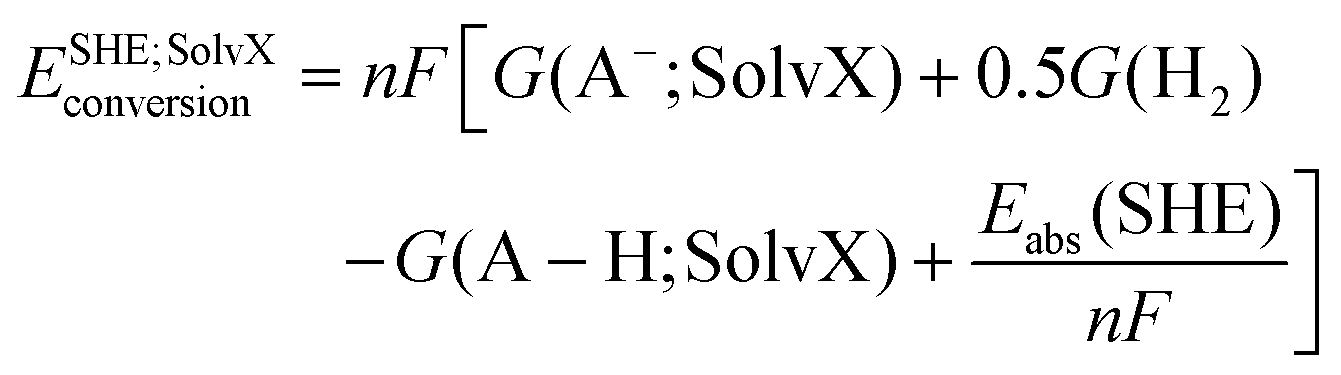 | (14) |
| E(ET; SolvX) = E(ET; H2O) + ESHE; SolvXconversion | (15) |
| E(PCET; H2O) = E(PCET; SolvX) − ESHE; SolvXconversion | (16) |
3.2 Absolute potential in non-aqueous solvents
Taking advantage of eqn (14) it is possible to compute conversion factors for the SHE in different solvents. Since this value corresponds to the relative stability of the proton in different solvents it is equivalent to the shift of the thermodynamic HER onset with respect to water. Combining this value with the absolute potential of water taken from earlier highly accurate CCSD(T)/SMD computations,42 the absolute potential in the solvent of interest is obtained. In Fig. 2a, the absolute potentials of the SHE in selected common solvents and aprotic ionic liquids are summarized. It is noteworthy that CCSD(T) and the two DFT methods predict almost identical values. This highlights, that the method dependence for the computation is solely accounted for in the effective absolute potential while the conversion factors are method independent. This is not unexpected from a purely formal perspective since the final absolute potentials only differ in the conversion factor computed through eqn (14). Thus, only the total energies of molecules with simple electronic structures in 2 different solvents, which are described well by state of the art electronic structure methods, are required. | ||
| Fig. 2 Method dependence of the absolute potential of the SHE in protic and aprotic solvents and ionic liquids. The solvents are ordered according to their polarisability as indicated by the relative static permittivity ε. (a) Dependence on the electronic structure method. CCSD(T) and DFT in combination with the SMD implicit solvation model were tested. (b) Influence of varying the cavity size within the SMD model. Used abbreviations: DMSO – dimethylsulfoxide; DMF – n,n-dimethylformamide; THF – tetrahydrofuran; EMIM – 1-ethyl-3-methylimidazolium cation; DCA – dicyanamide; BMIM – 1-butyl-3-methylimidazolium cation; MBPy – 4-methyl-N-butylpyridinium; HMIM – 1-hexyl-3-methylimidazolium cation; NTf2 – bis(trifluoromethylsulfonyl)imide. | ||
Besides the electronic structure method, also the implicit solvation model may have a significant influence. To test this possibility we modified the cavity size by varying the corresponding scaling value α between 1.0 and 1.2. These variations have a minor influence on the absolute potential of water, which decreases with increasing cavity size. E.g. it decreases from 4.14 V at α = 1.0, which comprises the default value used in all other computations, to 4.05 V for α = 1.2. In order to highlight the influence on the conversion factor between different solvents, the absolute potential of water obtained using CCSD(T) in combination with the Gaussian 16 default settings for SMD was used as reference point. Identical to the trends observed for the electronic structure methods, we find only minor variations of the order of 100 mV or less between the different systems (Fig. 2b). Note that the solvation energies of the acid and its corresponding base are still rather strongly affected by the modification of the cavity size. These changes are, however, systematic and thus, result in overall cancellation of errors. Switching on the other hand from SMD to the PCM model has a significantly larger effect. E.g. the predicted absolute potentials typically vary by several hundred mV. This is not unexpected when considering that using the SMD solvation model results in the inner potential whereas PCM predicts, at least in water, the experimentally accessible outer potential.39 Owing to the very limited information in the literature it is, however, unclear to which extend such a relationship also exists in non-aqueous solvents. But the rather good agreement between the surface potentials obtained experimentally and predicted from the difference between SMD and PCM indicates, that a similar relationship may also exist at least for some non-aqueous solvents. A more detailed discussion on this question can be found in the ESI.†
In Fig. 2 the absolute potentials of the HER in 21 selected solvents are summarized. The considered solvents are ordered according to their primary nature (protic and aprotic solvents, aprotic ionic liquids) and within these groups by decreasing polarisability as indicated by the dielectric constant ε. Comparing the data within the three groups, less polarisable solvents (placed at the bottom of the groups) qualitatively display a higher absolute potential. These trends are present both for protic and aprotic solvents. A notable exception is water which displays an anomalous behaviour in the sense that it possess a much too low absolute potential in comparison to formamide which, according to the dielectric constant, is significantly more polarisable. No comparable trends could be extracted from the set of ionic liquids owing to their very similar polarisability. Nevertheless, in line with the trends for the other solvents all ionic liquids display very similar absolute potentials. These qualitative trends are not surprising when considering that the absolute potential is closely tied to the stability of the proton in the respective solvent. Generally, a more polarisable solvent will allow for stronger dipole interactions and thus, be able to form a more stable solvent-proton complex. This in turn decreases the electrochemical onset potential of the HER. I.e. the cost for reducing the proton to H2 increases, which in turn corresponds to a lower absolute potential.
In addition to dipole interactions also hydrogen bonding is critical for the stability of the solvated proton.70 Accordingly, a solvent which is able to form a strong hydrogen bonding network should, assuming both materials are of comparable polarisability, display a lower absolute potential. This can for example be seen for diethylamine (protic solvent with ε = 3.651) and toluene (aprotic solvent with ε = 2.451). Owing to their low polarisability both compounds posses very high absolute potentials of 5.11 V (diethylamine) and 5.65 V (toluene). Among these molecules only diethylamine is able to form a noticeable hydrogen bonding network which in turn reduces the absolute potential. Similar trends are also present for all other solvents.
Considering the equal importance of polarisability and the ability for hydrogen bonding in describing the stability of the solvated proton it is not surprising that water, which displays a unique ability to form strong hydrogen bonds, does not follow the general trends between the solvents shown in Fig. 2. It is therefore clear that a single parameter approach which only includes the polarisability as a variable is an oversimplification and a more complex fitting approach which equally considers hydrogen bonding and polarisability is needed. Indeed, solvation models such as at the Kamlet–Taft equation55 generally include these parameters, i.e. the dependence of an arbitrary solvent dependent parameter X is given by:
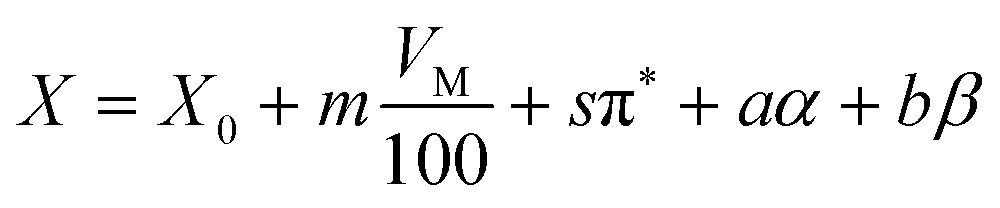 | (17) |
A strong correlation between the absolute potential of the HER in different solvents and the Kamlet–Taft polarisability and hydrogen bonding parameters is observed when fitting with respect to solvatochromic parameters of 35 solvents for which the relevant parameters could be obtained from the literature71,73 (eqn (18)).
 | (18) |
 | ||
| Fig. 3 (a) Comparison of the absolute potentials predicted using the Kamlet–Taft equation and CCSD(T). The solvatochromic parameters used as an input in eqn (18) were taken from ref. 71 and 73. (b) General trends between solvents and the absolute potential. The solvents' ability to stabilize protons through polarisability and hydrogen bonding is described using an effective solvatochromic parameter. | ||
The relationship between the absolute potential of 21 selected solvents (for complete data set see ESI†) is visualized using an effective solvatochromic parameter which is computed from eqn (18) under the assumption of X0 = 0 (Fig. 3b). Protic solvents in general tend to display very negative effective solvatochromic parameters whereas aprotic solvents display less negative ones. This correlates with the observed reactivity. The unique solvent properties of water are highlighted by the very high rating in hydrogen bond donation and polarisability/dipole interactions combined with a fair ability for hydrogen bond acceptance. This in turn results in a very negative effective solvatochromic parameter which places it, in line with the observed reactivity, at the far left of the plot where well stabilized protons and thus, a lower absolute potential must be expected. Formamide, on the other hand, which was placed on the top based on the dielectric constant, displays a somewhat lower polarisability and a significantly worse hydrogen bonding ability. Thus, it is placed close to ethanol and formic acid which posses comparable effective solvatochromic parameters. Also other solvents such as formic acid, acetic acid, DMSO or the considered ionic liquids display equally high or even higher values in one of the categories but none of them is comparable to water in all three categories. Formic acid or acetic acid for example display an equally high potential for hydrogen bonding. However, their polarisability is, owing to the presence of less polarisable C–H and CCH3 groups lower. Thus, they lack the ability to equally well stabilize the proton through additional dipole interactions. DMSO and the considered ionic liquids on the other hand are highly polarisable and thus, able to stabilize the proton through dipole interactions and as hydrogen bond acceptor. However, owing to their aprotic nature they are unable to contribute through hydrogen bond donation which in turn places them halfway between toluene and water. Toluene and triethylamine finally display the least negative effective solvatochromic parameters within the selected set. Note that triethylamine still possesses, according to the solvatochromic parameters, the ability to act as a hydrogen bond acceptor which is a result of the free electron pair at the nitrogen atom. Toluene on the other hand possesses, in contrast to what could be expected from the dielectric constant, a medium polarisability. This reflects toluene's ability to interact through its aromatic benzene ring. In line with this low ability for stabilizing the proton, the HER onset is shifted to very positive potentials. Even more positive HER onset potentials can be achieved when moving towards even less polar solvents which do not posses the ability for hydrogen bonding and are only able to interact through very weak dispersion interactions such as n-heptane or cyclohexane.
3.3 Comparison to literature
Despite the importance of the absolute potential for electrochemistry only few values have been reported either directly or indirectly in the literature.33,38,49,50,75 The data reported by Trasatti,33 Inerowicz et al.50 and Matsubara et al.75 have been determined from electrochemical measurements of the relative stability of the proton in different solvents. Trasatti reports an absolute potential which also includes the surface potential (e.g. the outer potential). These values can be connected to our computations through subtraction of the surface potential. Unfortunately, the proposed surface potentials vary strongly between different measurements. A comparison of our estimate and previous literature values76–81 can be found in the ESI.† Matsubara et al. and Inerowicz et al. on the other hand report only relative stabilities of the proton in different solvents referenced to the HER in water. Thus, any surface potential contribution cancel and these values can be compared directly to our computations. The absolute potentials reported by Kelly et al.49 finally were determined from the experimental pKa of selected acids or bases in the solvent of interest combined with DFT calculations. Owing to the computational setup of the DFT computations, the inner potential is reported (e.g. the surface potential is neglected) which renders them directly comparable to our computations.Let us start the discussion with the protic non-aqueous solvents. Here, we were able to identify data for ethanol, methanol and formamide (Fig. 4). The absolute potential of the SHE in methanol has been measured by Trasatti33 and calculated by Kelly et al.49 Trasatti reported a value of 4.19 V which is somewhat lower than the absolute potential in water whereas Kelly et al. found an increase from 4.28 V in water to 4.43 V. This qualitatively agrees with measurements by Inerowicz et al.50 which also indicate an increase of the absolute potential by 0.11 V. It is noteworthy that the difference between Trasatti's and Kelly's value is of a similar magnitude as the surface potential in methanol.77–82 Thus, all three studies indicate a slightly increased absolute potential which is in good agreement with our prediction. For ethanol an absolute potential similar to that of methanol was reported.33 Correcting again for the surface potential contribution, which is comparable to that of methanol,77–82 the deviations between our predictions and earlier measurements become less than 0.1 V. In formamide earlier measurements33 and our data are at a first glance in good agreement. But it must be cautioned that the literature value again includes the surface potential. It was proposed that the surface potential also in formamide has a negative sign,82 but no information regarding its magnitude is available. Assuming, however, a magnitude comparable to what has been found for the alcohols, Trasatti's and our data remain in good agreement.
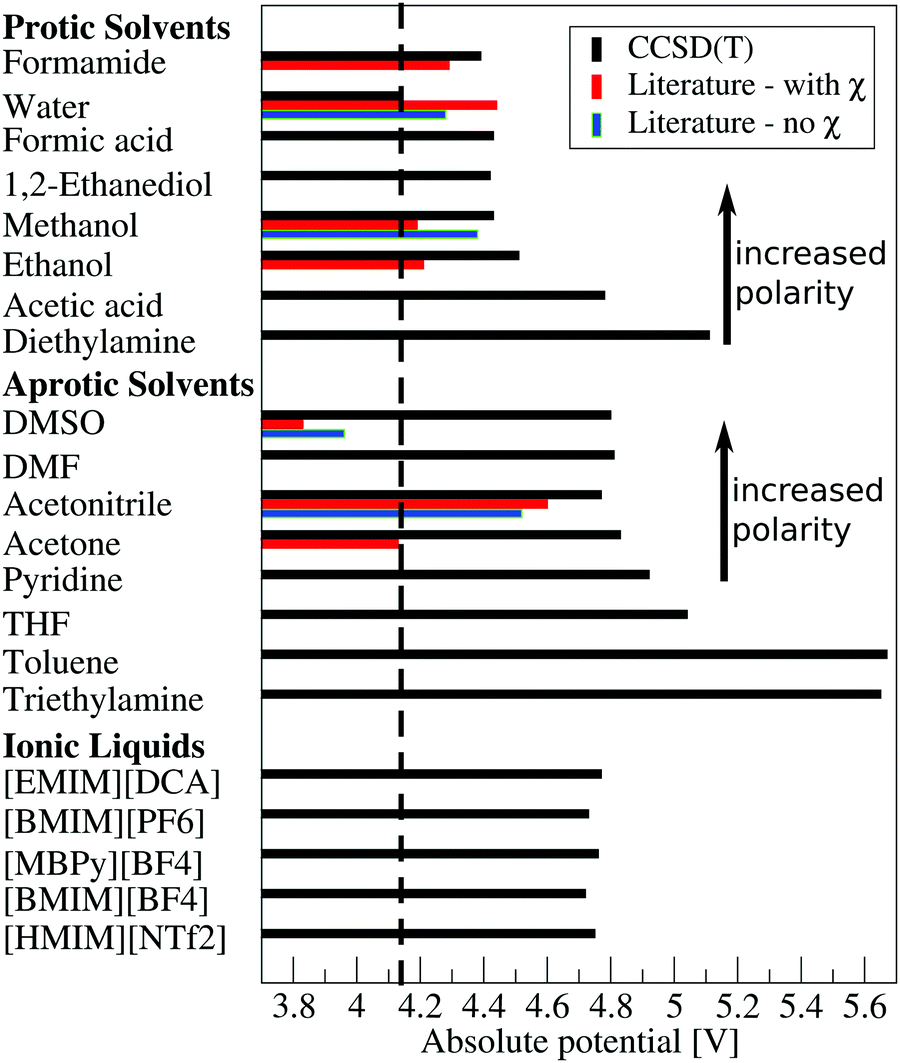 | ||
| Fig. 4 Comparison of computed absolute potentials obtained using the “effective absolute potential method”42 (CCSD(T)/SMD) with literature values including and excluding the surface potential (χ). The solvents are ordered according to their polarisability as indicated by the relative static permittivity ε. Used abbreviations: DMSO – dimethylsulfoxide; DMF – n,n-dimethylformamide; THF – tetrahydrofuran; EMIM – 1-ethyl-3-methylimidazolium cation; DCA – dicyanamide; BMIM – 1-butyl-3-methylimidazolium cation; MBPy – 4-methyl-N-butylpyridinium; HMIM – 1-hexyl-3-methylimidazolium cation; NTf2 – bis(trifluoromethylsulfonyl)imide. Literature data are taken from ref. 33, 34, 38 and 49. | ||
Moving to aprotic ionic liquids we were only able to identify data for the absolute potential in 1-ethyl-3-methylimidazolium [EMIM] bis(trifluoromethylsulfonyl)imide [NTf2].75 Considering the similarities between all imidazolium based ionic liquids independent from the choice of counterion it is reasonable to assume, that these measurements are at least qualitatively comparable to our data for similar ionic liquids. In line with our predictions (Fig. 4) also Matsubara et al. reported an increase of the relative absolute potential by 380 mV. This is in semi-quantitative agreement with our computations.
While our computed absolute potentials are in good agreement with literature for protic solvents and imidazolium based ionic liquids this is no longer the case when moving to aprotic solvents. Here, absolute potentials for dimethylsulfoxide (DMSO), acetonitrile and acetone have been reported directly33,49 and data regarding the relative stability of protons in pyridine50 are available (Fig. 4). In acetonitrile, an absolute potential of 4.52 V (without surface potential) has been found by Kelly et al.49 which corresponds to an increase by 240 mV compared to the absolute potential reported by the same group in water.34 This increase is comparable to the relative increase of the inner absolute potential by 160 mV to 4.6 V suggested by Trasatti33 Converting this value to the inner potential through subtraction of the slightly negative surface potential of approximately −0.1 V,76,79 this is in reasonable agreement with the 4.77 V predicted by us. Inerowicz et al.50 finally reported a relative increase by 479 mV which is still significantly smaller than the increase in the absolute potential by 630 mV to 4.77 V suggested by our computations.
Even more pronounced disagreements between our computations and earlier predictions are present for DMSO and acetone. Considering the somewhat lower polarisability of acetone compared to acetonitrile as indicated by the lower dielectric constant and the limited possibilities to act as hydrogen bond acceptor, a comparable or lower stability of the solvated proton could be expected. This is indeed predicted by our CCSD(T) calculations, i.e. the absolute potential increases slightly from 4.77 V for acetonitrile to 4.83 V for acetone. Trasatti on the other hand predicted with 4.13 V an absolute potential which is 300 mV lower than that of water in the same paper.33 This would, despite the lack of polarisability or ability for hydrogen bonding comparable to water,73 indicate, that the solvated proton in acetone would be more stable compared to its counterpart in water. This unexpected trend can partly be explained by the surface potential which is included in this value. In contrast to water the surface potential is negative in acetone by −0.3 V to −0.4 V.78–81 Thus, the directly comparable inner potential becomes roughly 4.5 V. While this is approximately 200 mV to 300 mV larger than the inner absolute potential in water it is still significantly lower than the 4.83 V suggested by our computations. Similar unexpected trends have also been reported for DMSO.38,49,50,83 Here, both the data reported by Kelly et al.38,49 and Fawcett38,83 suggest that a proton in DMSO should be several hundred mV more stable compared to H+ in water. Including the surface potential contributions of approximately −0.2 V to −0.3 V78–81 to the outer absolute potential reported by Fawcett,83 the inner absolute potential is still only of the same order of magnitude than that observed for water.34,42 A similar but less pronounced trend was also found by Inerowicz et al.50 who suggest that the absolute potential in DMSO is 195 mV lower than in water. Our calculations in contrast suggest the opposite trend. I.e. the absolute potential should increase by ≈650 mV. Inerowicz et al.50 additionally suggest an increased stability of protons in pyridine by 290 mV where our calculations again suggest an increase of the absolute potential by 780 mV compared to water.
These significant qualitative and quantitative deviations between literature and our predictions warrant a closer inspection of potential shortcomings. Considering the good agreement between our data and literature observed for protic solvents, it is unlikely that the new method used to predict absolute potentials in non-aqueous solvents is the source of the observed deviations. However, a major shortcoming of the present study is the strong reliance on the SMD implicit solvation model. While this model has been used extensively and is generally known to be sufficiently accurate,62 a failure for the systems at hand can naturally not be excluded. To test this possibility, we performed additional calculations for protic (water, ethanol, methanol) and aprotic (DMSO, n,n-dimethylformamide (DMF), acetonitrile, carbon tetrachloride, tetrahydrofuran (THF) and nitrobenzene) solvents using the pbf solvation model as implemented into Jaguar. Despite some minor deviations in the absolute values, we are able reproduce all major trends previously observed with SMD. Accordingly, also the calculations using the pbf solvation model suggest that protons in DMSO and other solvents with comparable polarisability should be significantly less stable compared to protons in water. Moving towards less polarisable solvents such as THF or carbon tetrachloride, we find identical to the trends present in the SMD based computations a further destabilisation. Moving on the other hand from DMSO to ethanol and methanol, no stabilisation, comparable to what was found with SMD, is observed. This may be a result of a worse performance of pbf in describing hydrogen bonding. Considering the rather good qualitative agreement between both sets of calculations, shortcomings in the solvation model can likely be excluded as the origin of the differences between our data and literature.
An additional potential source of errors, could lie in the experimental reference data used in earlier studies. While our predictions only require the pKa of an acid in water as reference these studies relied on parameters measured in non-aqueous solvents. Kelly et al.49 for example used the pKa measured in acetonitrile and DMSO as a basis for the prediction of the absolute potential while Trasatti33 and Inerowicz et al.50 relied on electrochemical measurements in non-aqueous solvents. However, recent work clearly showed that solvated protons are not necessarily relevant for acid base reactions in non-polar aprotic solvents.84 Expanding the argument to the more polar acetonitrile and DMSO, it can be questioned whether the data obtained from these experiments truly contain information regarding the proton's stability in solution. Furthermore, even in dried solvents traces of water can not be exclude.85,86 These impurities in turn can significantly stabilize the protons and thus, shift the absolute potential to lower values. The additional stabilisation observed in DMSO or pyridine could indicate a cooperative effect between the water impurities and the solvents which are significantly better hydrogen bond acceptors than water.73,87 However, further studies would be needed to verify this speculation. These experimental uncertainties can be expected to affect measurements in protic solvents such as alcohols or formamide to a lesser extend owing to the already present hydrogen bonding network. Thus, the observed good agreement between our data and earlier predictions in protic solvents is not unexpected.
4 Conclusions
We have developed an accurate and computationally efficient procedure to predict the conversion factors for the absolute potential of the SHE in water to any non-aqueous solvent or ionic liquids. In contrast to earlier work, our method only requires the experimental pKa of an arbitrary acid measured in water and few simple quantum-chemical calculations as input. Thus, experimental limitations in measuring properties in water free solvents will not affect our method adversely. Taking advantage of this procedure a new generalized computational standard hydrogen electrode for the computation of ET and PCET reactions in non-aqueous solvents was proposed. This enables for the first time the efficient and reliable prediction of redox potentials in solvents other than water. The method was tested by computing the absolute potential of the SHE in 36 solvents and ionic liquids. Taking advantage of the dataset, global trends in the absolute potential between all classes of solvents were established using the Kamlet–Taft linear solvation energy model. Our results indicate, that the proton stability equally depends on the solvents ability to act as H bond acceptor, H bond donor and its polarisability/dipolarity. Water which scores very high in all 3 categories is best able to stabilize solvated protons. For protic solvents the obtained absolute potentials were in reasonable agreement with earlier measurements but significant deviations were observed for aprotic solvents. In case of the latter earlier studies suggested a counter-intuitive increased proton stability in DMSO and pyridine compared to water. Our method on the other hand predicts, in line with the global trends between absolute potentials and the solvents' ability for hydrogen bonding and polarisability, an increased absolute potential and thus, a lower stability of the solvated proton compared to water. The predicted absolute potentials are almost completely independent from the quantum chemical method, while minor differences are observed between solvation models.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Calculations were performed at the Chalmers Centre for Computational Science and Engineering (C3SE) and the Finnish IT centre for science (CSC). M. B. and K. L. acknowledge financial support from the Jane and Aatos Erkko Foundation through the “Renewable energy storage to high value chemicals” project.Notes and references
- R. Francke and R. Little, Chem. Soc. Rev., 2014, 43, 2492–2521 RSC.
- T. Alhedabi, G. Herlem, H. Cattey, V. Blondeau-Patissier and T. Gharbi, J. Phys. Chem. C, 2014, 118, 25041–25050 CrossRef CAS.
- C. Wiberg, M. Busch, L. Evenäs and E. Ahlberg, Electrochim. Acta, 2020, 367, 137480 CrossRef.
- K. Exner, ChemSusChem, 2019, 12, 2330–2344 CAS.
- F. Nazneen, N. Tanwee, N. Shahed, S. Khanom, K. Hossain, J. Khandaker, F. Ahmed and M. Hossain, Mater. Today Commun., 2020, 25, 101409 CrossRef CAS.
- T. Liu, Z. Jin, D. Liu, C. Du, L. Wang, H. Lin and Y. Li, Sci. Rep., 2020, 10, 6897 CrossRef CAS PubMed.
- D. Wang, L. Liu, S. Zhao, B. Li, H. Liu and X. Lang, Phys. Chem. Chem. Phys., 2013, 15, 9075–9083 RSC.
- R. Fornari and P. de Silva, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2020, e1495 Search PubMed.
- K. Exner, J. Anton, T. Jacob and H. Over, Angew. Chem., Int. Ed., 2016, 55, 7501–7504 CrossRef CAS PubMed.
- K. Exner, J. Anton, T. Jacob and H. Over, Angew. Chem., Int. Ed., 2014, 53, 11032–11035 CrossRef CAS PubMed.
- E. Skulason, V. Tripkovic, M. Björketun, S. Gudmundsdottir, G. Karlberg, J. Rossmeisl, T. Bligaard, H. Jonsson and J. Nørskov, J. Phys. Chem. C, 2010, 114, 18182–18197 CrossRef CAS.
- M. Busch, N. Halck, U. Kramm, S. Siahrostami, P. Krtil and J. Rossmeisl, Nano Energy, 2016, 29, 126–135 CrossRef CAS.
- H. Hansen, I. Man, F. Studt, F. Abild-Pedersen, T. Bligaard and J. Rossmeisl, Phys. Chem. Chem. Phys., 2010, 12, 283–290 RSC.
- F. Song, M. Busch, B. Lassalle-Kaiser, C. Hsu, E. Petkucheva, M. Bensimon, H. Chen, C. Corminboeuf and X. Hu, ACS Cent. Sci., 2019, 5, 558–568 CrossRef CAS PubMed.
- M. Busch, E. Ahlberg and I. Panas, Phys. Chem. Chem. Phys., 2011, 13, 15069–15076 RSC.
- M. Vandichel, M. Busch and K. Laasonen, ChemCatChem, 2020, 12, 1436–1442 CrossRef CAS.
- V. Ortiz-Martinez, L. Gomez-Coma, G. Perez, A. Ortiz and I. Ortiz, Sep. Purif. Technol., 2020, 252, 117436 CrossRef CAS.
- T. Rüther, A. Bhatt, A. Best, K. Harris and A. Hollenkamp, Batteries Supercaps, 2020, 3, 793–827 CrossRef.
- J. Lehn and R. Ziessel, Proc. Natl. Acad. Sci. U. S. A., 1982, 79, 701–704 CrossRef CAS PubMed.
- R. Angamuthu, P. Byers, M. Lutz, A. L. Spek and E. Bouwman, Science, 2010, 327, 313–315 CrossRef CAS.
- J. K. Nørskov, J. Rossmeisl, A. Logadottir, L. Lindqvist, J. R. Kitchin, T. Bligaard and H. Jónsson, J. Phys. Chem. B, 2004, 108, 17886–17892 CrossRef.
- J. Rossmeisl, Z. Qu, H. Zhu, G. Kroes and J. K. Nørskov, J. Electroanal. Chem., 2007, 607, 83–99 CrossRef CAS.
- M. Busch, E. Ahlberg and I. Panas, Phys. Chem. Chem. Phys., 2011, 13, 15062–15068 RSC.
- G. Gritzner, in Reference Redox Systems in Nonaqueous Systems and the Relation of Electrode Potentials in Nonaqueous and Mixed Solvents to Standard Potentials in Water, ed. G. Inzelt, A. Lewenstam and F. Scholz, Springer Berlin Heidelberg, Berlin, Heidelberg, 2013, ch. 2, pp. 25–31 Search PubMed.
- L. Roy, E. Jakubikova, M. Guthrie and E. Batista, J. Phys. Chem. A, 2009, 113, 6745–6750 CrossRef CAS PubMed.
- S. Konezny, M. Doherty, O. Luca, R. Crabtree, G. Soloveichik and V. Batista, J. Phys. Chem. C, 2012, 116, 6349–6356 CrossRef CAS.
- M. Busch, A. Fabrizio, S. Luber, J. Hutter and C. Corminboeuf, J. Phys. Chem. C, 2018, 122, 12404–12412 CrossRef CAS.
- C. J. Cramer and D. G. Truhlar, Phys. Chem. Chem. Phys., 2009, 11, 10757–10816 RSC.
- A. Ghosh and P. R. Taylor, Curr. Opin. Chem. Biol., 2003, 7, 113–124 CrossRef CAS.
- H. Roth, N. Romero and D. Nicewiz, Synlett, 2016, 714–723 CAS.
- C. Andrieux, P. Hapiot, J. Pinson and J. Saveant, J. Am. Chem. Soc., 1993, 115, 7783–7788 CrossRef CAS.
- H. Reiss and A. Heller, J. Phys. Chem., 1985, 89, 4207–4213 CrossRef CAS.
- S. Trasatti, Pure Appl. Chem., 1986, 58, 955–966 CAS.
- C. Kelly, C. Cramer and D. Truhlar, J. Phys. Chem. B, 2006, 110, 16066–16081 CrossRef CAS PubMed.
- A. Isse and A. Gennaro, J. Phys. Chem. B, 2010, 114, 7894–7899 CrossRef CAS PubMed.
- P. Van Rysselberghe, J. Chem. Phys., 1953, 21, 1550–1551 CrossRef CAS.
- E. Lange and K. Miskenco, Z. Phys. Chem., 1930, 149, 1–41 CAS.
- A. Marenich, J. Ho, M. Coote, C. Cramer and D. Truhlar, Phys. Chem. Chem. Phys., 2014, 16, 15068–15106 RSC.
- J. Ho, M. Coote, C. Cramer and D. Truhlar, in Organic Electrochemistry, ed. O. Hammerich and B. Speiser, CRC Press, Boca Raton, 5th edn, 2015, Chapter 6, pp. 229–259 Search PubMed.
- J. Tomasi, B. Mennucci and R. Cammi, Chem. Rev., 2005, 105, 2999–3094 CrossRef CAS PubMed.
- J. Herbert, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2021, e1519 Search PubMed.
- M. Busch, K. Laasonen and E. Ahlberg, Phys. Chem. Chem. Phys., 2020, 22, 25833–25840 RSC.
- W. Hansen and D. Kolb, J. Electroanal. Chem., 1979, 100, 493–500 CrossRef CAS.
- W. Donald, R. Leib, J. O'Brien, M. Bush and E. Williams, J. Am. Chem. Soc., 2008, 130, 3371–3381 CrossRef CAS PubMed.
- W. Donald, M. Demireva, R. Leib, M. Aiken and E. Williams, J. Am. Chem. Soc., 2010, 132, 4633–4640 CrossRef CAS.
- T. Matsui, Y. Kitagawa, M. Okumura, Y. Shigeta and S. Sakaki, J. Comput. Chem., 2013, 34, 21–26 CrossRef CAS PubMed.
- T. Matsui, Y. Kitagawa, M. Okumura and Y. Shigeta, J. Phys. Chem. A, 2015, 119, 369–376 CrossRef CAS PubMed.
- Standard Potentials in Aqueous Solution, ed. A. Bard, R. Parsons and J. Jordan, CRC Press, Boca Raton, 1985, vol. 6, p. 848 Search PubMed.
- C. Kelly, C. Cramer and D. Truhlar, J. Phys. Chem. B, 2007, 111, 408–422 CrossRef CAS PubMed.
- H. Inerowicz, W. Li and I. Persson, J. Chem. Soc., Faraday Trans., 1994, 90, 2223–2234 RSC.
- CRC Handbook of Chemistry and Physics, ed. D. R. Lide, CRC Press, Boca Raton, 85th edn, 2004 Search PubMed.
- A. Kütt, S. Selberg, I. Kaljurand, S. Tshepelevitsh, A. Heering, A. Darnell, K. Kaupmees, M. Piirsalu and I. Leito, Tetrahedron Lett., 2018, 59, 3738–3748 CrossRef.
- T. Greaves and C. Drummond, Chem. Rev., 2008, 108, 206–237 CrossRef CAS.
- T. Ishii, S. Fujioka, Y. Sekiguchi and H. Kotsuki, J. Am. Chem. Soc., 2004, 126, 9558–9559 CrossRef CAS.
- M. Kamlet and R. Taft, Acta Chem. Scand., Ser. B, 1985, 39, 611–628 CrossRef.
- C. Lee, W. Yang and R. G. Parr, Phys. Rev. B: Condens. Matter Mater. Phys., 1988, 37, 785–789 CrossRef CAS PubMed.
- A. D. Becke, J. Chem. Phys., 1993, 98, 5648–5652 CrossRef CAS.
- F. J. Devlin, J. W. Finley, P. J. Stephens and M. J. Frisch, J. Phys. Chem., 1995, 99, 16883–16902 CrossRef CAS.
- Y. Zhao and D. Truhlar, Theor. Chem. Acc., 2008, 120, 215–241 Search PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. V. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery, Jr., J. E. Peralta, F. Ogliaro, M. J. Bearpark, J. J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, T. A. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman and D. J. Fox, Gaussian’16 Revision B.01, 2016, Gaussian Inc., Wallingford CT Search PubMed.
- S. Grimme, J. Antony, S. Ehrlich and H. Krieg, J. Chem. Phys., 2010, 132, 154104 CrossRef PubMed.
- A. Marenich, C. Cramer and D. Truhlar, J. Phys. Chem. B, 2009, 113, 6378–6396 CrossRef CAS.
- V. Bernales, A. Marenich, R. Contreras, C. Cramer and D. Truhlar, J. Phys. Chem. B, 2012, 116, 9122–9129 CrossRef CAS PubMed.
- C. Cortis, J. Langlois, M. Beachy and R. Friesner, J. Chem. Phys., 1996, 105, 5472–5484 CrossRef CAS.
- C. Cortis and R. Friesner, J. Comput. Chem., 1997, 18, 1570–1590 CrossRef CAS.
- C. Cortis and R. Friesner, J. Comput. Chem., 1997, 18, 1591–1608 CrossRef CAS.
- A. Bochevarov, E. Harder, T. Hughes, J. Greenwood, D. Braden, D. Philipp, D. Rinaldo, M. Halls, J. Zhang and R. Friesner, Int. J. Quantum Chem., 2013, 113, 2110–2142 CrossRef CAS.
- R. Kronberg, H. Lappalainen and K. Laasonen, Phys. Chem. Chem. Phys., 2020, 22, 10536–10549 RSC.
- Compendium of Chemical Terminology – Gold Book, International union of pure and applied chemistry technical report, 2014 Search PubMed.
- D. Marx, M. Tuckerman, J. Hutter and M. Parrinello, Nature, 1999, 397, 601–604 CrossRef CAS.
- P. Jessop, D. Jessop, D. Fu and L. Phan, Green Chem., 2012, 14, 1245–1259 RSC.
- Y. Altun and F. Koseoglu, J. Solution Chem., 2013, 42, 1691–1705 CrossRef CAS.
- Y. Marcus, J. Solution Chem., 1991, 20, 929–944 CrossRef CAS.
- Y. Dodge, Least Absolute Deviation Regression, in The Concise Encyclopedia of Statistics, Springer, New York, NY, 2008, pp. 299–302 DOI:10.1007/978-0-387-32833-1_225.
- Y. Matsubara, D. Grills and Y. Koide, ACS Omega, 2016, 1, 1393–1411 CrossRef CAS.
- B. Case, N. Hush, R. Parsons and M. Peover, J. Electroanal. Chem., 1965, 10, 360–370 CAS.
- J. Randles, Phys. Chem. Liq., 1977, 7, 107–179 CrossRef CAS.
- S. Trasatti, Electrochim. Acta, 1987, 32, 843–850 CrossRef CAS.
- V. Parfenyuk, Colloid J., 2004, 66, 466–469 CrossRef CAS.
- L. Krishtalik, Russ. J. Electrochem., 2008, 44, 43–49 CrossRef CAS.
- L. Krishtalik, N. Alpatova and E. Ovsyannikova, J. Electroanal. Chem., 1992, 329, 1–8 CrossRef CAS.
- B. Case and R. Parsons, Trans. Faraday Soc., 1967, 63, 1224–1239 RSC.
- W. Fawcett, Langmuir, 2008, 24, 9868–9875 CrossRef CAS.
- E. Paenurk, K. Kaupmees, D. Himmel, A. Kütt, I. Kaljurand, I. Koppel, I. Krossing and I. Leito, Chem. Sci., 2017, 8, 6964–6973 RSC.
- C. Xiao, A. Weremfo, C. Wan and C. Zhao, Electroanalysis, 2014, 26, 596–601 CrossRef CAS.
- B. Jensen and V. Parker, J. Chem. Soc., Chem. Commun., 1974, 367–368 RSC.
- J. Li, Y. Zhang, A. Dallas and P. Carr, J. Chromatogr. A, 1991, 550, 101–134 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Summary of the used Kamlet–Taft parameters, the computed and experimental absolute potentials for different solvents, and total Gibbs free energies. See DOI: 10.1039/d1cp00499a |
| This journal is © the Owner Societies 2021 |