Proton transfer in bulk water using the full adaptive QM/MM method: integration of solute- and solvent-adaptive approaches†
Received
10th January 2021
, Accepted 18th March 2021
First published on 1st April 2021
Abstract
The quantum mechanical/molecular mechanical (QM/MM) method is a hybrid molecular simulation technique that increases the accessibility of local electronic structures of large systems. The technique combines the benefit of accuracy found in the QM method and that of cost efficiency found in the MM method. However, it is difficult to directly apply the QM/MM method to the dynamics of solution systems, particularly for proton transfer. As explained in the Grotthuss mechanism, proton transfer is a structural interconversion between hydronium ions and solvent water molecules. Hence, when the QM/MM method is applied, an adaptive treatment, namely on-the-fly revisions on molecular definitions, is required for both the solute and solvent. Although several solvent-adaptive methods have been proposed, a full adaptive framework, which is an approach that also considers adaptation for solutes, remains untapped. In this paper, we propose a new numerical expression for the coordinates of the excess proton and its control algorithm. Furthermore, we confirm that this method can stably and accurately simulate proton transfer dynamics in bulk water.
Introduction
Proton transfer (PT) is an important phenomenon in biology, engineering, and solution chemistry. Although the first PT model was proposed 200 years ago by von Grotthuss, details of the mechanism of PT and solvation structure of the hydronium ion (whether it is an Eigen vs. Zundel cation) have not been fully understood because experimental observations cannot be straightforwardly interpreted, and such cations cannot be uniquely distinguished. However, in the late 1990s and in the first decade of the 21st century, computational advancements have facilitated the development of plausible simulations and advanced knowledge of the mechanism.1–10 PT is accompanied by the creation/annihilation of covalent bonds between oxygen and hydrogen, which is known as “structural diffusion”, and which requires a quantum mechanical (QM) description that can explicitly count the electronic structures. Therefore, ab initio molecular dynamics (AIMD) has played a major role in the progress of PT studies. In AIMD, the electronic structure of the entire system is evaluated to obtain the potential energy and forces acting on the respective atoms.2,3,7,8,11 However, AIMD is severely limited in terms of the system size to be evaluated as well as the duration of the molecular dynamics (MD) simulation because of the significant computational cost. For instance, the relaxation of the solvation structure, including the second solvation shell, is assumed to be a rate-limiting process for PT. Therefore, the solvation shell should interact properly with the surrounding environment in the PT dynamics. Indeed, a recent study demonstrated that the radial distribution around hydroxide ions depends on the system size.10 However, the extension of the system size results in a significant increase in computational cost. Obviously, large molecules such as proteins are also beyond the scope of AIMD, despite the high demand for them in fields such as chemical engineering and biochemistry. Car–Parrinello MD (CPMD),12 which is one of the most popular AIMDs, introduced a fictitious mass for electrons. However, an anomalously large value has been employed for the fictitious mass to ensure adiabaticity, which can distort hydrogen dynamics as deuterium.9
It should be noted that divide-and-conquer (DC) treatment,13,14 in which the entire system is fragmented into a number of subsystems. Because DC drastically reduced CPU time and memory usage, it has been successfully applied to macromolecular researches.15–17 In particular, it reduces the CPU time (for instance, to O(n1.2)) when used in combination with the density functional tight-binding (DFTB) method18–20 for homogeneous water systems, where n represents the number of water molecules. However, DC treatment still requires a large number of CPUs in accordance with the number of subsystems. In addition, DC treatment can cause discontinuities when particles diffuse across subsystem borders, although it is assumed to be mitigated to some extent by the introduction of buffer zones. Hence, there remain challenges for quantitative investigations and applications in the PT mechanism.
On the other hand, a hybrid simulation, namely the quantum mechanics/molecular mechanics (QM/MM) method, may be an appealing alternative because it can reduce the computational cost by partially applying QM calculations to the system.21 Because the computational cost of the QM/MM method depends primarily on the size of the local QM region as opposed to that of the whole system, the QM/MM method has been widely employed in research on large molecular systems. However, the QM/MM method cannot be directly applied to PT dynamics simulations, requiring adaptive treatment for both the solute and solvent. In conventional QM/MM simulations for solutions, a solute of interest is placed at the center of the QM region so that it is surrounded by QM solvent molecules, and molecular definitions are fixed throughout the MD simulation. However, owing to diffusion, the surrounding QM water molecules are replaced by other water molecules (MM). Furthermore, the transferred proton itself is not consistent throughout the MD simulation because of structural diffusion, as explained by the Grotthuss mechanism. In such a case, the position of the hydronium ion can deviate from the QM center and diffuse across the QM/MM border, leading to the collapse of the MD simulation. Therefore, both definitions of the excess proton and the surrounding solvent molecules should be adaptively updated during the MD simulation so that the hydronium ion is constantly located at the QM center surrounded by QM water molecules.
Regarding the solvent diffusion problem, to date, several solvent-adaptive QM/MM methods have been proposed.22–41 To understand solvent-adaptive QM/MM methods, it is useful to introduce the concept of QM/MM partitioning, which describes how the entire system is divided into QM and MM regions. In general, adaptive QM/MM methods are based on multi-partitioning approaches, where multiple QM/MM partitions are considered for every MD time step. While these partitions share the same QM solute molecule, they have different numbers and combinations of QM solvent molecules. Potential energy and forces are independently evaluated for respective QM/MM partitions. Finally, the resulting potentials or forces are linearly combined to evaluate the effective potential or forces that are used to update the coordinates for the MD simulation. In general, the adaptive QM/MM Hamiltonian can be represented as:
| | 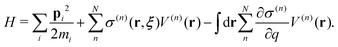 | (1) |
where the subscript
ξ represents the QM center, and alphabetic subscripts represent particles.
V(n)(
r) is the potential energy of the
nth partition and weight functions
σ(n) of coordinates of the respective solvent molecules and the QM center
ξ and the respective solvent molecules of the
nth partition. The second term, which is called “effective potential”, is the weighted sum of the potential energies of all partitions. The third term is the “bookkeeping term”, which was introduced to cancel out the artificial forces that arise from the derivatives of the weight function.
26,42 We defined the Hamiltonian with the bookkeeping term as the potential-oriented Hamiltonian and the one without the bookkeeping term as the force-oriented Hamiltonian. Note the potential-oriented Hamiltonian is based on the conserved force and corresponding effective potential,
| | 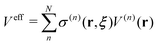 | (2) |
On the other hand, the derivative of the effective potential is
| | 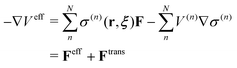 | (3) |
where
Feff is effective force, while
Ftrans is artifact termed transition force dependent on the weight function. As a result, the resulting dynamics and ensembles are severely distorted. In contrast, the force-oriented Hamiltonian is free from the transition force, while the potential that derives effective force
Feff does not exist. Thus, the internal energy which is the sum of the first and second term in
eqn (1) is not conserved during MD simulations, causing corresponding temperature drifts as confirmed in the present study. These drawbacks in both approaches are inextricably linked together and originates from spatial discontinuity.
43 Although adaptive QM/MM approaches enable the incorporation of quantum chemical effects of solvation into the MD simulation, most approaches suffer from severe artifacts that are termed “temporal” and “spatial” discontinuities. Because we have discussed the discontinuities elsewhere,
43,44 we will briefly introduce them here. A temporal discontinuity is related to the differentiability of the weight function
σ. With discontinuous weight function, the effective potential is not linked to conserved force. In other words,
Feff is not a conserved force. As a result, the Hamiltonian is not conserved in course of MD time evolution. Some solvent-adaptive QM/MM methods, such as the sorted adaptive partitioning (SAP)
24 and size-consistent multipartitioning (SCMP)
34 methods, are free from temporal discontinuity. On the other hand, the spatial discontinuity is linked to
Ftrans and inevitable even in the force-oriented approach, in which
Ftrans does not explicitly appear, but manifest as the monotonic drift of the bookkeeping term during the course of the MD simulation. Trivially, if the potential energy does not depend on partitioning, the transition force does not appear as
| | 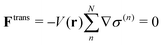 | (4) |
In other words,
Ftrans arises from the systematic energy difference among respective QM/MM partitioning potential
V(n) between compact and fragmented forms of the QM region. Although some
ad hoc corrections have been proposed,
36,39,43 spatial discontinuities are inevitable for any QM/MM method because they arise from the unnatural manipulation of dividing a homogeneous solution into multiple layers, where each layer is characterized by a different molecular model. Therefore, for fairness, the static QM/MM method should also be subject to spatial discontinuities, and not only adaptive QM/MM. However, spatial discontinuities have not been extensively discussed because they are not considered a critical factor in static simulations. Note that compared to multi-size approaches such as SAP and difference-based adaptive solvation (DAS),
26 a spatial discontinuity can be suppressed by size-consistent treatment, in which the number of QM solvents is consistent among partitions.
43
Towards a solute-adaptive method, the first step is to numerically express the position of the excess proton, which is called an “excess proton indicator (EPI)”. To date, several groups have addressed the development and application of EPI.45–49 Previously, Chakrabarti et al. proposed the expression of the EPI ξ as
| | 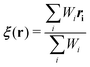 | (5) |
where
ri is the coordinate of the
ith oxygen atom.
47 The weight function for the
ith oxygen atom can be written as
| | 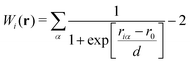 | (6) |
where
riα is the distance between the
ith oxygen and the
αth hydrogen atoms, and
r0 and
d are parameters. The weight function
Wi defined by
eqn (6) indicates that the hydronium ion is more weighted in the EPI estimation in
eqn (5) than ordinary water molecules whose
Wi values are close to zero. Although this indicator has been used as the reaction coordinate in the post-processing of the MD trajectory for PT in membrane proteins, it cannot be used for an adaptive simulation of the bulk solution because of its discontinuity, scale-dependency, instability, and computational cost. The problems above mainly arise from the fact that the weights for ordinary water molecules distant from the hydronium ion in (5) are not necessarily zero. Note that the residual contribution is not negligible, and can cause critical errors when accumulated. Let us assume that a simulation cell is filled with water molecules, including a solvated hydronium ion. In the case of a small simulation cell, EPI in
eqn (5) and (6) may be suitable, with EPI corresponding to the position of the hydronium ion throughout the simulation. However, as the system size increases, the indicator points to the geometric center of the simulation cell regardless of the position of the hydronium ion. This is because the accumulated residual contributions from ordinary water become comparable to that of the hydronium ion (scale-dependency). In addition, when the weighted water molecules move across the periodic boundary condition, the EPI becomes discontinuous, causing it to violate the Hamiltonian conservation. As a result, the temperature unnaturally increases, destabilizing MD simulations (discontinuity). Furthermore, even ordinary water distant from the hydronium ion can temporarily have a value as large as that of the hydronium ion when it forms strong hydrogen bonds. This results in an extraordinary displacement of the QM center, destabilizing the MD simulation (instability). It should also be noted that the computational cost to evaluate the weight of
N water molecules is almost 2
N2 considering all possible oxygen-hydrogen pairs, and thus, its computational cost will become as large as the force evaluation, which is the bottleneck of MD simulations (computational cost). It is also notable that Pezeshki
et al. formulated EPI as
| | 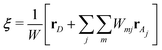 | (7) |
where
rD and
rAj are the coordinates of the proton donor and the
jth acceptor oxygen atoms, respectively.
50 Here,
W is a normalization factor and
Wmj is a weight function of
ρmj, which is defined as
| | 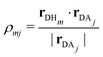 | (8) |
Here,
Hm represents the
mth hydrogen atom bonded to the donor. Although the EPI is based on a continuous weight function
Wmj, the distinction between the donor and acceptor can cause discontinuities when the definitions are swapped, corresponding to PT. In addition, the EPI employs a threshold distance for acceptors to be considered, and hence becomes discontinuous when the acceptor list within the threshold is updated. Thus, when the EPI is used for MD control,
e.g., the QM center position in the adaptive QM/MM method, the dynamics is not based on the continuous potential energy surface, resulting in a violation of Hamiltonian conservation. To avoid such problems, the proton indicator should consist of continuous functions of the coordinates of all particles, and it should also be able to detect local structural attributes around the hydronium ion. Although it is obvious that the molecular geometries of solvent water and the solvation structure in the vicinity of the hydronium ion differ from that of bulk water, it is not clear whether there exists any index represented by a continuous function (which can identify the position of the hydronium ion without noise and error throughout the MD simulation).
In this paper, we first propose a new EPI expression, which could be introduced as one method to achieve an accurate and stable MD simulation and implement the EPI expression into the SCMP method to achieve full adaptive QM/MM in combination of solute- and solvent-adaptive approaches. It should also be noted that in the adaptive QM/MM method, the conservation of the effective potential Veff is important. In the potential-oriented approach, this conservation is obviously linked to the Hamiltonian conservation. In contrast, in the force-oriented approach, Feff derived from the pseudo-potential (the sum of Veff and the bookkeeping term) is not conserved. Nonetheless, the conservation of Veff is still valuable in practice because with the conserved effective potential, we can quantitatively evaluate the spatial discontinuity by the drift of the bookkeeping term coupled to the change of the effective internal energy. For conservation force, the partial derivative of Veff should be exchangeable as
| | 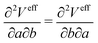 | (9) |
where
a,
b ∈ {
x,
y,
z}. In most adaptive QM/MM methods, the weight
σ(n) is a function of distance between the QM center
ξ and solvent molecules, and the QM center
ξ is represented by any particle, such as an atom in a solute molecule. In this case, the effective potential
Veff is linked to conserved force as long as the weight function
σ(n) is differentiable. In contrast, the EPI used in solute adaptive QM/MM is supposed to be a function of the coordinates of some particles. When the weight function depends on the EPI, it is not trivial whether the effective potential
Veff satisfies the condition of the conservation, where the differentiability of
ξ(
r) is an important factor (Appendix A). Also, note that the EPI is desirable to be controllable in enhanced MD sampling such as umbrella sampling and meta dynamics, in which the artificial force is applied to a target molecule to reproduce rare event within limited simulation time. However, it is not trivial how to control the EPI on enhanced sampling. Thus, we also propose a new protocol to control the indicator during the MD simulation, in which we introduce a dummy particle representing the QM center using constraint dynamics, which is called the RATTLE method.
51 We suppose that there are two advantages for treatment of a virtual particle. In the following sections, we first review the SCMP method and its weight function. Second, we describe a new form of the EPI. Next, we also detail a protocol of the EPI control in MD simulation by introducing new variables and using constraint. Finally, we demonstrate the benchmark simulation for PT in bulk water, in which the Hamiltonian is numerically conserved, achieving a stable and durable MD simulation. Note that, throughout this paper, alphabetic and Greek subscripts represent oxygen and hydrogen atoms, respectively.
Theory and method
Weight function in size-consistent multipartitioning (SCMP) method
Here, we briefly review the SCMP method. In the SCMP method, all partitions have a consistent number of QM solvent molecules in the QM region, but different combinations. The weight function σ(n) in eqn (1) is defined as below for the SCMP method.| | 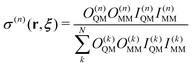 | (10) |
Here, the EPI ξ represents the QM center. O(n)QM and O(n)MM are fade-out functions for the QM and MM solvent molecules in the nth partitioning, respectively, and they are defined as| | 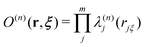 | (11) |
where riξ is a distance between the ith solvent and the QM center. λ(n)j is the progress function of the jth QM or MM solvent, which continuously ranges between zero and one. (details in Appendix B) I(n)QM and I(n)MM are fade-in functions for the QM and MM solvent molecules, respectively, which are defined asNote that the QM and MM fade-out functions become zero when λ(n) = 0 for a QM and MM solvent molecule become zero in the nth partitioning, respectively. On the other hand, the QM and MM fade-in functions take non-zero value when λ(n) < 1 for a QM and MM solvent molecule in the nth partitioning, respectively. As a result, σ(n) = 0 when the QM region of the nth partitioning is compact or fragmented. Hence, the partitioning whose QM region is fragmented is replaced by one with a compact QM region without discontinuity in Veff. The details of the SCMP method are provided in the literature.34,43,52
Excess proton indicator
In the present study, EPI ξ, which was used as the QM center in the SCMP simulation is written as,| | 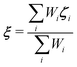 | (13) |
where ζi is an EPI precursor, and Wi is a EPI weight function of the ith oxygen atom, which are detailed separately below (Fig. 1). First, let us clarify the conditions required for an EPI precursor ζ. As shown in the Results section, the proton is transferred for a time period of 10 fs. When ζ coincides with the oxygen atoms, the EPI is rapidly displaced over a distance of 2.4 Å (oxygen–oxygen distance) in 10 fs according to PT. This extraordinarily rapid displacement of EPI can result in an abrupt change in the balance of weight σ among QM/MM partitionings, which destabilizes the MD simulation. On the other hand, when ζ coincides with the transferred proton, the moving distance of the EPI will become smaller during PT, reflecting the actual distance that the proton moved across. Hence, we introduced a virtual hydrogen site ηi that reflects a candidate for transferred hydrogen. Then, ζi is defined as the dividing point between the ith oxygen atom ri and its virtual hydrogen site ηi and expressed aswhere c is a rating scale parameter.
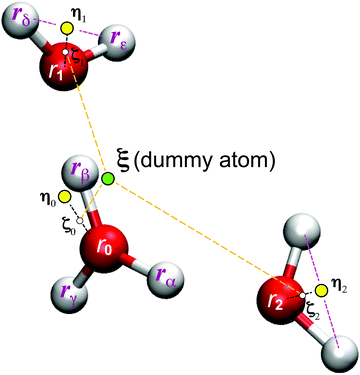 |
| | Fig. 1 Illustration for the excess proton indicator ξ. | |
Virtual hydrogen site
The virtual hydrogen site ηi for the ith oxygen atom was defined using the hydrogen coordinates rα as| | 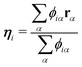 | (15) |
where ϕiα is a score function of αth hydrogen. Note that ηi is a function of all hydrogen atoms. The virtual hydrogen site ηi should reflect a hydrogen atom whose distance from the ith oxygen is the longest among hydrogen atoms covalently-bonded to the oxygen. On the other hand, other hydrogen in the distance from the ith oxygen should not contribute to ηi. Hence, we employed Gaussian-type function for ϕ(r) as.| | 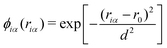 | (16) |
where riα is the distance between the ith oxygen atom and the αth hydrogen atom. Here, d and r0 are the parameters which are tuned to realize the conditions above.
In the present study, the EPI was defined as the weighted sum of internally dividing points ζ between the oxygen coordinate ri and the virtual site ηi over all solvent oxygen atoms. When the parameter c = 1 in eqn (14), the EPI is a linear combination of virtual sites ηi with weight Wi. In contrast, when c = 0, the EPI is a linear combination of water oxygen coordinates ri. Even in this case, the EPI is indirectly subject to the water hydrogen coordinate through weight Wi, as described below. Thus, when the value of c falls between 0 and 1, the EPI becomes a dividing point between the two positions (Fig. 1). In contrast to the previous EPI in eqn (8), the present EPI treats atoms without distinction between solute H3O+ and solvent H2O.
EPI weight function
Here, we define the EPI weight W in eqn (13). W should reflect hydronium ion and its neighboring water molecules, otherwise the problems will manifest as described in the introduction. To this end, we introduced a function ψi for the ith oxygen as the summation of the spline function S of the distance riα between the ith oxygen and the αth hydrogen:| | 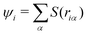 | (17) |
Here, the spline function S satisfies the following boundary conditions:
| | 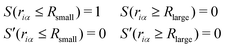 | (18) |
where
Rsmall and
Rlarge are the bonding parameters that satisfy
Rsmall <
Rlarge. Here,
S(
riα) is the bonding score between the
ith oxygen and the
αth hydrogen, which ranges from zero to one. Thus,
ψi can be regarded as a continuous expression of the number of covalent bonds of the
ith oxygen atom (see
Fig. 1). Although we applied the spline curve here, note that the function
S is arbitrary unless it satisfies
eqn (18). Finally, the EPI weight function
Wi of the
ith EPI precursor was defined using another spline function of
ψi, which monotonically increases between 2.0 and 3.0, and satisfies the following boundary conditions:
| | 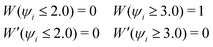 | (19) |
Since
Wi increases with the number of OH covalent bonds
ψ, oxygen, and hydrogen atoms, the EPI mainly reflects the coordinates of atoms in a hydronium ion moiety. Note that the computational cost for both
ζ and
W are seemingly
O(
N2). However, we found that
Wi could become zero for oxygen atoms except for several molecules located close to the hydronium ion by tuning parameters
r0 and
α in
eqn (16) (see Discussions). This implies that when proper values are set as
Rsmall and
Rlarge in
eqn (18), the contribution to the EPI can be limited to only two or three oxygen atoms in addition to several hydrogen atoms close to the oxygen atom. This fact helps to exclude most solvent molecules from the EPI evaluation by saving computational cost, as discussed below.
Control of QM center
To control the EPI in MD simulation, we placed a dummy atom at the position corresponding to ξ. Note that the dummy atom does not directly interact with other particles but indirectly interacts through a constraint, as described below. In other words, the potential function V(r) is independent of ξ. Hence, the coordinate and conjugate momentum of the dummy atom are treated as independent variables of the Hamiltonian. As a result, regardless of the differentiability of ξ with respect to other particles, Veff in eqn (2) is conserved if the weight function satisfies the symmetric conditions expressed as| | 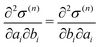 | (20) |
where ai, bi ∈ {xi, yi, zi} and subscript i stand for any particle. Since ξ is defined as a continuous form, this feature does not bring benefits in the present study, but may allow more variety in EPI expression. We chose the mass of the dummy atom to be 1.0 × 10−8 a.u., which is small enough to well satisfy the constraint condition but does not affect other particles. The velocity Verlet integrator was employed for MD in the present study, and thus, the RATTLE algorithm51 was applied to control the constraint. In addition to eqn (13), the constraint condition with respect to velocity is given as| | 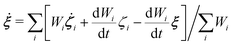 | (21) |
The MD algorithm for the ith particle is as follows:
(i) 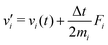
(ii) 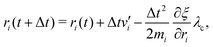
(iii) 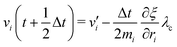
(iv) 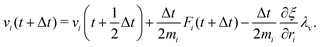 where λc and λv are Lagrange multipliers determined from iterations to satisfy eqn (13) and (21), respectively The mass of the dummy atom was set to 1 × 10−8 a.u., which is sufficiently smaller than other particles. Thus, this artificial treatment does not affect the dynamics. Indeed, we found that the use of a dummy atom smaller than the one set in the present study did not make any significant difference in the dynamics. However, a dummy atom with a larger mass may slow down the motion of the EPI and make the RATTLE iterations unstable, which can lead to the collapse of the MD simulation. In Algorithm 1, the entire workflow is illustrated by pseudocode.
where λc and λv are Lagrange multipliers determined from iterations to satisfy eqn (13) and (21), respectively The mass of the dummy atom was set to 1 × 10−8 a.u., which is sufficiently smaller than other particles. Thus, this artificial treatment does not affect the dynamics. Indeed, we found that the use of a dummy atom smaller than the one set in the present study did not make any significant difference in the dynamics. However, a dummy atom with a larger mass may slow down the motion of the EPI and make the RATTLE iterations unstable, which can lead to the collapse of the MD simulation. In Algorithm 1, the entire workflow is illustrated by pseudocode.
Adaptive Langevin thermostat
We also carried out Langevin dynamics simulation to maintain a constant temperature, where the coupling strength with the thermostat adapts to the QM profile,34,44 which is an index of the degree to which a solvent molecule behaves as a QM molecule. Using the velocity Verlet integrator for Langevin dynamics,53 the coordinate r and velocity v are propagated as| | 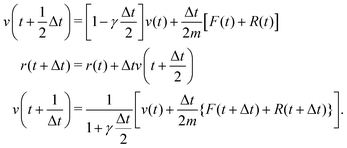 | (22) |
where m and γ are the mass and friction coefficients, respectively. F is a deterministic force derived from the potential function V, and R is the Gaussian random force, which is defined as:| | 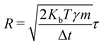 | (23) |
where τ is a random number that satisfies 〈τ〉 = 0 and 〈τ2〉 = 1. In the limit γ → 0, eqn (22) reduces to the ordinary velocity Verlet algorithm for Hamiltonian dynamics. In the SCMP method, the QM profile ωi for the ith solvent molecule can be represented as:| | 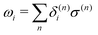 | (24) |
where δ(n)i = 1 if the ith solvent molecule is QM in the nth QM/MM partitioning, and δ(n)i = 0 if the solvent molecule is MM. As a result, ωi = 1 when the ith solvent molecule behaves as a pure QM model, which can be rephrased as the ith solvent molecule, is defined as QM throughout all weighted partitions. ωi = 0 when the ith solvent molecule behaves as a pure MM, which can be rephrased similarly. In the present study, we associated the QM profile and friction coefficient of the i-th solvent molecule aswhere γ0 is a parameter. Because ωi changes at every MD time step, γi for the respective solvent molecule also changed. This enabled the MM solvent molecules to be fully linked to the thermostat, and thus, the coupling strength gradually attenuated when the solvent molecule approached the QM center.
| Algorithm 1 MD flow |
initialization; 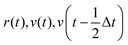 |
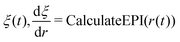
|
|
repeat
|
| PARTITIONINGUPDATE(r(t)) |
| F(n)(t),V(t)(n) = CALCUALTEFORCE(r(t), ξ(t)) |
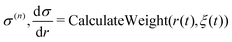 |
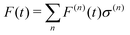 |
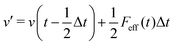 |
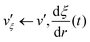 |
|
repeat
|
| *** RATTLE ITERATION *** |
| λv ← vξ′ |
| v′ ← λv |
| vξ′ ← v′ |
|
until convergence |
| v(t) = v′; vξ(t) = vξ′; vξ(t) = vξ′ |
| B(t) ← B(t − Δt), v(t), vξ(t), V(t) |
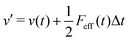 |
| r′ = r(t) + v′*Δt |
| ξ′ = ξ(t) + vξ(t)Δt |
|
repeat
|
| *** SHAKE ITERATION *** |
| λc ← ξ′ |
| r′, ← λc |
| ξ′ = CALCULATEEPI(r′) |
|
until convergence |
| r(t + Δt) = r′; ξ(t + Δt) = ξ′ |
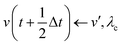 |
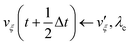 |
| evaluted dξ/dr(t) |
|
until reach to max MD step = 0 |
Computational details
The SCMP method was implemented in a local version of the GROMACS 5.0.7 package.54–56 In all SCMP simulations, a total of 80 QM/MM partitions were considered, where the forces and energy calculations were carried out based on different partitions. The system consisted of one hydronium ion and 2047 water molecules in a periodic cubic box with a side length of 40.28 Å. Each partitioning has one QM solute hydronium ion and 32 QM solvent water molecules. In the SCMP, the transition parameters sQM, tQM, sMM, and tMM were set to 6.4, 8.4, 4.0, and 6.4 Å, respectively (see ref. 34 and 44 for details). In the partitioning updating protocol, we allowed the updated partitioning to have a degree of order of 75% for efficiency, as detailed in our previous work.44 We employed the SPC-Fw water model57 for the MM water. For the QM part, we employed DFTB318–20 implemented in GROMACS, as reported previously58 using standard 3OB parameter sets.20 The electrostatic interactions in the MM-MM and QM-MM models were calculated using the particle-mesh Ewald method.59 There have been proposed several practical implementations of PME for QM/MM,60–63 we employed “separated PME” as implemented in GROMACS.58 Both electrostatic interactions and van der Waals interactions were damped to zero in the range of 8.5% to 9.0 Å. The electrostatic potentials on the QM atoms induced by the charges of the MM atoms were obtained using the smooth particle-mesh Ewald method with a switching function for electrostatic interactions (electrostatic embedding). After equilibration for several picoseconds (ps), all MD simulations were conducted for 100 ps with time steps of 0.25 and 0.50 fs for hydronium ion solution and bulk water simulations, respectively. For the control of the EPI, we chose r0 = 1.3 Å and α = 0.129 Å for eqn (16). In addition, we set Rsmall = 1.20 Å and Rlarge = 1.32 Å for eqn (18) and νsmall = 2.0. For temperature control, we employed the friction coefficients γ0 = 100 ps−1 and 10 ps−1 for eqn (25).
Results
Energy and bookkeeping term
For stable MD simulation and the production of an accurate ensemble, the MD simulation needs to numerically conserve the Hamiltonian (total energy) throughout its entire course. As shown in Fig. 2, we evaluated the Hamiltonian with different values of c in eqn (13) under the microcanonical (NVE) condition in the present simulation. The Hamiltonian was numerically conserved in all simulations over 2.0 ps from the beginning in each simulation. Based on the fact that PT between the hydronium ion and water molecules was observed several times during this time period, we assume that the PT was simulated on a continuous energy surface for several ps. However, the simulations with c = 0.2 and c = 0.8 collapsed at 3.7 and 1.2 ps, respectively, because either the partitioning weight σ in eqn (10) or EPI weights W in eqn (13) became zero. On the other hand, the simulations with c = 0.0 and 0.5 were sustainable for more than 4.0 ps, but the energy conservation is violated after 2.0 ps in the respective simulations. Although it is not straightforward to attribute the cause of the energy violation, we observed the extraordinary fast displacement observed of the QM center ξ at those time point. This rapid diffusion of the QM center allows approximation of MM solvent and separation of QM solvent molecules. As a result, the weights of most, but not all, QM/MM partitioning went zero, which maybe numerically destabilized the MD simulation. Note that these problems were never observed for the first few picoseconds in the simulations, but they commonly occurred in the next several picoseconds, which implies that some errors are implicitly accumulated during the course of the simulation time. To clarify this point, we evaluated the bookkeeping term in eqn (1). As we have reported in our previous study,43 the drift of the bookkeeping term is related to spatial discontinuity. Notably, when c = 0.8, it drifted by 1700 kJ mol−1 for 1 ps under microcanonical conditions, which is approximately 200 times greater than the shift of the bookkeeping term for pure water or several monoatomic ion solutions. This discontinuity arises from the systematic difference in potential energies between QM/MM partitioning, whose QM regions are compact and fragmented. Therefore, we suppose that the large drift may reflect the dynamics of the hydronium ion because the diffusion of hydronium ions is much faster than that of water molecules and other ions, and the deformation of the QM regions occurs sooner. Taking into account the numerical conservation of the Hamiltonian, the decrease in the bookkeeping term should correspond to the increase in internal energy, in line with the temperature increase observed under the microcanonical condition in Fig. 2. To be precise, the released energy manifests as the acceleration of solvent molecules around the QM/MM border. Then, if the energy release is faster than its dissipation, the solvation structure and dynamics are significantly distorted, leading to the failure of the MD simulation. We found that the drift of the bookkeeping term became more distinct as the parameter c increased. Indeed, MD simulations with larger values of c tend to become stuck earlier.
 |
| | Fig. 2 Hamiltonian (total energy), bookkeeping term, and temperature in the course of MD simulation time under a microcanonical condition. The black, red, green, and blue lines represent the SCMP simulation with EPI parameters of c = 0.0, 0.2, 0.5, and 0.8, respectively. | |
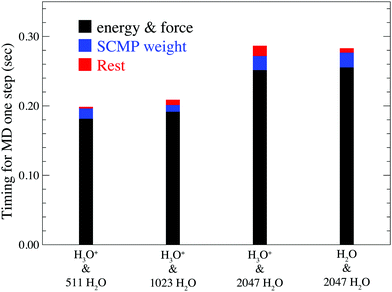
As we proposed,43 the drift of the bookkeeping term can be alleviated by introducing a correction potential U to cancel out the artificial diffusive force. However, the ad hoc approach is not promising for finding the optimal form, although it requires additional computational cost. As an alternative to both stabilizing the simulation and reproducing plausible PT dynamics, we employed the Langevin thermostat in an adaptive manner so that the coupling strength with solvent molecules gradually changes in accordance with the changes in molecular definition based on its distance from the QM center. To this end, we introduced the QM profile ωi as defined in (eqn (24)), which indicates how much a solvent molecule behaves as a QM molecule. In the SCMP method, the QM profile averaged over the MD simulation smoothly shifted from 1 to 0 as the distance from the QM center increased, indicating that the molecular properties of solvent water gradually alternated between QM and MM.34,44Fig. 4 shows the temperature over the course of the SCMP simulation with c = 0.0 employing an adaptive Langevin thermostat with the friction coefficient γ0 = 100 ps−1. It can be observed that the adaptive thermostat controls the temperature well, maintaining it at the reference temperature of 300 K, enabling the MD simulation to be durable over several hundreds of picoseconds. It should also be noted that the spatial discontinuity distorted the dynamics of the solvent molecules located near the QM/MM border rather than the QM center.43 The thermostat can also alter the dynamics, but under the present adaptive usage, the solute and solvent molecules in the vicinity of the QM center are free from the Langevin thermostat. Therefore, it is plausible to evaluate the dynamical properties from the trajectories obtained in the present simulation, as discussed below. As will be discussed later, the friction coefficient γ0 does not affect the results of the hydronium ion simulations. Thus, most analyses in the present study are based on simulations with c = 0.0 and γ0 = 10 ps−1, unless otherwise stated.
Computational cost
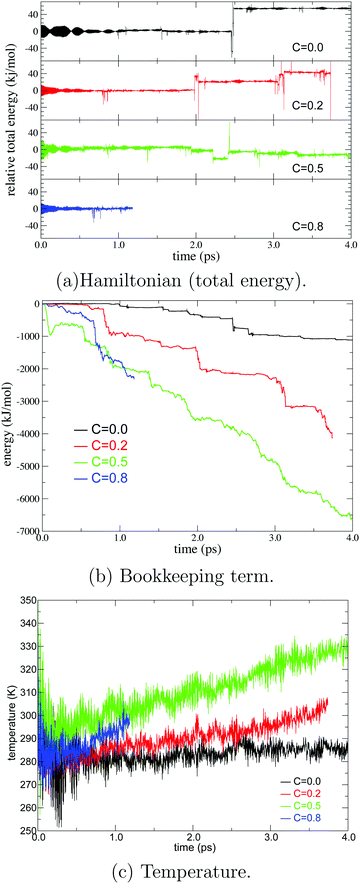
Fig. 3 shows the computational time for one MD step of the full adaptive QM/MM method compared with the solvent adaptive QM/MM method, both of which are based on the SCMP framework. Here, the solute molecule is H3O+ in the full adaptive QM/MM and H2O in the solvent adaptive QM/MM. Note that although the system size increases twice, the entire computational time remains at a comparable scale. This shows the advantage of the QM/MM-based approach over QM (ab initio) MD simulation, which faces a drastic increase in computational time with system size. Fig. 3 indicates that force and energy calculations, which include QM(DFTB3), MM, and QM/MM interactions amounts to ca. 80%. Because the SCMP method is based on multi partitioning approach, the computation cost for this part linearly increases according to the number of partitionings. Note, however, that these parts of the calculation on respective partitionings are independent of each other. Hence, it is suitable for parallel computation. In addition, because all partitionings in the SCMP method have a QM region of the same size, it shows higher scalability in parallelization than the other solvent-adaptive QM/MM. As a result, the MD performance of the proposed method is comparable to that of the conventional QM/MM MD simulation. The size dependency of this part mainly arises from the MM part calculation, which is somewhat moderate compared to that of the QM calculation. The second bottleneck is the evaluation of the SCMP weight function σ in eqn (10), which is shared by both solvent-adaptive and full-adaptive QM/MM. Note that the computational cost for this process can be independent of the system size because the progress function λ in eqn (11) becomes constant when the solvent distance from the QM center is larger than the threshold, and thus it is sufficient to count a limited number of solvent molecules to evaluate the SCMP weight σ. The residual cost contains the EPI evaluation and RATTLE calculation, which are specific to the full adaptive QM/MM method. Notably, these specific parts are so small that the computation costs did not vary significantly between the solvent and full adaptive QM/MM methods.
 |
| | Fig. 3 Relation between system size and computational time for one MD step. The black column represents the energy and force calculation times. The blue column represents the calculation time for the SCMP weight function σ. The red line represents the calculation time for the rest, including EPI evaluations and the RATTLE algorithm. | |
 |
| | Fig. 4 Temperature in the course of MD simulation time using an adaptive Langevin thermostat with a friction coefficient γ0 = 0.1 ps−1. Red solid and green dash lines represent a sliding 5 ps average and reference temperature 300 K, respectively. | |
Solvation structure
Hereafter, O* denotes the oxygen atom nearest to the QM center. While O* represents the oxygen atom in solute H2O in the ordinary solvent adaptive method, in the case of hydronium ion simulation, O* is assumed to be a part of the hydronium ion. Fig. 5 shows the radial distribution function (RDF) around the oxygen atom O* in the solute molecule. Compared to the bulk water simulation, the first peak of the O*–O RDF around the hydronium ion shifted from 2.8 Å to 2.6 Å, while the first peak in the experiments was observed at 2.5 Å.64 In contrast, DFTB2 simulations for hydronium ions showed bimodal peaks at approximately 2.4 Å and 2.8 Å, indicating that a Zundel-type structure was the dominant component.65 On the other hand, the present O*–O RDF showed a distinct single peak, which is consistent with the DFTB3 simulations.66 According to the previous AIMD simulation,2 such a result implies that an Eigen-type structure is the major component. The height of the first peak of oxygen atoms in bulk water was 4.1, which was significantly larger than the experimental value of 2.5.67 The first peak of the O*–O RDF is broader than the empirical potential structural refinement (EPSR) of the experimental data.64 As a result, the coordination number was estimated to be as large as 5.4 by integrating the first solvation shell. As DFTB3, which was employed for the QM region, causes oversolvation and high density for bulk water, such properties appear to be carried over to the hydronium ion simulation. With respect to the O*–H RDF, the first peak reflects the hydrogen atoms that are covalently bonded to the hydronium oxygen. While the bulk water simulation showed a second peak of O*–H RDF at 1.8 Å, it was not observed in the hydronium ion simulations, indicating that the hydronium ion did not accept hydrogen bonds. Note that the previous AIMD simulation showed that the O*–H RDF of the Eigen-type cation had an additional peak at approximately 1.6 Å.7 Although an Eigen-type cation was more probable in the present simulation, as shown in the next section, we could not find any additional peak in that distance range.
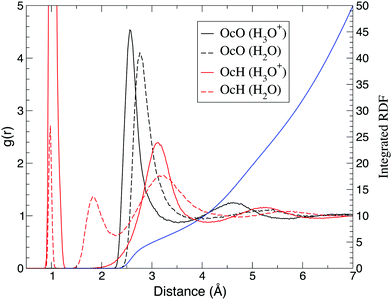 |
| | Fig. 5 Radial distribution function of oxygen (black) and hydrogen (red) atoms around the oxygen O*. Solid and dash lines represent simulations of hydronium and water as solute, respectively. The blue line represents the integrated radial distribution function of the oxygen atom around the hydronium ion. | |
Energetics and dynamics
Fig. 6 shows the potential of mean force for PT compared with the potential energy in the gaseous phase, which is projected on two reaction coordinates, the distance between two oxygen atoms RO*O′ and hydrogen displacement δ. The hydrogen displacement δ is defined as the difference between the two distances as δ = RO′H* − RO*H*, where the third nearest hydrogen atom to O* is denoted as H*, and the oxygen atom nearest to H* other than O* is defined as O′. Note that the EPI parameter c and friction coefficient γ0 did not affect the potential mean force (see ESI†). While the Zundel cation was more stable than the Eigen cation in vacuo, the balance was inverted in the aqueous phase, which is in agreement with previous studies.8,65Fig. 6 gives a clear picture of the PT mechanism in the Eigen to Zundel to Eigen sequence, where the energy barrier for the PT disappears as RO*O′ becomes smaller than 2.43 Å. The present SCMP simulation estimated the energy barrier for PT to be approximately 2.7 kJ mol−1, which was in good agreement with the DFTB3/3OB simulation.66 Although DFTB3 leads to the overbinding of OH covalent bonds and the underestimation of hydrogen bonds as previously reported,20,43 the estimated barrier was in agreement with previous studies of AIMD with DFT using BLYP7 and HCTH functionals;68 however, it was significantly smaller than those of MS-EVB2 and 3, which were estimated to be 8.4 kJ mol−1.69 It should be noted that the nuclear quantum effect lowered the PT barrier.7,9,70,71 Moreover, the energy barrier of approximately 1KBT at room temperature is supposed to disappear by the incorporation of the nuclear quantum effect, causing the topological defect to delocalize and transfer at a rate faster than 100 fs.7,9 In contrast, the present profile showed a distinct free energy minimum corresponding to the Eigen structure observed during the resting state of PT. Fig. 7 shows the time evolution of PT projected on two reaction coordinates. Here, the PT event was centered around a moment with δ= 0, and the trajectories were averaged over more than 3000 PT events. Note that the PT appeared as an event within 100 fs. It should also be noted that concerted oscillations were observed along the two reaction coordinates with a period of approximately 15 fs, where RO*O′ decreases and |δ| increases are clearly coupled.
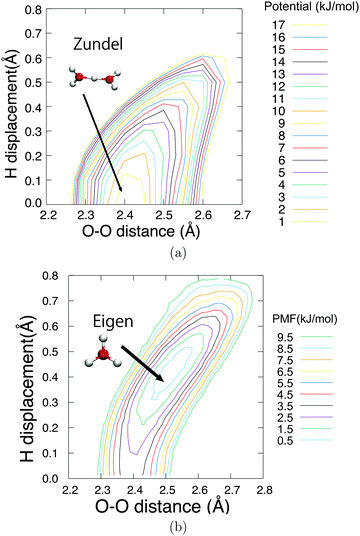 |
| | Fig. 6 Potential energy in gas phase (a) and potential of mean force in the aqueous phase (b) of hydronium ion. The horizontal axis represents the distance between the oxygen atom O* nearest to the QM center and the second nearest oxygen O′. The vertical axis represents the transferred hydrogen displacement, δ = RO′H − RO*H. The baseline value was set to zero. The contour is displayed for every 1.0 kJ mol−1. The gas phase potential is also evaluated with DFTB3/3OB method. | |
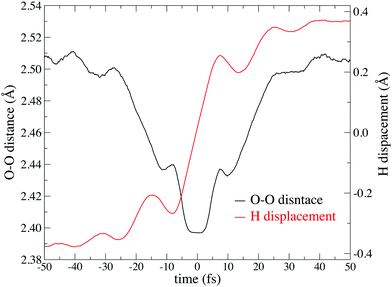 |
| | Fig. 7 Time evolution centered on the hydrogen bond switching event. The black line represents the distance between the two oxygen O* and O′. The red line represent δ = ROH − RO*H, where the oxygen indices for O* and O′ is fixed before and after the PT event. | |
Diffusion coefficient
In general, a thermostat can affect dynamical properties. To reproduce plausible dynamics, we employed the adaptive Langevin thermostat, where the coupling strength to the thermostat of the respective solvent molecule is adaptively updated according to the distance from the QM center. To verify the influence of the thermostat, we benchmarked the diffusion coefficient of the QM water molecules in the MM water system in the conventional solvent adaptive QM/MM method by employing two approaches: linear fitting of the mean-square displacement (MSD) and the integration of the velocity autocorrelation function (VAC) of the EPI. As shown in Table 1, when γ0 = 10 ps−1, the diffusion coefficient of H2O shows good agreement with the NVE and QM-MD simulations with the DFTB3 model, while it is underestimated when γ0 = 100 ps−1. This implies that the dynamics with γ0 < 10 ps−1 are almost free from artifacts from the Langevin thermostat.
Table 1 Diffusion coefficient (×10−1 Å2 ps−1)
|
γ
0 (ps−1) |
SCMP(DFTB3/3OB) |
DFTB3 |
DC-DFTB3 (diag)f |
Exp.g |
| 100 |
10 |
NVEc |
| MSDa |
VACb |
MSDa |
VACb |
MSDa |
|
Evaluation by linear fitting of MSD.
Evaluation by integration of velocity autocorrelation function.
Microcanonical condition.
128 water molecules with DFTB3/3OB (diag) by Goyal et al. (2011).65
2048 water molecules by Watanabe et al. (2018).44
Nakai et al. (2016).83
Roberts et al. (1988).84
|
| H2O |
2.7 ± 0.3 |
3.2 ± 0.3 |
4.8 ± 0.7 |
4.5 ± 0.3 |
4.6 ± 0.4 |
3.8d, 4.5e |
1.9 |
2.3 |
| H3O+ |
5.7 ± 1.0 |
5.8 ± 0.6 |
10.5 ± 1.1 |
9.7 ± 0.9 |
|
6.6 ± 2.0d |
9.1 |
9.4 ± 0.1 |
Next, we calculated the diffusion coefficient of hydronium ions with full adaptive QM/MM using MSD and VAC, where the EPI of the present study was used to estimate the position and velocity of the hydronium ion. For evaluation, we employed six and fifteen independent 100 ps-trajectories with γ0 = 100 and 10 ps−1, respectively. We emphasize that the availability of VAC analysis indicates the advantage of the present full adaptive QM/MM simulation, which makes the velocity of the hydronium ion directly accessible. The resulting values obtained by the two analyses showed agreement and smaller statistical error, which implies that sufficient sampling was achieved by the stable MD framework.
The obtained diffusion coefficients varied according to the friction coefficient, where 0.58 Å2 ps−1 with γ0 = 100 ps−1, and 1.0 Å2 ps−1 with γ0 = 10 ps−1. Considering H2O, the result with γ0 = 10 ps−1 is more plausible and in good agreement with the experimental value of 0.94 Å2 ps−1, although we suppose that there appears to be error cancellation. This agreement is in stark contrast with previous studies, where there can be found a systematic underestimation as 0.40 and 0.36 Å2 ps−1 by MS-EVB2,46,69 0.29 Å2 ps−1 by classical MS-EVB3,69 0.50 Å2 ps−1 by quantum MS-EVB3,69 and 0.33 Å2 ps−1 by CPMD with HCTH functional68 These underestimations may be attributed to the limited size effect in the DFTB3-diag simulation.69,72 On the other hand, the limited size effect is less likely in the present simulation because of the sufficiently large system consisting of 2048 H2O molecules employed by taking advantage of the QM/MM method. It is also insightful to compare the previous DFTB studies, although they employed a modified parameter 3OB(diag) for the OH repulsive potential, while 3OB was used in the present study. In this study, the obtained values are larger than (0.66 Å ± 0.20 Å2 ps−1) in the previous paper based on QM-MD simulation using DFTB3-diag,65 while it is in good agreement with DC-DFTB3 (diag). The disagreement between the DFTB3-diag and DC-DFTB3-diag was also expected to result from the limited size effect in the DFTB3-diag simulation. However, this discrepancy cannot be accounted for by the size-limiting effect alone because the DFTB3-diag for the 128 water system resulted in a H2O diffusion coefficient of 0.38 Å2 ps−1, which was larger than the diffusion coefficient of 0.19 Å2 ps−1 for DC-DFTB3-diag with 513 water molecules (Table 1). The deviation may also have resulted from either/both the protocol of the diffusion coefficient calculation and/or DC treatment, such as the discontinuity caused by particles crossing the subsystems. In a previous study, the hydronium diffusion coefficient was indirectly evaluated by the summation of the vehicular and Grotthuss diffusion coefficients, where the former corresponds to the water diffusion coefficient, and the latter is estimated using the PT pitch and rate, and the two dynamics are assumed to be independent. On the other hand, the present approach with the use of EPI allowed the direct evaluation of diffusion without the assumption of independence. In addition, DFTB3/3OB and DC-DFTB3-diag showed different diffusion coefficients of H2O, while they had a comparable PT barrier in free energy (Table 1) Hence, the agreement between the present result and DC-DFTB3-diag implies that the correlation of solvent-PT dynamics and/or the other factor of the rate-limiting process of proton diffusion in this model.
Previous simulations have proposed that the rate-limiting step is the change in the coordination number of water molecules in the solvation shell.2,9,73 In addition, water reorientation related to molecular rotation occurred together with the formation/deformation of hydrogen bonds.74Table 2 shows the resulting values obtained by the explicit integration of the second rank auto-correlation function of the OH bond orientation of H2O using the solvent adaptive QM/MM (SCMP) method. Although the result appears to be free from thermostat artifacts, it showed that DFTB3/3OB significantly underestimated the relaxation time when compared to the experimental value. This result is in agreement with the fact that the underestimation of the hydrogen bond energy by DFTB3 results in a faster relaxation of the bond orientation of water compared with that in the experiments.66 Consistently, the diffusion coefficient of DFTB3 water was significantly overestimated.66,75 Therefore, we assume that the overestimation of the free energy barrier and the underestimation of the orientational relaxation canceled out, which is in agreement with experiments regarding the diffusion coefficient of the hydronium ion. Because the nuclear quantum effect is assumed to lower the free energy barrier, the balance of error will be altered when it is incorporated. However, it is difficult to precisely predict that it can indirectly have competing effects on water diffusion with respect to surrounding water molecules.76
Table 2 Orientational relaxation time from P2 correlation function (ps) with different friction coefficients
|
γ
0 (ps−1) |
SCMP(DFTB3/3OB) |
DFTB3/3OBa |
Exp.85–88 |
| 100 |
10 |
1 |
|
MD simulation for 124 water system.66
|
| Correlation time |
0.53 ± 0.05 |
0.50 ± 0.08 |
0.58 ± 0.07 |
0.7 |
1.7–2.6 |
Discussions
To achieve a solute-adaptive QM/MM, it is essential to numerically express the hydronium position. The indicator should be capable of distinguishing the solvation structure specific to hydronium ions from ordinary bulk water. Intuitively, the number of covalent bonds appears to be such a structural measure, but it cannot be directly used as an EPI because it is discretized. On the other hand, ψ found in eqn (17) can be regarded as a continuous expression of the number of covalent bonds of an oxygen atom. Fig. 8(a) visualize the time evolution of ψ of the first and second nearest oxygen to the QM center. The intersects of the black and red lines indicate the PT events, where the ψ indices also vary according to the displacement of the QM center. Before the PT, the value of ψ2 gradually increased, which was sometimes accompanied by a decrease in ψ1. Then, after PT with the index switches, the “original” ψ1 and ψ2 decrease and increase, respectively. Fig. 8(b) shows the distribution of the ψi value of oxygen atoms sampled in the course of MD simulations, where the subscript represents its positional order from the QM center. With respect to the nearest neighboring oxygen, which is assumed to be a part of the hydronium ion, ψ1 almost took the value of 3.0, although it was observed to be below 3.0, in agreement with Fig. 8(a). The second nearest oxygen to the QM center, which forms the nearest neighboring water, showed a bimodal distribution with respect to ψ2. The first majority was at ψ2 = 2.0, which occupied 88%, while the second majority appeared at 3.0, which amounted to 4%. The ψ2 value of 2.0 corresponds to the resting state of PT, where the hydronium ion takes the Eigen form. In contrast, ψ3 equaled 2.0, most of the time, and it rarely became larger. It is obvious that oxygen atoms farther away from the QM center than the third nearest oxygen atom have a ψi value of only 2.0. Because of eqn (19), the EPI weight Wi of the oxygen atoms with ψ= 2 becomes zero, which indicates that the EPI was insensitive to distant water molecules from the hydronium ion. This is the most important attribute of an EPI to avoid scale-dependency and instability, as described above. Furthermore, this also helps to drastically reduce the computational cost of evaluating the proton indicators because the number of water molecules that need to be considered is very small.
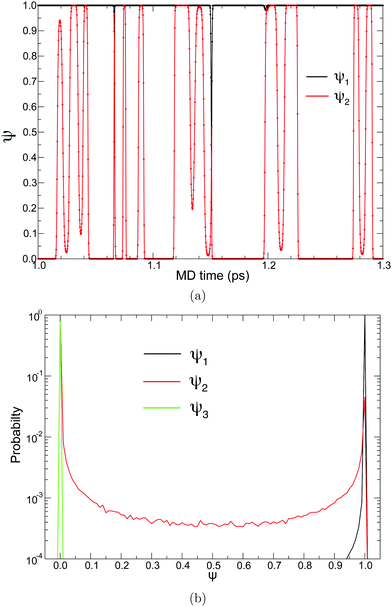 |
| | Fig. 8 (a) ψ in course of MD simulation time and (b) normalized probability distribution of ψ sampled in course of MD simulation. Black, red, and green lines represent the first, second, and third nearest oxygen atoms to the QM center, respectively. | |
While the EPI in previous studies was a function of oxygen coordinates as defined in eqn (5), the EPI in the present study was expressed as a linear combination of dividing points ζ between water oxygen and virtual site coordinates η, as defined in eqn (13). Note that when r = r0 in eqn (16), the Gaussian-type function ϕ′ has the maximum value. Thus, r0 was tuned to be larger than the ordinary OH covalent bond length. As a result, as shown in eqn (15), ηi mainly reflects the hydrogen position that has the longest covalent bond with the ith oxygen atom. If the oxygen coordinates are used in eqn (13), that is, c = 0, the EPI during the resting state of PT (which constitutes most of the MD simulation) would correspond to the oxygen in the hydronium ion. Hence, the EPI is displaced between two oxygen atoms at an anomalously high speed when the proton transfers. However, the EPI with c > 0 fluctuated even during the resting state of PT, reflecting the stretching of OH covalent bonds. On the other hand, when the proton is transferred, it is displaced at a moderate speed, which likely reflects the speed of the hydrogen transfer because the EPI was mainly dominated by a longest-covalently bonded hydrogen atom, that is, the transferred proton. Fig. 9 shows the velocity distribution of the EPI sampled in course of MD simulation with c = 0.0 and 0.2. Indeed, when c = 0.2, the probability of a large velocity is smaller than that with c = 0.0, although the probability of the middle-range velocity increased. Because anomalously rapid displacement of the EPI can destabilize the simulation causing discontinuity, the non-zero value of c appears to be more suitable. However, this contradicts Fig. 2(a), where c = 0.0 showed the best stability and sustainability. To account for this, Fig. 2(b and c) is insightful, where c = 0.0 resulted in the smallest drifts of the bookkeeping term. Therefore, small but constant fluctuations of the EPI coordinates can frequently cause the deformation of the QM region and the corresponding partitioning update, which accelerates the drift of the bookkeeping term and distorts the dynamics. The distorted dynamics can also induce anomalously rapid displacements of the EPI, which is more critical than that observed when c = 0.
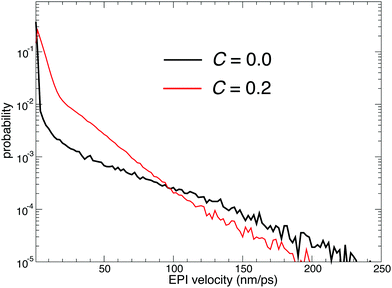 |
| | Fig. 9 Probability distribution of the EPI velocity sampled in course of MD simulation. The black and red lines represent the simulation results with c = 0.0 and 0.2, respectively. | |
Conclusions
In this study, we proposed a numerical expression for the excess proton indicator for hydronium ion simulations in bulk water, and implemented it in the SCMP code. Based on the results, we achieved a QM/MM simulation that was both solute- and solvent-adaptive, which we refer to as a “full adaptive QM/MM method”, and we successfully demonstrated its stability and efficiency. Based on this new framework, we were able to confirm the numerical conservation of the total energy of the PT simulation, that is, temporal continuity, with Hamiltonian dynamics under microcanonical conditions. However, the bookkeeping term and the temperature constantly varied owing to spatial discontinuities, for which we next conducted a Langevin dynamics simulation by employing the thermostat in an adaptive manner to retain plausible dynamics of the PT. We emphasize that the present computational approach is advantageous for evaluating the dynamical properties of PT in bulk water for the following three reasons. First, the position of the excess proton is defined with numerical stability and obtained on-the-fly through MD simulations. This provides direct access to various physical properties, such as the diffusion coefficient through the MSD or velocity autocorrelation function, which have been indirectly evaluated in previous studies. Moreover, the EPI may be used as a reaction coordinate for enhanced sampling, such as umbrella sampling, by imposing an artificial force on a dummy atom. Notably, the present EPI can also be used for post-MD analysis, as long as the coordinate information is retained. This may be useful to reassess previous AIMD simulations by removing artificial noise. Second, the computational cost required for the present approach based on the QM/MM method is moderate compared to AIMD, making it accessible to longer dynamic trajectories. As a result, statistical errors can be significantly suppressed compared to conventional studies using the AIMD method. The linear shape of the obtained MSD and the agreement between diffusion coefficients obtained by MSD and velocity autocorrelation function provide strong evidence that statistical errors are greatly suppressed (see ESI†). We assume that this feature will further facilitate the analysis of inhomogeneous systems, such as the one used in the present study, rather than homogeneous systems such as pure water. Because the number of trajectories of interest obtained by a single MD run is very limited, longer production runs are required for higher accuracy in the case of simulations for inhomogeneous systems. Hence, the computational cost becomes a critical factor. Also note that, the adaptive treatment may be extended to quantum mechanical forcefield methods such as X-pol77,78 and mDC,79,80 which draw an increasing attention in application to macromolecular system. Although both Linear-scaling quantum mechanical method and quantum forcefield are based on the fragmentation of the entire system into subsystem, the main difference lies in treatment of inter-fragment coupling. Instead of buffer regions in DC16 and many-body expansion in fragmented molecular orbital method,81,82 quantum forcefield employs the empirical parameters to realize faster calculations. However, there still remains room for improvement in treatment of reacting molecules and fragments. The adaptive treatment QM/MM for different models may be extended to inter-fragment treatment of the quantum forcefield. Third, this full adaptive QM/MM method also provides access to large systems that cannot be treated with a full QM method. In the present study, we considered one hydronium ion and 2047 water molecules in a cubic box with side lengths of 39.5 Å, which is beyond the range of applications of ordinary AIMD simulations. It is known that the diffusion coefficient is subject to a limited size effect under periodic boundary conditions, resulting in a drastic underestimation in previous studies using AIMD simulations. On the other hand, the SCMP method can suppress the artifacts, as we have previously reported,43 although it is not sufficient. Therefore, in the present study, a major part of the deviation of the obtained diffusion coefficient from the experimental value can be attributed to either/both the shortcoming of the employed QM model (DFTB3/3OB) or/and the missing nuclear quantum effect.
Owing to these advantages, the present full adaptive QM/MM method makes the hydronium ion simulation accessible with a plausible cost and moderate computation time, which has been a long-standing challenge in molecular simulations. It is clear that this will become an effective tool in advancing the theoretical analysis of hydronium ions in the subsequent stage.
Conflicts of interest
There are no conflicts to declare.
Let the effective potential Veff be defined as,| | 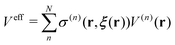 | (26) |
where V is a potential energy that is a function of particle coordinates r.| | 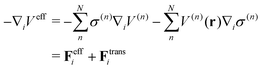 | (27) |
Thus, the derivative of effective function with respect to xi is
| | 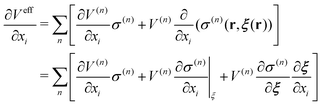 | (28) |
Then, the second derivative of effective function with respect to xi and yi is
| |  | (29) |
For the conservation of Veff, ξ(r) as well as σ should be differentiable, and the derivative should have the symmetry with respect to exchange of xi and yi.
Appendix B
The Spline curve employed in Seqn (18) and QM progress function λQM in eqn (11) is written as,| | 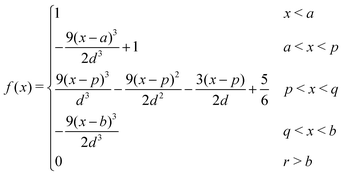 | (30) |
where 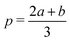 ,
, 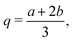 and d = b − a. For S in eqn (18), we employ a = Rsmall = 12.0 Å and b = Rlarge = 13.2 Å. For λMM in eqn (11), we employ a = sQM = 6.4 Å and b = tQM = 8.4 Å.
and d = b − a. For S in eqn (18), we employ a = Rsmall = 12.0 Å and b = Rlarge = 13.2 Å. For λMM in eqn (11), we employ a = sQM = 6.4 Å and b = tQM = 8.4 Å.
Likewise, the Spline curve employed in Weqn (19) and the MM progress function λMM is detailed as,
| | 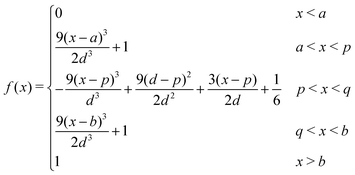 | (31) |
In the case of W in eqn (19), we employ a = 2 and b = 3. For λMM, we employ a = sMM = 4.2 Å and b = tMM = 6.4 Å.
Acknowledgements
We thank Dr Yutaka Shikano and Dr Taisuke Hasegawa for their constructive comments. H. C. W. was supported by JSPS Grant Numbers 20K03885 and 20H05518, and JST PRESTO Grant number JPMJPR17GC. In addition, H. C. W. and Y. S. were supported by the MEXT Quantum Leap Flagship Program Grant Number JPMXS0118067285. This study was conducted using Fujitsu PRIMEGY CX600M1/CX1640M1 (Oakforest-PACS) at the CCS, University of Tsukuba and SGI Rackable C1102-GP8 (Reedbush).
References
- T. Komatsuzaki and I. Ohmine, Energetics of proton transfer in liquid water. I, Ab initio study for origin of many-body interaction and potential energy surfaces, Chem. Phys., 1994, 180, 239–269 CrossRef CAS.
- M. Tuckerman, K. Laasonen, M. Sprik and M. Parrinello, Ab initio molecular dynamics simulation of the solvation and transport of H3O+ and OH-ions in water, J. Phys. Chem., 1995, 99, 5749–5752 CrossRef CAS.
- M. Tuckerman, K. Laasonen, M. Sprik and M. Parrinello, Ab initio molecular dynamics simulation of the solvation and transport of hydronium and hydroxyl ions in water, J. Chem. Phys., 1995, 103, 150–161 CrossRef CAS.
- J. Lobaugh and G. A. Voth, The quantum dynamics of an excess proton in water, J. Chem. Phys., 1996, 104, 2056–2069 CrossRef CAS.
- R. Vuilleumier and D. Borgis, Molecular dynamics of an excess proton in water using a non-additive valence bond force field, J. Mol. Struct., 1997, 436, 555–565 CrossRef.
- U. W. Schmitt and G. A. Voth, Multistate empirical valence bond model for proton transport in water, J. Phys. Chem. B, 1998, 102, 5547–5551 CrossRef CAS.
- D. Marx, M. E. Tuckerman and M. Parrinello, Solvated excess protons in water: quantum effects on the hydration structure, J. Phys.: Condens. Matter, 2000, 12, A153 CrossRef CAS.
- M. E. Tuckerman, D. Marx and M. Parrinello, The nature and transport mechanism of hydrated hydroxide ions in aqueous solution, Nature, 2002, 417, 925–929 CrossRef CAS PubMed.
- D. Marx, A. Chandra and M. E. Tuckerman, Aqueous basic solutions: hydroxide solvation, structural diffusion, and comparison to the hydrated proton, Chem. Rev., 2010, 110, 2174–2216 CrossRef CAS PubMed.
- A. W. Sakti, Y. Nishimura, H. Sato and H. Nakai, Divide-and-conquer density-functional tight-binding molecular dynamics study on the formation of carbamate ions during CO2 chemical absorption in aqueous amine solution, Bull. Chem. Soc. Jpn., 2017, 90, 1230–1235 CrossRef CAS.
- M. E. Tuckerman, A. Chandra and D. Marx, Structure and dynamics of OH-(aq), Acc. Chem. Res., 2006, 39, 151–158 CrossRef CAS PubMed.
- R. Car and M. Parrinello, Unified approach for molecular dynamics and density-functional theory, Phys. Rev. Lett., 1985, 55, 2471 CrossRef CAS PubMed.
- W. Yang and T.-S. Lee, A density-matrix divide-and-conquer approach for electronic structure calculations of large molecules, J. Chem. Phys., 1995, 103, 5674–5678 CrossRef CAS.
- M. Kobayashi, T. Kunisada, T. Akama, D. Sakura and H. Nakai, Reconsidering an analytical gradient expression within a divide-and-conquer self-consistent field approach Exact formula and its approximate treatment, J. Chem. Phys., 2011, 134, 034105 CrossRef PubMed.
- D. M. York, T.-S. Lee and W. Yang, Quantum mechanical study of aqueous polarization effects on biological macromolecules, J. Am. Chem. Soc., 1996, 118, 10940–10941 CrossRef CAS.
- T.-S. Lee, D. M. York and W. Yang, Linear-scaling semiempirical quantum calculations for macromolecules, J. Chem. Phys., 1996, 105, 2744–2750 CrossRef CAS.
- D. M. York, T.-S. Lee and W. Yang, Quantum mechanical treatment of biological macromolecules in solution using linear-scaling electronic structure methods, Phys. Rev. Lett., 1998, 80, 5011 CrossRef CAS.
- M. Elstner, D. Porezag, G. Jungnickel, J. Elsner, M. Haugk, T. Frauenheim, S. Suhai and G. Seifert, Self-consistent-charge density-functional tight-binding method for simulations of complex materials properties, Phys. Rev. B: Condens. Matter Mater. Phys., 1998, 58, 7260 CrossRef CAS.
- M. Gaus, Q. Cui and M. Elstner, DFTB3: extension of the self-consistent-charge densityfunctional tight-binding method (SCC-DFTB), J. Chem. Theory Comput., 2011, 7, 931–948 CrossRef CAS PubMed.
- M. Gaus, A. Goez and M. Elstner, Parametrization and benchmark of DFTB3 for organic molecules, J. Chem. Theory Comput., 2013, 9, 338–354 CrossRef CAS PubMed.
- A. Warshel and M. Levitt, Theoretical studies of enzymic reactions: dielectric, electrostatic and steric stabilization of the carbonium ion in the reaction of lysozyme, J. Mol. Biol., 1976, 103, 227–249 CrossRef CAS PubMed.
- T. Kerdcharoen, K. R. Liedl and B. M. Rode, A QM/MM simulation method applied to the solution of Li+ in liquid ammonia, Chem. Phys., 1996, 211, 313–323 CrossRef CAS.
- T. Kerdcharoen and K. Morokuma, ONIOM-XS: an extension of the ONIOM method for molecular simulation in condensed phase, Chem. Phys. Lett., 2002, 355, 257–262 CrossRef CAS.
- A. Heyden, H. Lin and D. G. Truhlar, Adaptive partitioning in combined quantum mechanical and molecular mechanical calculations of potential energy functions for multiscale simulations, J. Phys. Chem. B, 2007, 111, 2231–2241 CrossRef CAS PubMed.
- H. Lin and D. G. Truhlar, QM/MM: what have we learned, where are we, and where do we go from here?, Theor. Chem. Acc., 2007, 117, 185 Search PubMed.
- R. E. Bulo, B. Ensing, J. Sikkema and L. Visscher, Toward a practical method for adaptive QM/MM simulations, J. Chem. Theory Comput., 2009, 5, 2212–2221 CrossRef CAS PubMed.
- S. Pezeshki and H. Lin, Adaptive-partitioning redistributed charge and dipole schemes for QM/MM dynamics simulations: on-the-fly relocation of boundaries that pass through covalent bonds, J. Chem. Theory Comput., 2011, 7, 3625–3634 CrossRef CAS PubMed.
- C. N. Rowley and B. Roux, The solvation structure of Na+ and K+ in liquid water determined from high level ab initio molecular dynamics simulations, J. Chem. Theory Comput., 2012, 8, 3526–3535 CrossRef CAS PubMed.
- N. Bernstein, C. Várnai, I. Solt, S. A. Winfield, M. C. Payne, I. Simon, M. Fuxreiter and G. Csányi, QM/MM simulation of liquid water with an adaptive quantum region, Phys. Chem. Chem. Phys., 2012, 14, 646–656 RSC.
- N. Takenaka, Y. Kitamura, Y. Koyano and M. Nagaoka, An improvement in quantum mechanical description of solute-solvent interactions in condensed systems via the number-adaptive multiscale quantum mechanical/molecular mechanical-molecular dynamics method: application to zwitterionic glycine in aqueous solution, J. Chem. Phys., 2012, 137, 024501 CrossRef PubMed.
- R. E. Bulo, C. Michel, P. Fleurat-Lessard and P. Sautet, Multiscale modeling of chemistry in water: are we there yet?, J. Chem. Theory Comput., 2013, 9, 5567–5577 CrossRef CAS PubMed.
- M. Shiga and M. Masia, Boundary based on exchange symmetry theory for multilevel simulations. I. Basic theory, J. Chem. Phys., 2013, 139, 044120 CrossRef PubMed.
- M. P. Waller, S. Kumbhar and J. Yang, A density-based adaptive quantum mechanical/molecular mechanical method, ChemPhysChem, 2014, 15, 3218–3225 CrossRef CAS PubMed.
- H. C. Watanabe, T. Kubař and M. Elstner, Size-consistent multipartitioning QM/MM: a stable and efficient adaptive QM/MM method, J. Chem. Theory Comput., 2014, 10, 4242–4252 CrossRef CAS PubMed.
- M. Shiga and M. Masia, Quasi-boundary based on exchange symmetry theory for multilevel simulations, Mol. Simul., 2015, 41, 827–831 CrossRef CAS.
- J. M. Boereboom, R. Potestio, D. Donadio and R. E. Bulo, Toward Hamiltonian adaptive QM/MM: accurate solvent structures using many-body potentials, J. Chem. Theory Comput., 2016, 12, 3441–3448 CrossRef CAS PubMed.
- M. J. Field, An algorithm for adaptive QC/MM simulations, J. Chem. Theory Comput., 2017, 13, 2342–2351 CrossRef CAS PubMed.
- T. Jiang, S. Simko and R. E. Bulo, Accurate Quantum Mechanics/Molecular Mechanics Simulation of Aqueous Solutions with Tailored Molecular Mechanics Models, J. Chem. Theory Comput., 2018, 14, 3943–3954 CrossRef CAS PubMed.
- A. W. Duster, C.-H. Wang and H. Lin, Adaptive QM/MM for molecular dynamics simulations: 5. On the energy-conserved permuted adaptive-partitioning schemes, Molecules, 2018, 23, 2170 CrossRef PubMed.
- H. Takahashi, H. Kambe and A. Morita, Calculation of solvation free energy utilizing a constrained QM/MM approach combined with a theory of solutions. The, J. Chem. Phys., 2019, 150, 114109 CrossRef PubMed.
-
Z. Yang, On-the fly determination of active region centers in adaptive-partitioning QM/MM, 2020 Search PubMed.
- B. Ensing, S. O. Nielsen, P. B. Moore, M. L. Klein and M. Parrinello, Energy conservation in adaptive hybrid atomistic/coarse-grain molecular dynamics, J. Chem. Theory Comput., 2007, 3, 1100–1105 CrossRef CAS PubMed.
- H. C. Watanabe and Q. Cui, Quantitative analysis of QM/MM boundary artifacts and correction in adaptive QM/MM simulations, J. Chem. Theory Comput., 2019, 15, 3917–3928 CrossRef CAS PubMed.
- H. C. Watanabe, Improvement of performance, stability and continuity by modified size-consistent multipartitioning quantum mechanical/molecular mechanical method, Molecules, 2018, 23, 1882 CrossRef PubMed.
- R. Pomès and B. Roux, Free energy profiles for H+ conduction along hydrogen-bonded chains of water molecules, Biophys. J., 1998, 75, 33–40 CrossRef.
- T. J. Day, A. V. Soudackov, M. Čuma, U. W. Schmitt and G. A. Voth, A second generation multistate empirical valence bond model for proton transport in aqueous systems, J. Chem. Phys., 2002, 117, 5839–5849 CrossRef CAS.
- N. Chakrabarti, E. Tajkhorshid, B. Roux and R. Pomès, Molecular basis of proton blockage in aquaporins, Structure, 2004, 12, 65–74 CrossRef CAS PubMed.
- P. König, N. Ghosh, M. Hoffmann, M. Elstner, E. Tajkhorshid, T. Frauenheim and Q. Cui, Toward theoretical analyis of long-range proton transfer kinetics in biomolecular pumps, J. Phys. Chem. A, 2006, 110, 548–563 CrossRef PubMed.
- A. W. Duster and H. Lin, Tracking Proton Transfer through Titratable Amino Acid Side Chains in Adaptive QM/MM Simulations, J. Chem. Theory Comput., 2019, 15, 5794–5809 CrossRef CAS PubMed.
- S. Pezeshki and H. Lin, Adaptive-partitioning QM/MM for molecular dynamics simulations: 4. Proton hopping in bulk water, J. Chem. Theory Comput., 2015, 11, 2398–2411 CrossRef CAS PubMed.
- H. C. Andersen, Rattle: a velocity version of the shake algorithm for molecular dynamics calculations, J. Comput. Phys., 1983, 52, 24–34 CrossRef CAS.
- H. C. Watanabe, M. Banno and M. Sakurai, An adaptive quantum mechanics/molecular mechanics method for the infrared spectrum of water: incorporation of the quantum effect between solute and solvent, Phys. Chem. Chem. Phys., 2016, 18, 7318–7333 RSC.
- A. Brünger, C. L. Brooks III and M. Karplus, Stochastic boundary conditions for molecular dynamics simulations of ST2 water, Chem. Phys. Lett., 1984, 105, 495–500 CrossRef.
- B. Hess, C. Kutzner, D. Van Der Spoel and E. Lindahl, GROMACS 4: algorithms for highly efficient, load-balanced, and scalable molecular simulation, J. Chem. Theory Comput., 2008, 4, 435–447 CrossRef CAS PubMed.
- P. Bjelkmar, P. Larsson, M. A. Cuendet, B. Hess and E. Lindahl, Implementation of the CHARMM force field in GROMACS: analysis of protein stability effects from correction maps, virtual interaction sites, and water models, J. Chem. Theory Comput., 2010, 6, 459–466 CrossRef CAS PubMed.
- M. J. Abraham, T. Murtola, R. Schulz, S. Páll, J. C. Smith, B. Hess and E. Lindahl, GROMACS: high performance molecular simulations through multi-level parallelism from laptops to supercomputers, SoftwareX, 2015, 1, 19–25 CrossRef.
- Y. Wu, H. L. Tepper and G. A. Voth, Flexible simple point-charge water model with improved liquid-state properties, J. Chem. Phys., 2006, 124, 024503 CrossRef PubMed.
- T. Kubař, K. Welke and G. Groenhof, New QM/MM implementation of the DFTB3 method in the gromacs package, J. Comput. Chem., 2015, 36, 1978–1989 CrossRef PubMed.
- T. Darden, D. York and L. Pedersen, Particle mesh Ewald: an N
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/i_char_2009.gif) log(N) method for Ewald sums in large systems, J. Chem. Phys., 1993, 98, 10089–10092 CrossRef CAS.
log(N) method for Ewald sums in large systems, J. Chem. Phys., 1993, 98, 10089–10092 CrossRef CAS.
- K. Nam, J. Gao and D. M. York, An efficient linear-scaling Ewald method for longrange electrostatic interactions in combined QM/MM calculations, J. Chem. Theory Comput., 2005, 1, 2–13 CrossRef CAS PubMed.
- K. Nam, Acceleration of Ab Initio QM/MM Calculations under periodic boundary conditions by multiscale and multiple time step approaches, J. Chem. Theory Comput., 2014, 10, 4175–4183 CrossRef CAS PubMed.
- T. J. Giese and D. M. York, Ambient-potential composite Ewald method for ab initio quantum mechanical/molecular mechanical molecular dynamics simulation, J. Chem. Theory Comput., 2016, 12, 2611–2632 CrossRef CAS PubMed.
- H. Nishizawa and H. Okumura, Rapid QM/MM approach for biomolecular systems under periodic boundary conditions: combination of the density-functional tight-binding theory and particle mesh Ewald method, J. Comput. Chem., 2016, 37, 2701–2711 CrossRef CAS PubMed.
- A. Botti, F. Bruni, M. Ricci and A. Soper, Eigen versus Zundel complexes in HCl–water mixtures, J. Chem. Phys., 2006, 125, 014508 CrossRef CAS PubMed.
- P. Goyal, M. Elstner and Q. Cui, Application of the SCC-DFTB method to neutral and protonated water clusters and bulk water, J. Phys. Chem. B, 2011, 115, 6790–6805 CrossRef CAS PubMed.
- P. Goyal, H.-J. Qian, S. Irle, X. Lu, D. Roston, T. Mori, M. Elstner and Q. Cui, Molecular simulation of water and hydration effects in different environments: challenges and developments for DFTB based models, J. Phys. Chem. B, 2014, 118, 11007–11027 CrossRef CAS PubMed.
- A. K. Soper, The radial distribution functions of water as derived from radiation total scattering experiments: is there anything we can say for sure?, ISRN Phys. Chem., 2013, 2013 Search PubMed.
- C. M. Maupin, B. Aradi and G. A. Voth, The self-consistent charge density functional tight binding method applied to liquid water and the hydrated excess proton: benchmark simulations, J. Phys. Chem. B, 2010, 114, 6922–6931 CrossRef CAS PubMed.
- Y. Wu, H. Chen, F. Wang, F. Paesani and G. A. Voth, An improved multistate empirical valence bond model for aqueous proton solvation and transport, J. Phys. Chem. B, 2008, 112, 467–482 CrossRef CAS PubMed.
- H. Azzouz and D. Borgis, A quantum molecular-dynamics study of proton-transfer reactions along asymmetrical H bonds in solution, J. Chem. Phys., 1993, 98, 7361–7374 CrossRef CAS.
- K. Kosugi, H. Nakano and H. Sato, SCC-DFTB-PIMD method to evaluate a multidimensional quantum free-energy surface for a proton-transfer reaction, J. Chem. Theory Comput., 2019, 15, 4965–4973 CrossRef CAS PubMed.
- I.-C. Yeh and G. Hummer, System-size dependence of diffusion coefficients and viscosities from molecular dynamics simulations with periodic boundary conditions, J. Phys. Chem. B, 2004, 108, 15873–15879 CrossRef CAS.
- K. Andot and J. T. Hynes, HCl acid ionization in water: a theoretical molecular modeling, J. Mol. Liq., 1995, 64, 25–37 CrossRef.
- D. Laage and J. T. Hynes, A molecular jump mechanism of water reorientation, Science, 2006, 311, 832–835 CrossRef CAS PubMed.
- H. C. Watanabe, M. Kubillus, T. Kubař, R. Stach, B. Mizaikoff and H. Ishikita, Cation solvation with quantum chemical effects modeled by a size-consistent multipartitioning quantum mechanics/molecular mechanics method, Phys. Chem. Chem. Phys., 2017, 19, 17985–17997 RSC.
- S. Habershon, T. E. Markland and D. E. Manolopoulos, Competing quantum effects in the dynamics of a flexible water model, J. Chem. Phys., 2009, 131, 024501 CrossRef PubMed.
- J. Gao, Toward a molecular orbital derived empirical potential for liquid simulations, J. Phys. Chem. B, 1997, 101, 657–663 CrossRef CAS.
- J. Gao, D. G. Truhlar, Y. Wang, M. J. Mazack, P. Löffler, M. R. Provorse and P. Rehak, Explicit polarization: a quantum mechanical framework for developing next generation force fields, Acc. Chem. Res., 2014, 47, 2837–2845 CrossRef CAS PubMed.
- T. J. Giese, M. Huang, H. Chen and D. M. York, Recent advances toward a general purpose linear-scaling quantum force field, Acc. Chem. Res., 2014, 47, 2812–2820 CrossRef CAS PubMed.
- T. J. Giese and D. M. York, Quantum mechanical force fields for condensed phase molecular simulations, J. Phys.: Condens. Matter, 2017, 29, 383002 CrossRef PubMed.
- K. Kitaura, E. Ikeo, T. Asada, T. Nakano and M. Uebayasi, Fragment molecular orbital method: an approximate computational method for large molecules, Chem. Phys. Lett., 1999, 313, 701–706 CrossRef CAS.
- D. G. Fedorov and K. Kitaura, The importance of three-body terms in the fragment molecular orbital method, J. Chem. Phys., 2004, 120, 6832–6840 CrossRef CAS PubMed.
- H. Nakai, A. W. Sakti and Y. Nishimura, Divide-and-conquer-type density-functional tight-binding molecular dynamics simulations of proton diffusion in a bulk water system, J. Phys. Chem. B, 2016, 120, 217–221 CrossRef CAS PubMed.
- N. K. Roberts and H. L. Northey, Proton and deuteron mobility in normal and heavy water solutions of electrolytes, J. Chem. Soc., Faraday Trans. 1, 1974, 70, 253–262 RSC.
- K. Winkler, J. Lindner, H. Bürsing and P. Vöhringer, Ultrafast Raman-induced Kerr- effect of water: single molecule versus collective motions, J. Chem. Phys., 2000, 113, 4674–4682 CrossRef CAS.
- C. Lawrence and J. Skinner, Vibrational spectroscopy of HOD in liquid D2O. III. Spectral diffusion, and hydrogen-bonding and rotational dynamics, J. Chem. Phys., 2003, 118, 264–272 CrossRef CAS.
- H.-S. Tan, I. R. Piletic and M. Fayer, Orientational dynamics of water confined on a nanometer length scale in reverse micelles, J. Chem. Phys., 2005, 122, 174501 CrossRef PubMed.
- Y. Rezus and H. Bakker, On the orientational relaxation of HDO in liquid water, J. Chem. Phys., 2005, 123, 114502 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1cp00116g |
|
| This journal is © the Owner Societies 2021 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a  *ab,
Masayuki
Yamada
*ab,
Masayuki
Yamada
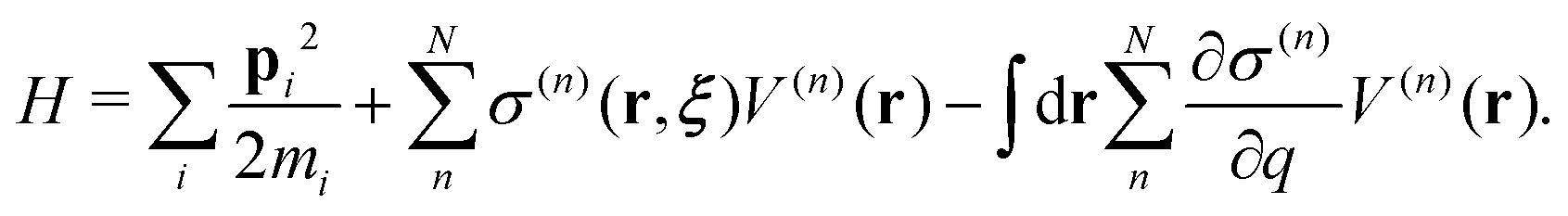
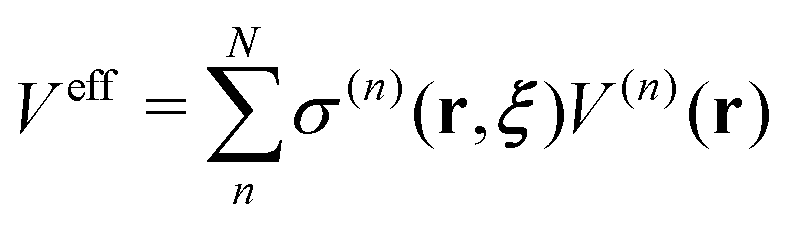
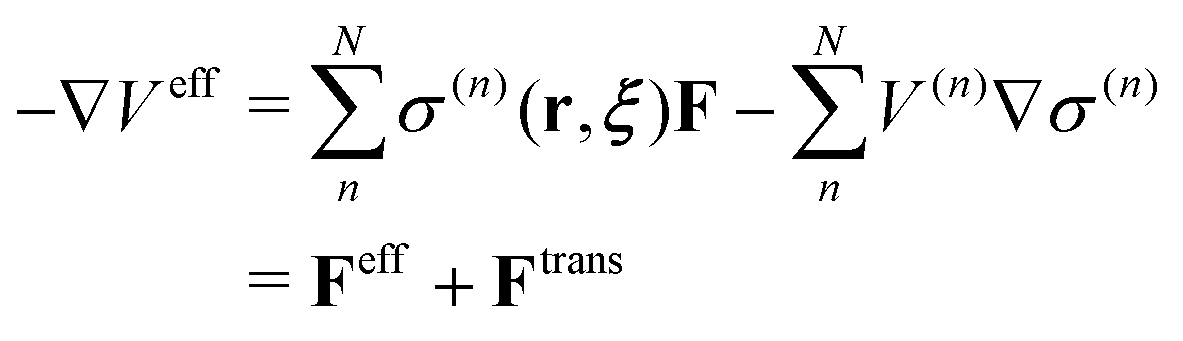
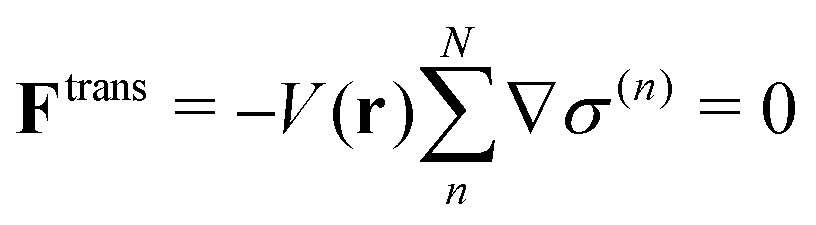
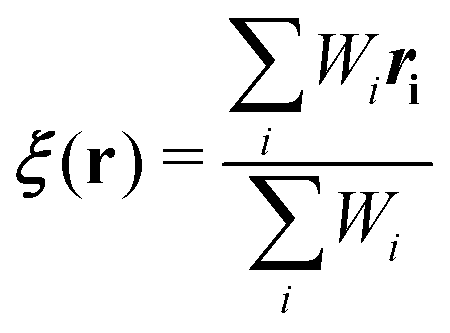
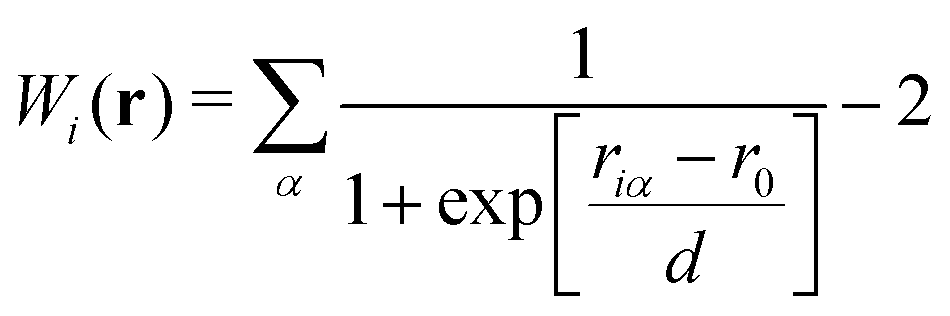
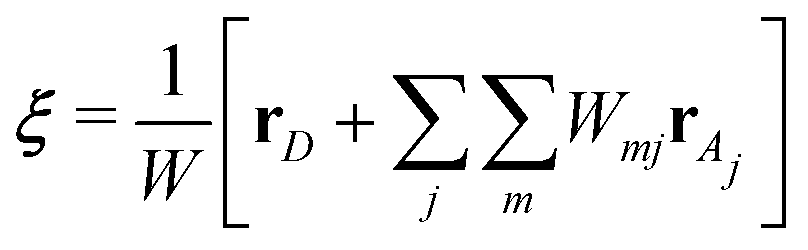
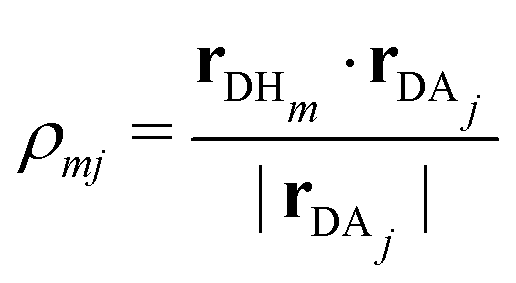
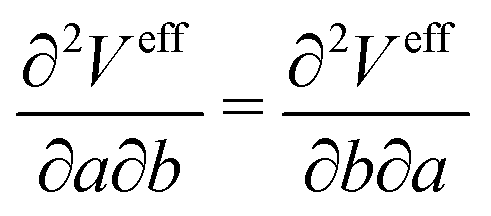
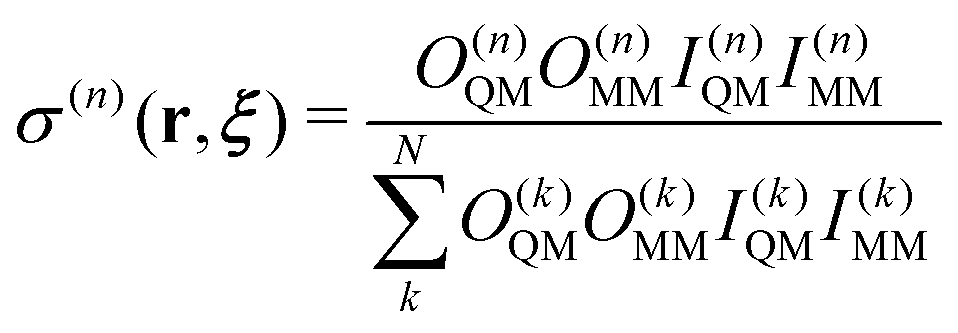
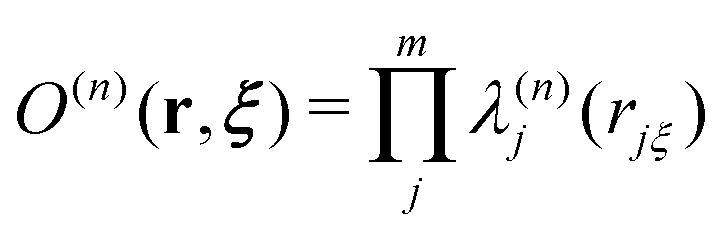
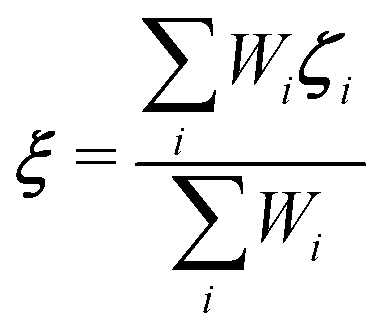
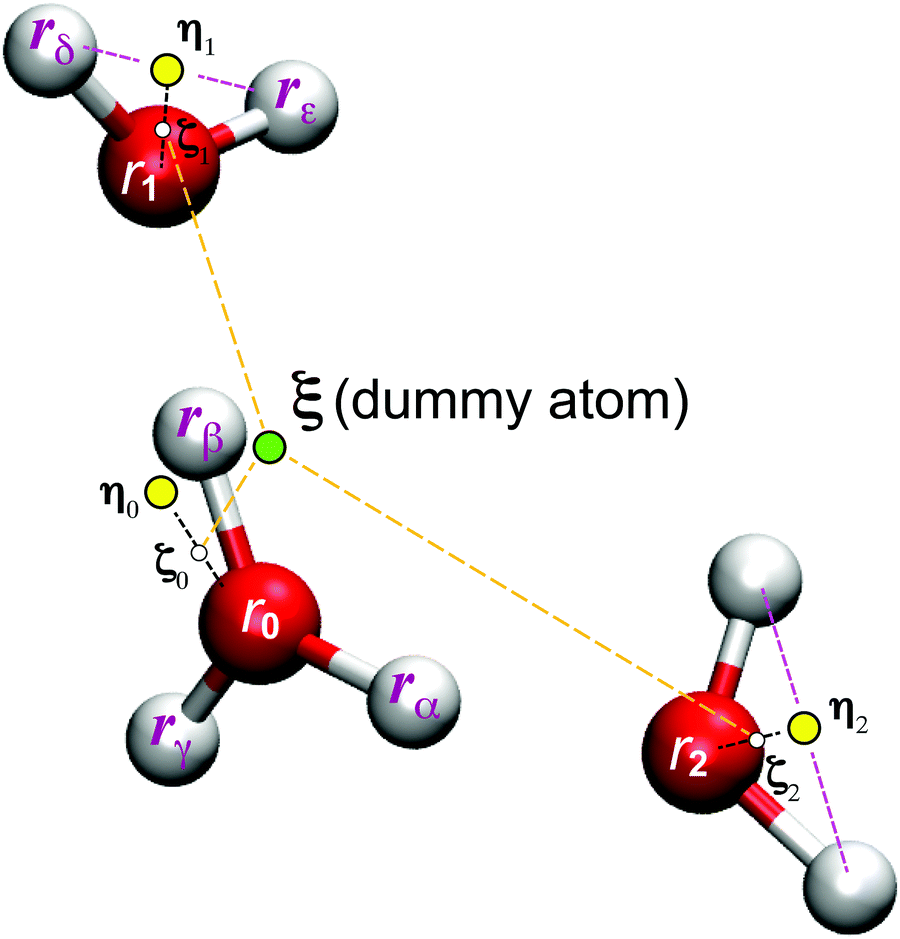
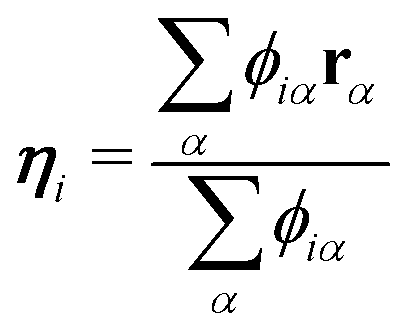
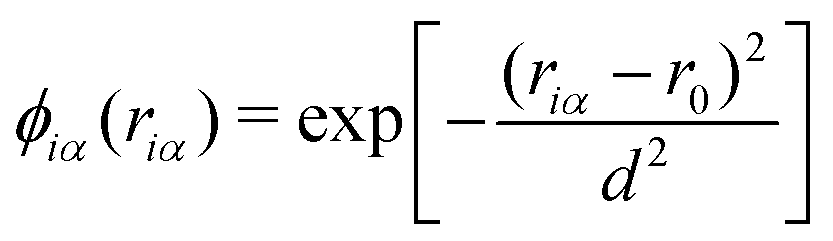
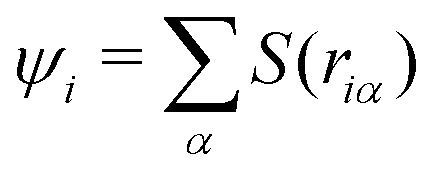
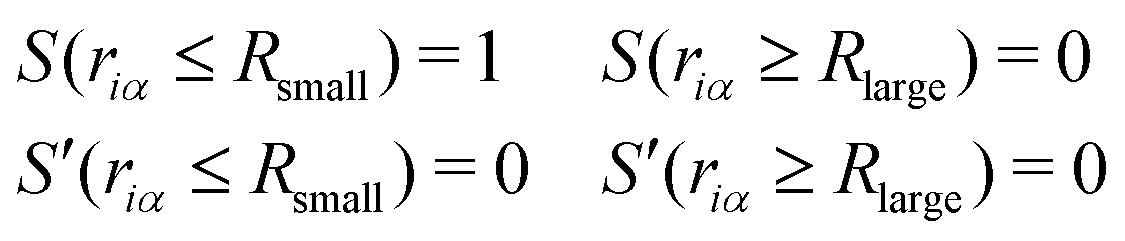
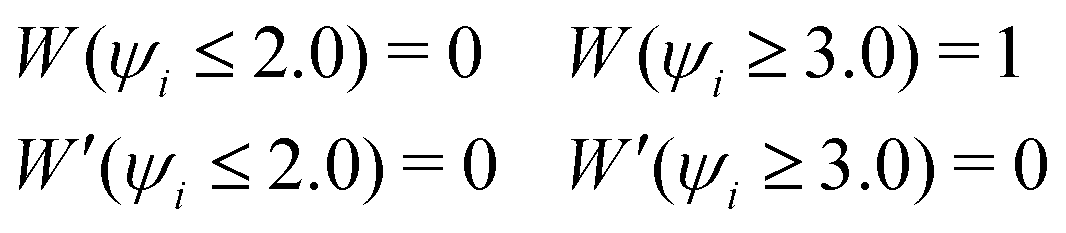
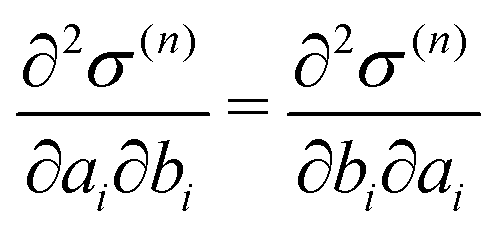
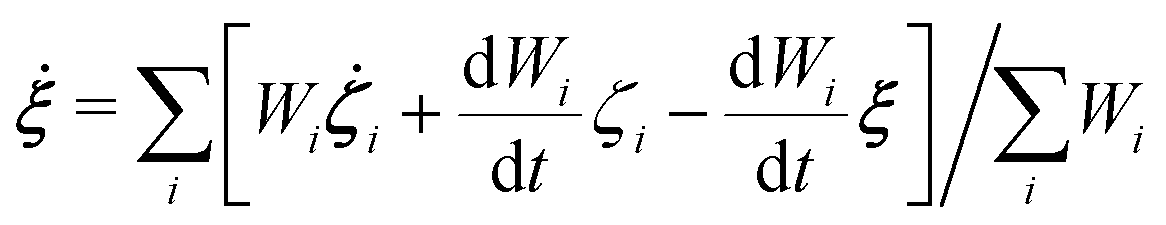
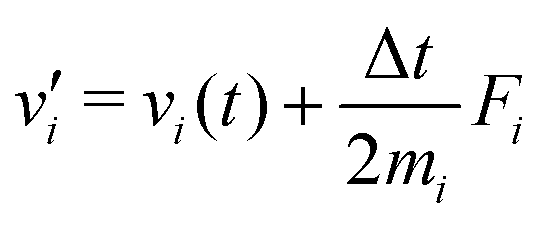
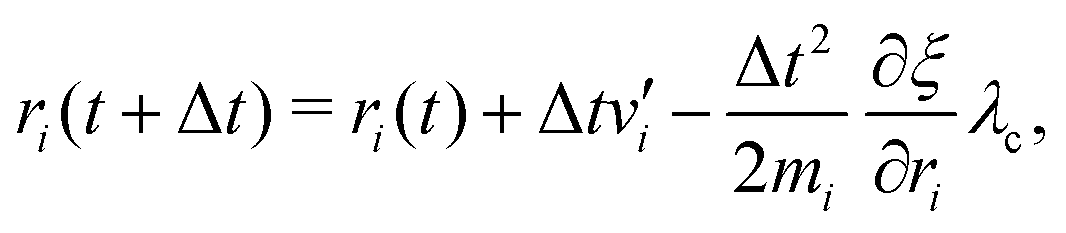
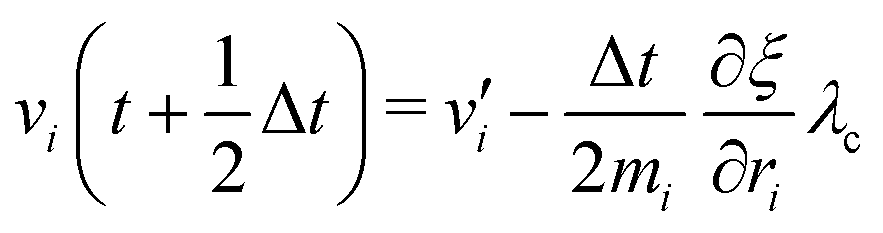
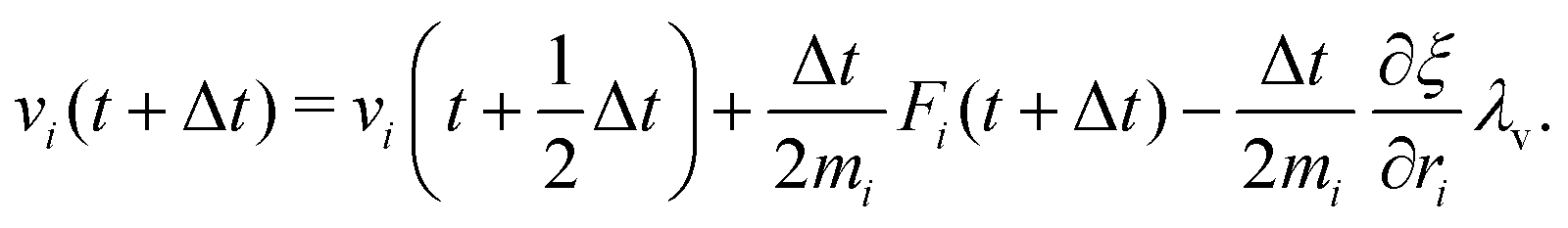 where λc and λv are Lagrange multipliers determined from iterations to satisfy eqn (13) and (21), respectively The mass of the dummy atom was set to 1 × 10−8 a.u., which is sufficiently smaller than other particles. Thus, this artificial treatment does not affect the dynamics. Indeed, we found that the use of a dummy atom smaller than the one set in the present study did not make any significant difference in the dynamics. However, a dummy atom with a larger mass may slow down the motion of the EPI and make the RATTLE iterations unstable, which can lead to the collapse of the MD simulation. In Algorithm 1, the entire workflow is illustrated by pseudocode.
where λc and λv are Lagrange multipliers determined from iterations to satisfy eqn (13) and (21), respectively The mass of the dummy atom was set to 1 × 10−8 a.u., which is sufficiently smaller than other particles. Thus, this artificial treatment does not affect the dynamics. Indeed, we found that the use of a dummy atom smaller than the one set in the present study did not make any significant difference in the dynamics. However, a dummy atom with a larger mass may slow down the motion of the EPI and make the RATTLE iterations unstable, which can lead to the collapse of the MD simulation. In Algorithm 1, the entire workflow is illustrated by pseudocode.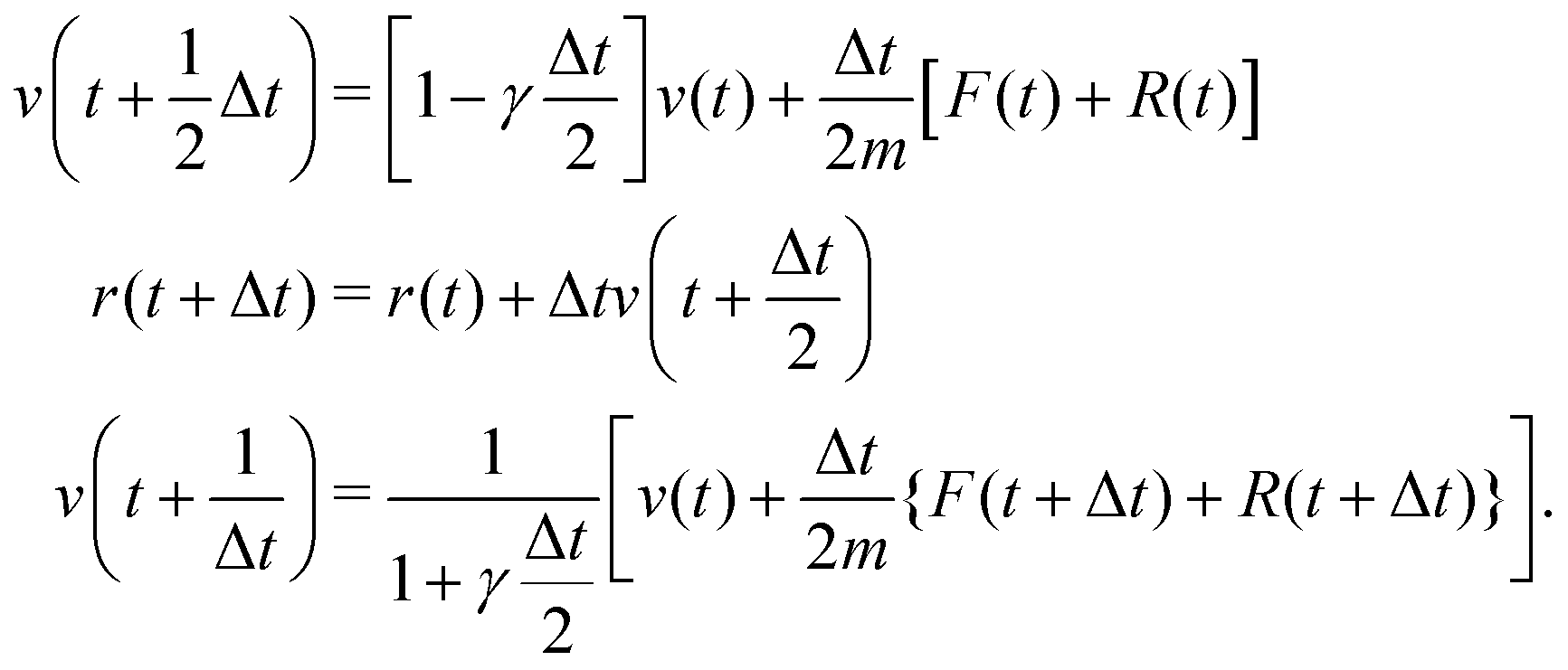
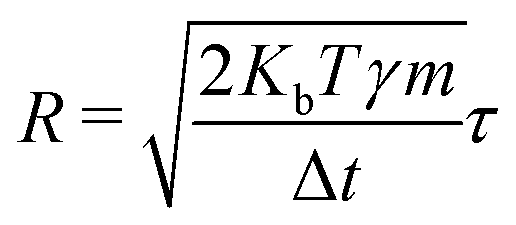
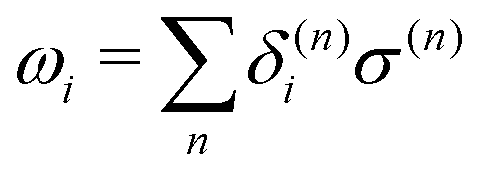
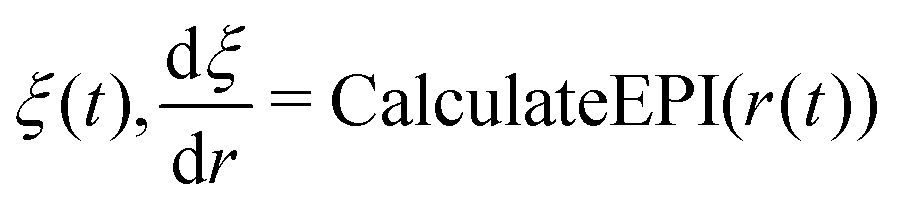

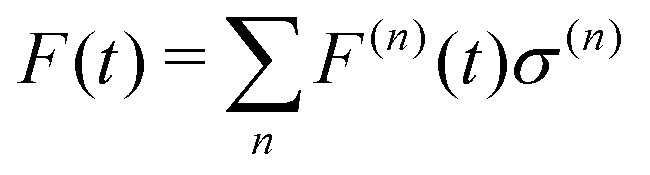
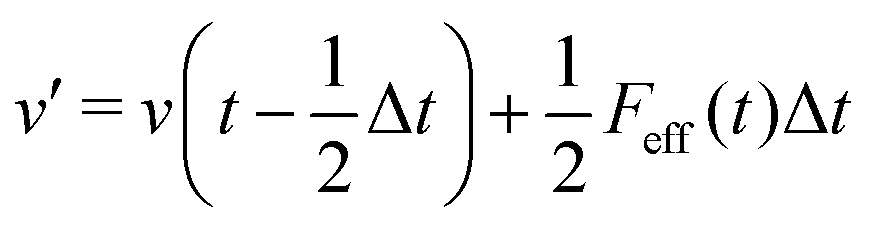
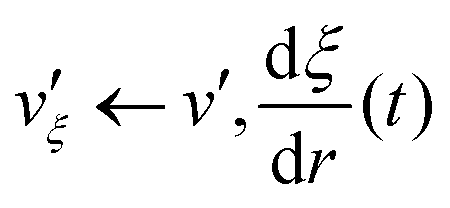
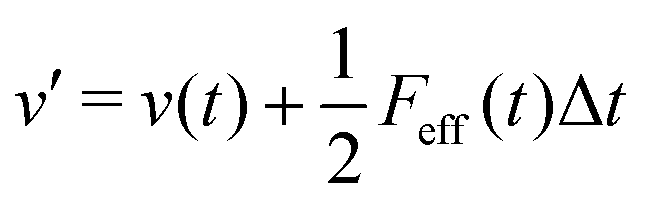
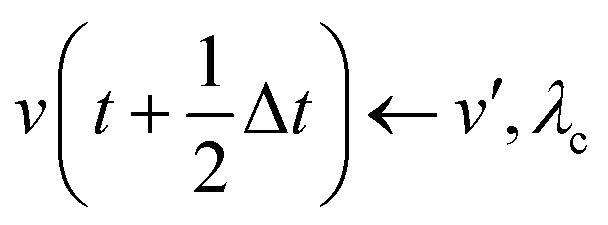
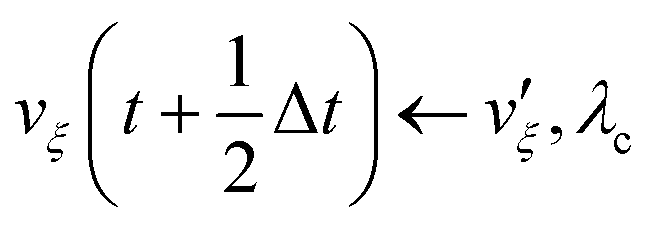
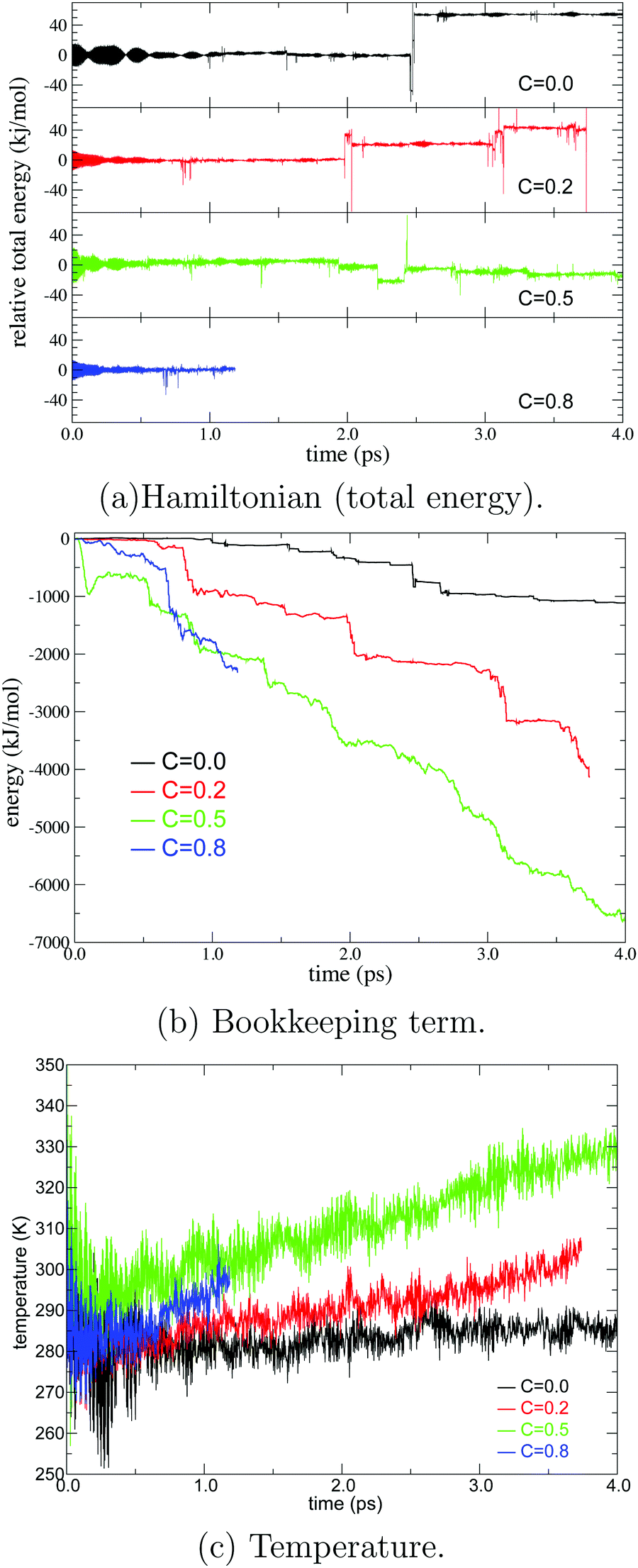
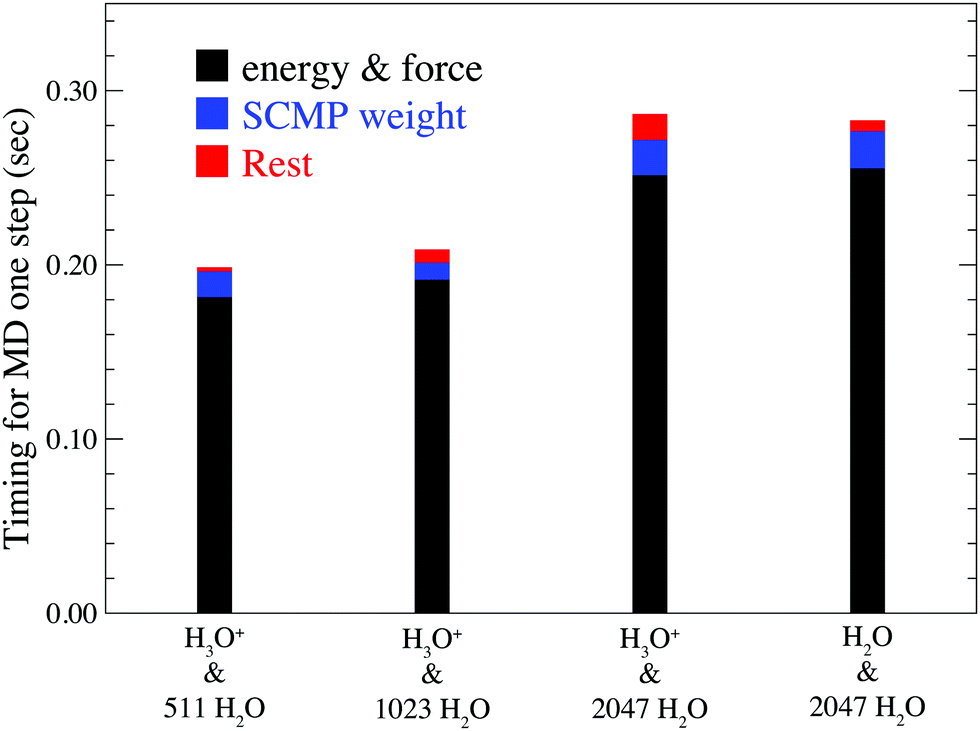
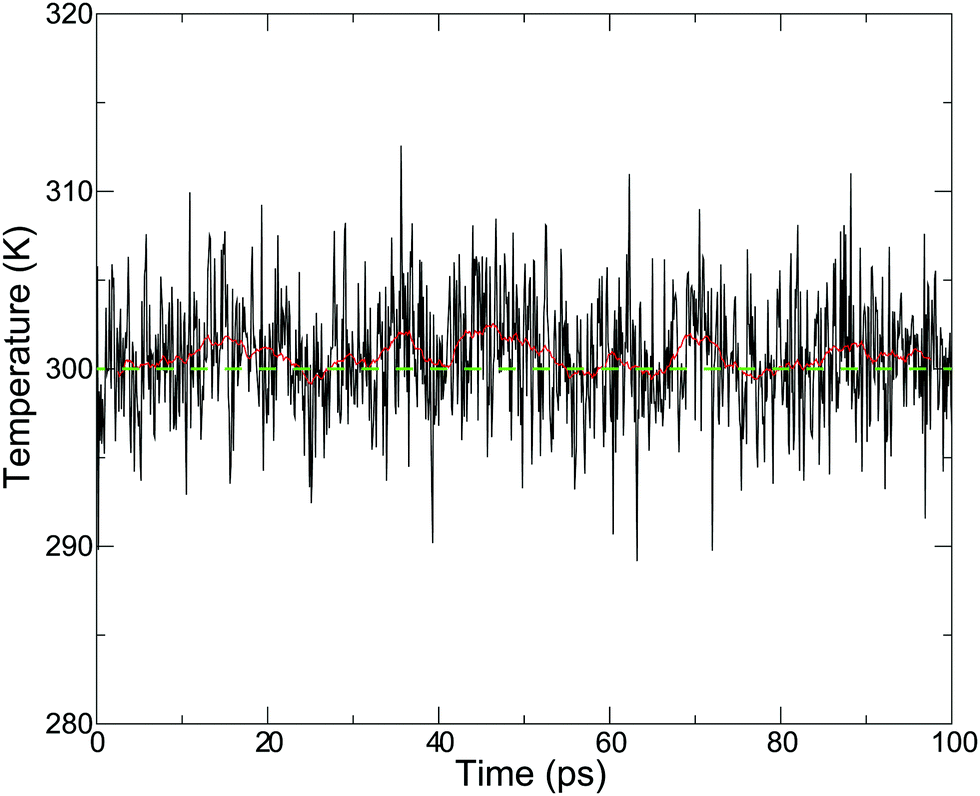
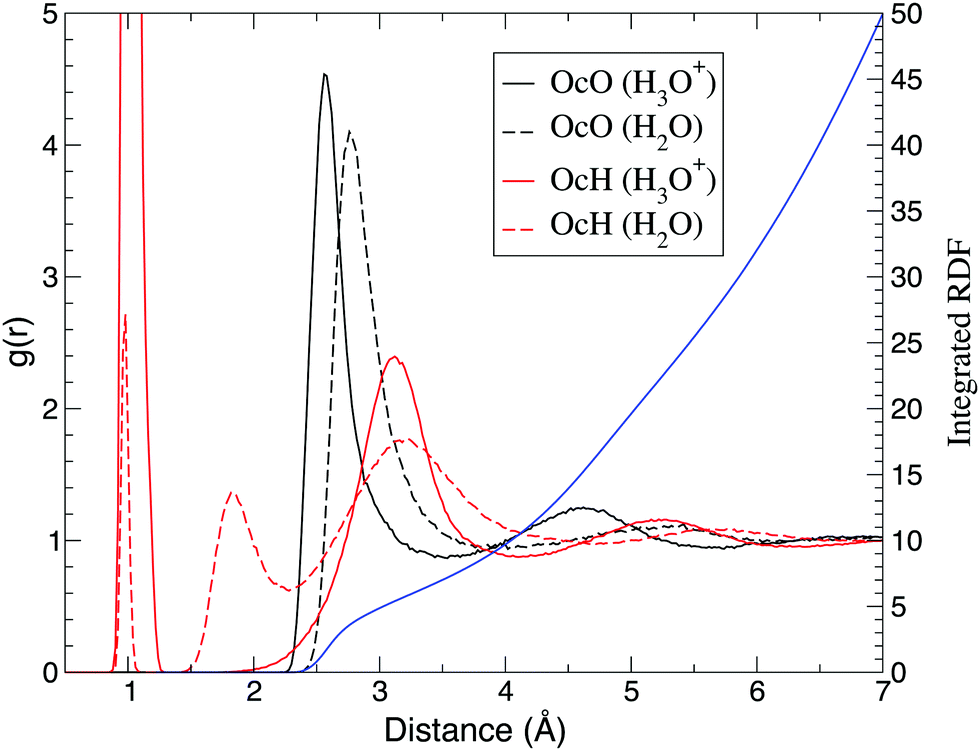
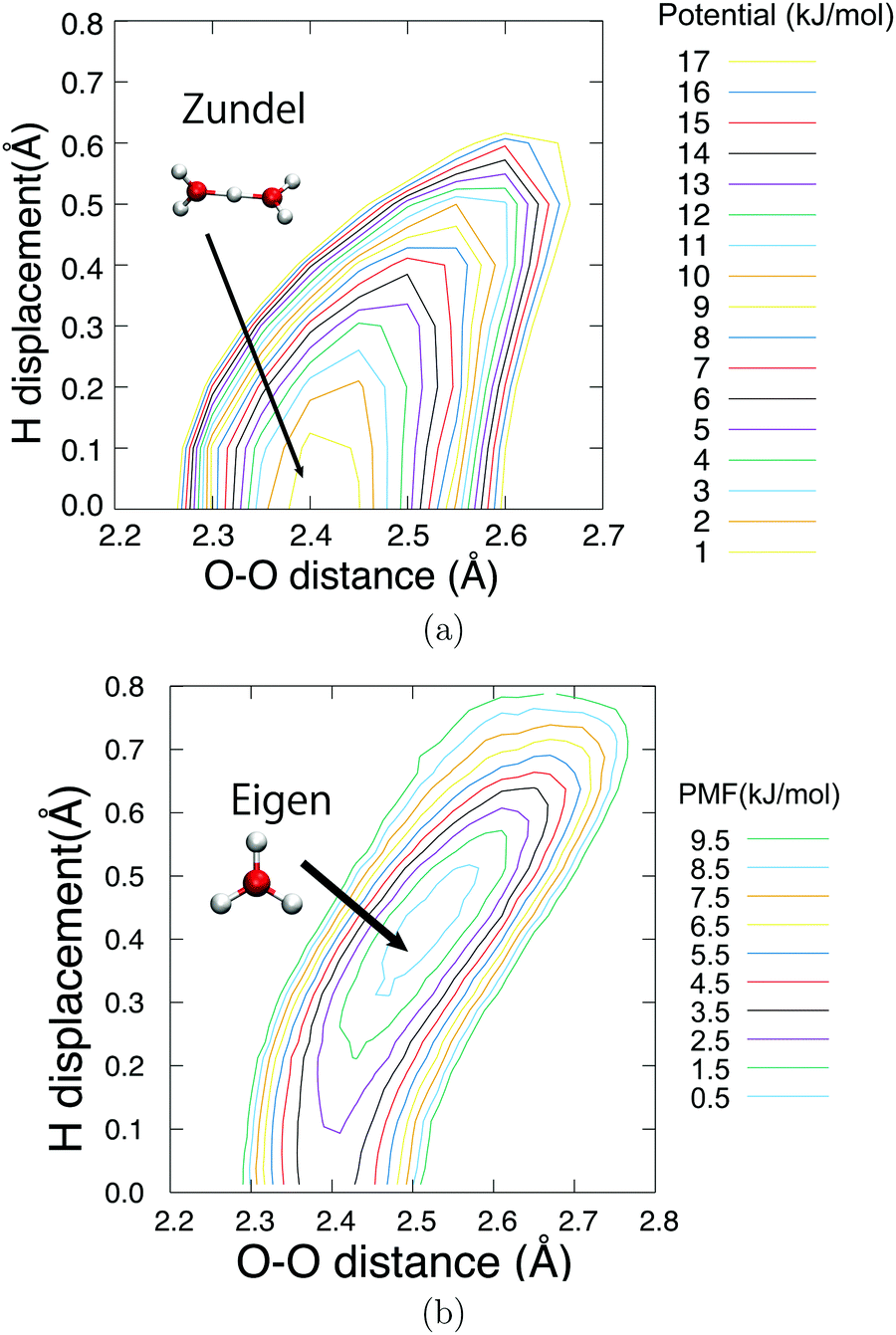
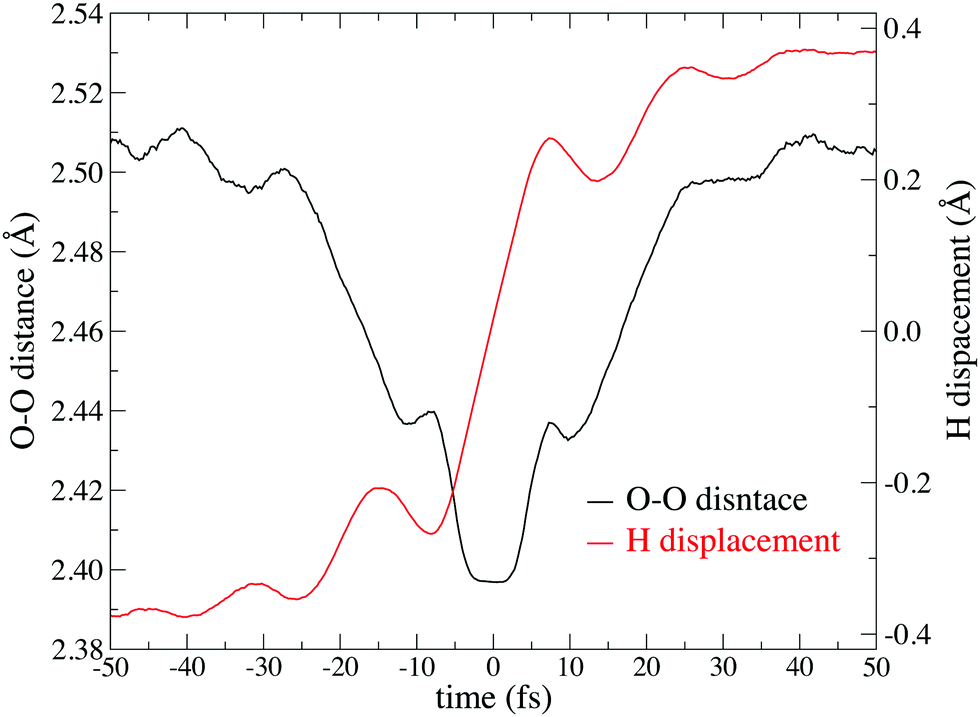
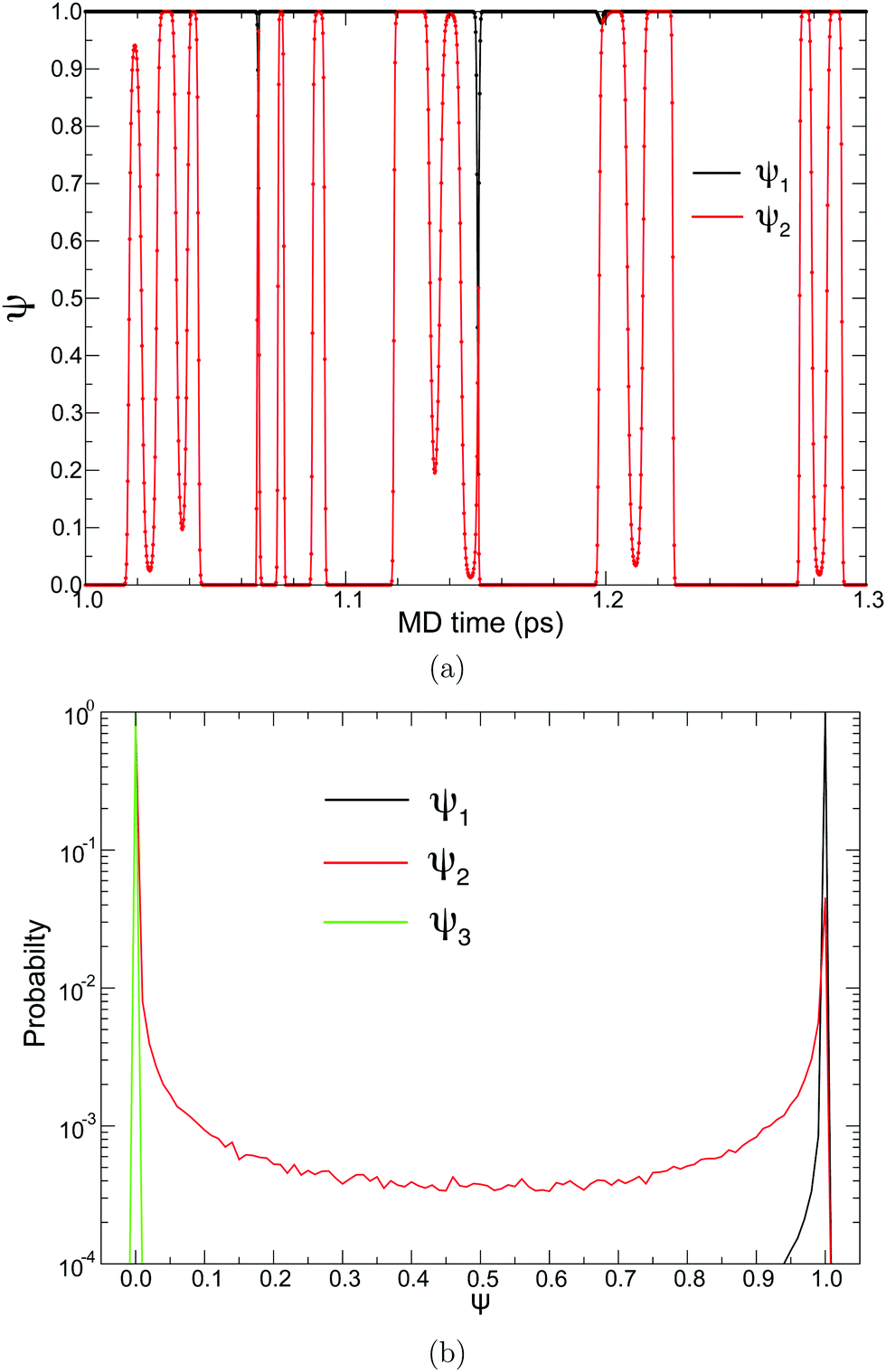
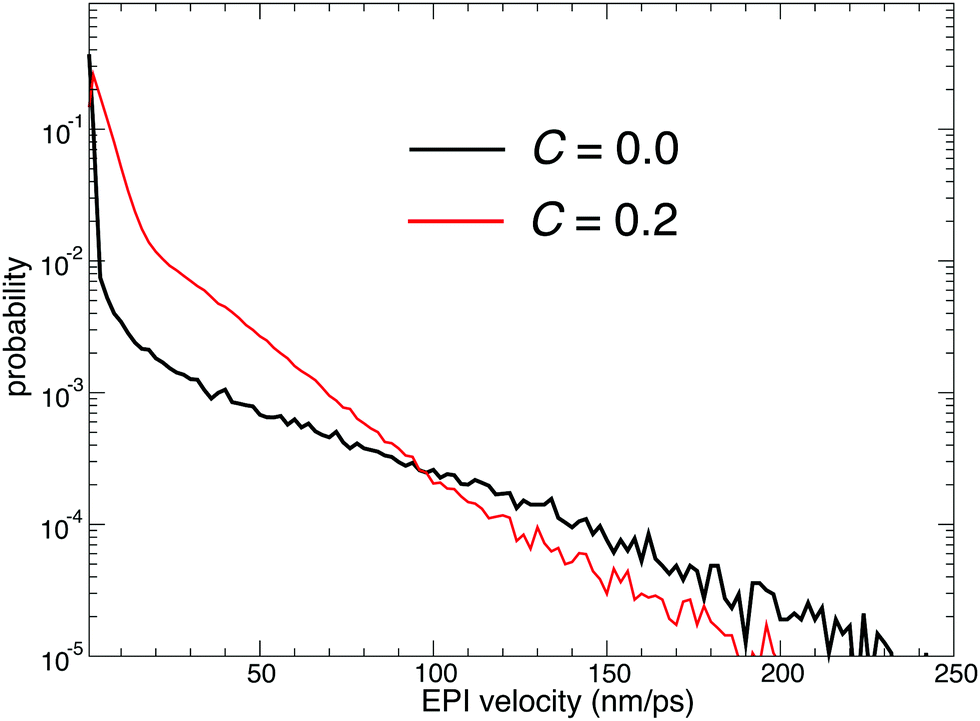
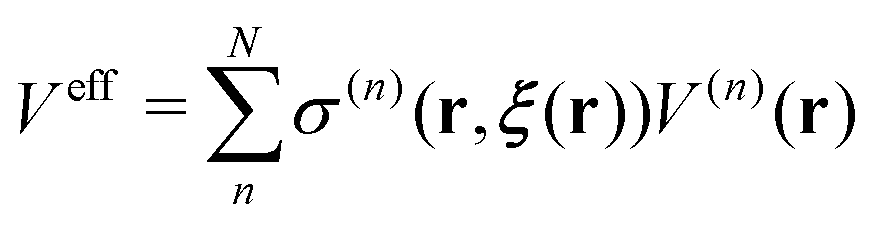
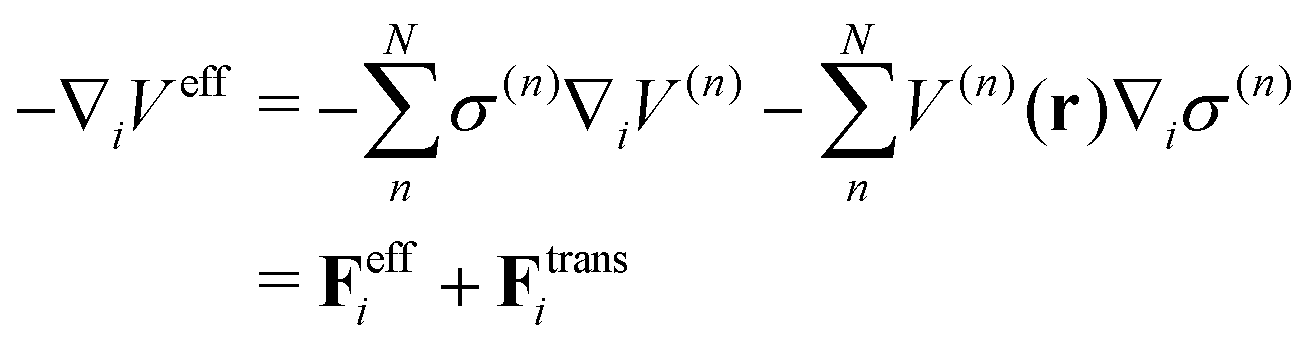
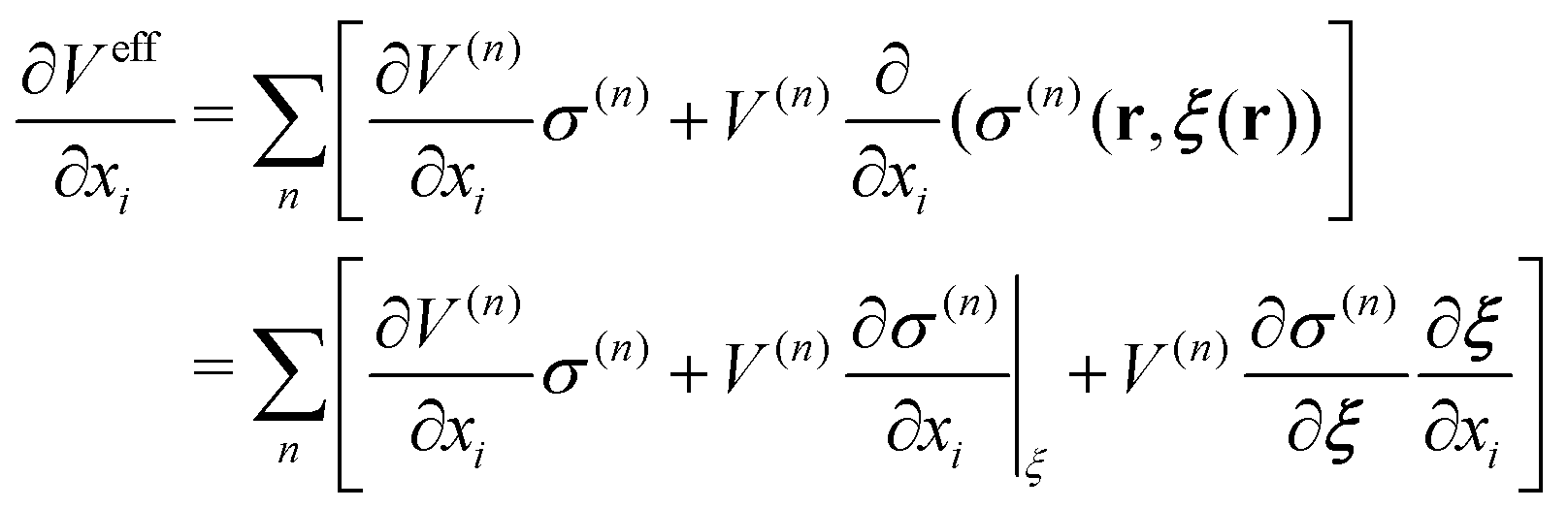
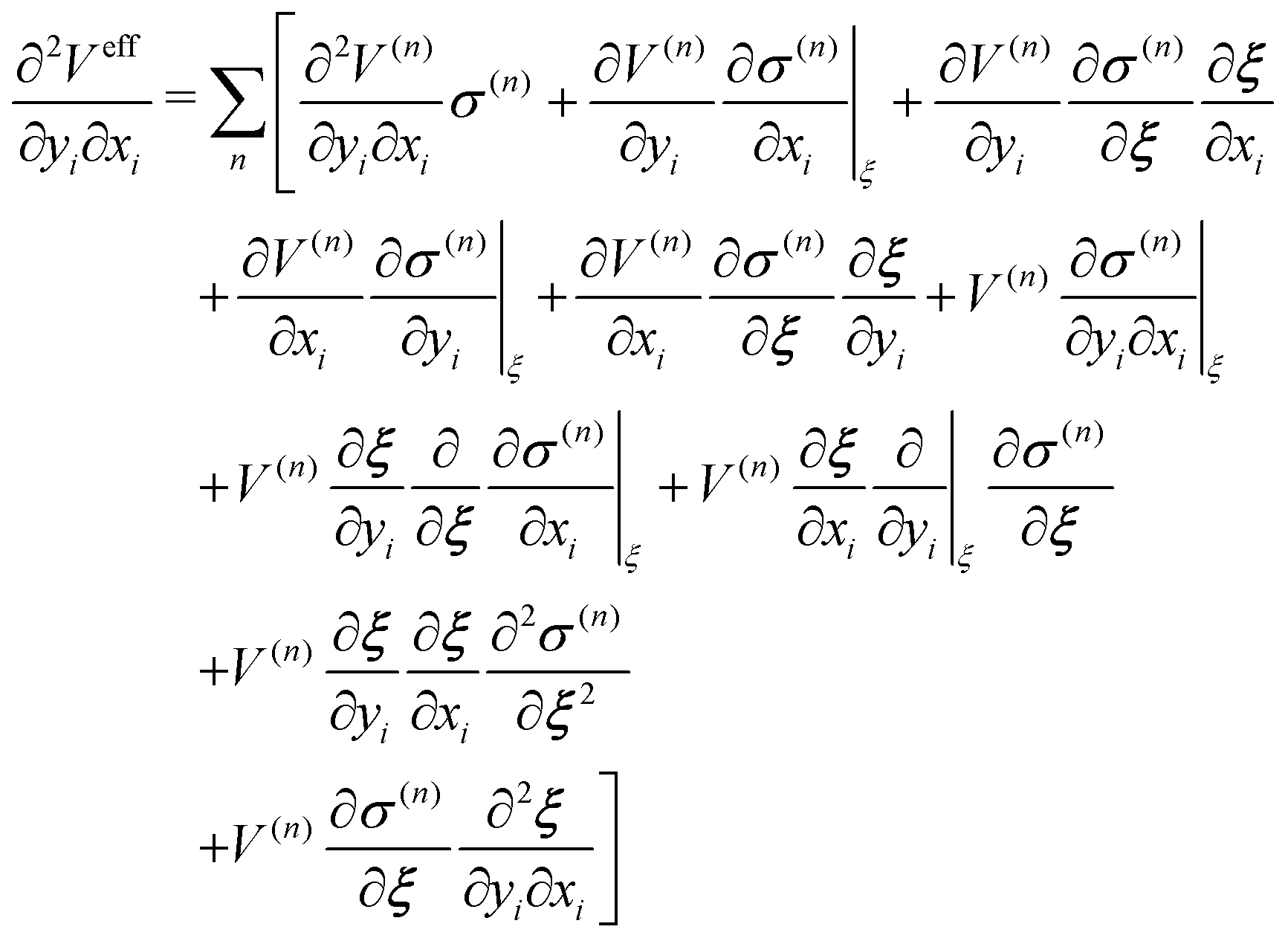
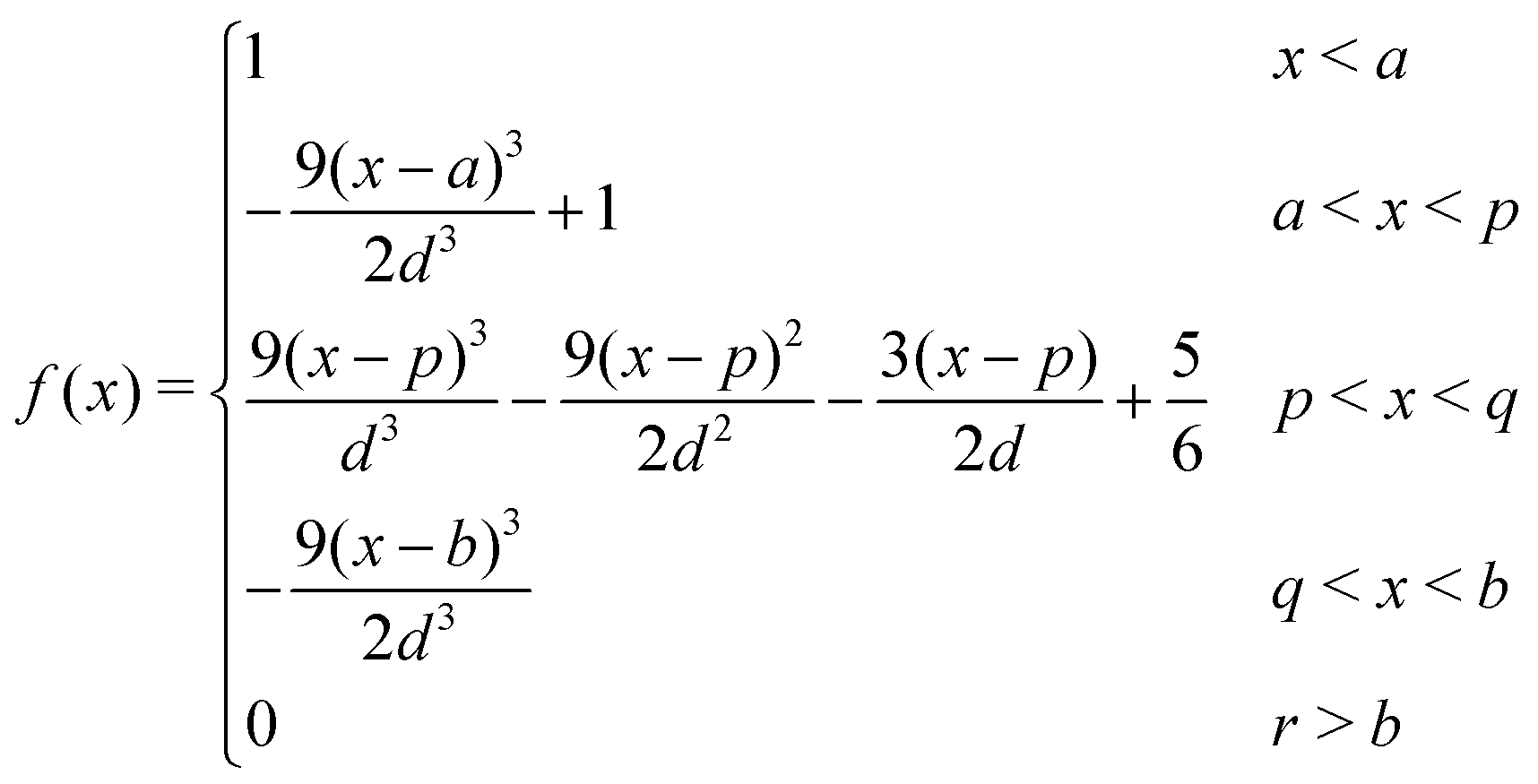
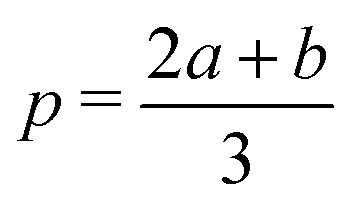 ,
, 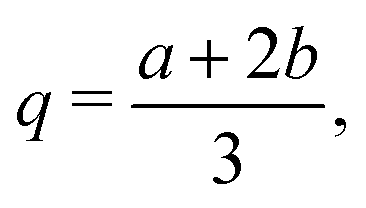 and d = b − a. For S in eqn (18), we employ a = Rsmall = 12.0 Å and b = Rlarge = 13.2 Å. For λMM in eqn (11), we employ a = sQM = 6.4 Å and b = tQM = 8.4 Å.
and d = b − a. For S in eqn (18), we employ a = Rsmall = 12.0 Å and b = Rlarge = 13.2 Å. For λMM in eqn (11), we employ a = sQM = 6.4 Å and b = tQM = 8.4 Å.