
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceBenchmark ab initio stationary-point characterization of the complex potential energy surface of the multi-channel Cl + CH3NH2 reaction
Tímea
Szűcs
 and
Gábor
Czakó
*
and
Gábor
Czakó
*
MTA-SZTE Lendület Computational Reaction Dynamics Research Group, Interdisciplinary Excellence Centre and Department of Physical Chemistry and Materials Science, Institute of Chemistry, University of Szeged, Rerrich Béla tér 1, Szeged H-6720, Hungary. E-mail: gczako@chem.u-szeged.hu
First published on 3rd April 2021
Abstract
We characterize the exothermic low/submerged-barrier hydrogen-abstraction (HCl + CH2NH2/CH3NH) as well as, for the first time, the endothermic high-barrier amino-substitution (CH3Cl + NH2), methyl-substitution (NH2Cl + CH3), and hydrogen-substitution (CH2ClNH2/CH3NHCl + H) pathways of the Cl + CH3NH2 reaction using an accurate composite ab initio approach. The computations reveal a CH3NH2⋯Cl complex in the entrance channel, nine transition states corresponding to different abstractions, Walden-inversion substitution, and configuration-retaining front-side attack substitution pathways, as well as nine post-reaction complexes. The global minima of the electronic and vibrationally adiabatic potential energy surfaces correspond to the pre-reaction CH3NH2⋯Cl and post-reaction CH2NH2⋯HCl complexes, respectively. The benchmark composite energies of the stationary points are obtained by considering basis-set effects up to the correlation-consistent polarized valence quadruple-zeta basis augmented with diffuse functions (aug-cc-pVQZ) using the explicitly-correlated coupled-cluster singles, doubles, and perturbative triples CCSD(T)-F12b method, post-(T) correlation up to CCSDT(Q) including full triples and perturbative quadruples, core correlation, and scalar relativistic and spin–orbit effects, as well as harmonic zero-point energy corrections.
I. Introduction
Reactions of atoms such as H, F, and Cl with molecules from H2via H2O, NH3, and CH4 to C2H6 have attracted significant scientific attention from the 1970s to the present day.1–18 Moving beyond these systems that have only equivalent H atoms, one may consider molecules with two different functional groups, thereby opening multiple hydrogen-abstraction reaction pathways. One possibility is to study the reactions of methanol (CH3OH), which combines the functional groups of H2O and CH4. The F/Cl + CH3OH reactions have been investigated recently by several experimental and theoretical groups.19–23 The present study aims to combine the functional groups of NH3 and CH4 and investigates the reaction pathways of the Cl + CH3NH2 multi-channel reaction. The H-abstraction channels of this reaction forming HCl and CH2NH2 or CH3NH were investigated in a joint experimental−theoretical study in 2003.24 Experimentally, the HCl rotational distributions were measured, while theoretically the stationary points of the H-abstraction channels were characterized by the G2//MP2/6-311G(d,p) level of theory.24 In 2009 a theoretical study applied W1’ theory, which utilizes UQCISD/6-31+G(d,p) geometries, HF, CCSD, and (T) extrapolations as well as core and scalar relativistic corrections, to map the reaction pathway of the HCl + CH2NH2 channel.25 In 2013 the F + CH3NH2 reaction was investigated with the CCSD(T) method using the aug-cc-pVnZ [n = D, T, Q] basis sets.26 Going back to Cl + CH3NH2, in 2015 the rate coefficients were measured and computed with a master equation model.27 This latest study used the MP2/cc-pVTZ level of theory to determine the stationary-point geometries for the H-abstraction pathways, which was followed by single-point energy computations at the CCSD(T)-F12a/aug-cc-pVTZ level.In this work we plan to provide additional insights into the mechanisms of the Cl + CH3NH2 reaction by searching for new reaction pathways besides the previously studied H-abstraction channels. One may consider substitution of the CH3 and NH2 groups by the Cl atom or one of the H atoms of either the CH3 or NH2 group. Previous work on atom/OH + CH4/C2H6 reactions16,28,29 showed that the substitution may occur via Walden-inversion and/or front-side attack pathways; thus, we aim to seek these mechanisms in the case of the title reaction as well. Furthermore, we plan to improve the accuracy of the previous studies24,25,27 on the title reaction by using the explicitly-correlated CCSD(T)-F12b method for geometry optimizations and frequency computations as well as considering basis set effects up to aug-cc-pVQZ, post-(T) electron correlation effects up to CCSDT(Q), core correlation corrections, scalar relativistic effects, and geometry-dependent spin–orbit corrections. This complete and accurate characterization of the stationary points of the title reaction may provide guidance for future global potential energy surface developments and reaction dynamics studies.
II. Computational details
To determine the important stationary points of the reaction channels we start the search with the restricted open-shell second-order Møller–Plesset perturbation theory (RMP2)30 with the correlation-consistent aug-cc-pVDZ basis set31 using initial structures based on chemical intuition and previous studies.16,24–27,29 Utilizing the minima and saddle-point geometries obtained at the RMP2/aug-cc-pVDZ level, we optimize with the restricted open-shell Hartree–Fock (ROHF)-based unrestricted explicitly-correlated coupled-cluster singles, doubles and perturbative triples (CCSD(T)-F12b) method,32 and apply two different basis sets: aug-cc-pVDZ and aug-cc-pVTZ.31 With the above-mentioned methods and basis sets, the harmonic vibrational frequencies are computed as well. Gradients and Hessians are obtained numerically usually using the default settings of Molpro.33 For optimizations this means a gradient threshold of 3 × 10−4 a.u., which is tightened to be 10−5 a.u. in a few cases where low-lying frequencies occur. It is computationally too expensive (and also unnecessary, see later) to perform geometry optimization at the CCSD(T)-F12b level with the aug-cc-pVQZ basis set; therefore we compute CCSD(T)-F12b/aug-cc-pVQZ single-point energies at the geometries obtained with the aug-cc-pVTZ basis set.To reach sub-chemical accuracy, the following energy contributions should be taken into account with single-point-energy computations, using the most accurate CCSD(T)-F12b/aug-cc-pVTZ geometries: post-CCSD(T) correlation, core-core and core-valence electron correlation (in short, core correlation), scalar relativistic effect, and spin–orbit coupling correction. For the correction of post-CCSD(T) correlation, unrestricted CCSD(T),34 CCSDT35 and CCSDT(Q)36 methods are used with unrestricted Hartree–Fock (UHF) reference and the cc-pVDZ basis set. The corrections are defined as:
| δ[CCSDT] = ΔE(CCSDT/cc-pVDZ) − ΔE(CCSD(T)/cc-pVDZ) | (1) |
| δ[CCSDT(Q)] = ΔE(CCSDT(Q)/cc-pVDZ) − ΔE(CCSDT/cc-pVDZ) | (2) |
| Δcore = ΔE(AE-CCSD(T)-F12b/cc-pCVTZ-F12) − ΔE(FC-CCSD(T)-F12b/cc-pCVTZ-F12) | (3) |
| Δrel = ΔE(DK-AE-CCSD(T)/aug-cc-pwCVTZ-DK) − ΔE(AE-CCSD(T)/aug-cc-pwCVTZ) | (4) |
| ΔSO = SO1(MRCI+Q/aug-cc-pwCVTZ) − non-SO1(MRCI+Q/aug-cc-pwCVTZ) | (5) |
| ΔEclassical = CCSD(T)-F12b/aug-cc-pVQZ + δ[CCSDT] + δ[CCSDT(Q)] + Δcore + Δrel + ΔSO | (6) |
| ΔEadiabatic = CCSD(T)-F12b/aug-cc-pVQZ + δ[CCSDT] + δ[CCSDT(Q)] + Δcore + Δrel + ΔSO + ΔZPE | (7) |
Computations of the MP2, CCSD(T)-F12b, AE, DK, MRCI, and SO results are carried out with the Molpro33 program package. For CCSD(T)-F12b the default auxiliary basis sets and accuracy thresholds are used as implemented in Molpro. The frozen-core CCSD(T), CCSDT, and CCSDT(Q) energies are obtained with MRCC44 interfaced to Molpro.
III. Results and discussion
1. Reaction pathways
Six different channels of the Cl + CH3NH2 reaction are investigated:(1) Methyl hydrogen-abstraction leading to HCl + CH2NH2 (CH3 HA)
(2) Amino hydrogen-abstraction leading to HCl + CH3NH (NH2 HA)
(3) Methyl hydrogen-substitution leading to H + CH2ClNH2 (CH3 HS)
(4) Amino hydrogen-substitution leading to H + CH3NHCl (NH2 HS)
(5) Methyl-substitution leading to NH2Cl + CH3 (MS)
(6) Amino-substitution leading to CH3Cl + NH2 (AS)Furthermore, the hydrogen-substitution in the methyl group and the amino-substitution can proceed via a Walden inversion (W) transition state (TS) or a front-side-attack (FS) TS.
The eight reaction pathways are shown in Fig. 1, with the computed benchmark classical(vibrationally adiabatic) energies relative to the reactants. Some characteristic structural parameters of the geometries displayed on this diagram are detailed at three different levels of theory in Fig. 2. In the entrance channel we have found a deep minimum stabilizing a CH3NH2⋯Cl complex with substantial De(D0) dissociation energies of 15.30(14.22) kcal mol−1. This pre-reaction minimum (PREMIN) may play an important steering role in the entrance channel and may make the reaction indirect, especially at low collision energies. The two hydrogen-abstraction reactions occur with small (CH3 HA) and negative (NH2 HA) barriers. The thermodynamically favored HA from the methyl-group is exothermic with −6.29(−10.79) kcal mol−1, while from the amino-group the reaction is slightly endothermic (with 1.36 kcal mol−1) classically and exothermic (−3.88 kcal mol−1) adiabatically.
 | ||
| Fig. 1 Energy diagram of the Cl + CH3NH2 reaction pathways showing benchmark classical(vibrationally adiabatic) relative energies, acquired from eqn (6) (eqn (7)). See Table 1. * denotes the use of MP2/aug-cc-pVDZ geometry and ** denotes MP2/cc-pVDZ relative energy. Intrinsic reaction coordinate (IRC) computations show that PREMIN is along the amino hydrogen-substitution pathway, but it is not connected, because PREMIN may play significant roles in the other pathways as well. | ||
 | ||
| Fig. 2 Structures of reactants, stationary points, and products with the most important distances (Å) and angles (degree) at the MP2/aug-cc-pVDZ (blue), CCSD(T)-F12b/aug-cc-pVDZ (green), and CCSD(T)-F12b/aug-cc-pVTZ (red) levels of theory. * denotes distances at MP2/cc-pVDZ. PREMIN is shown at amino hydrogen-substitution based on IRC computations, but PREMIN may also play significant roles in the other pathways. | ||
In the case of methyl hydrogen-abstraction, two transition-state geometries (CH3 HA TS′ and CH3 HA TS′′) are found. We report both, because we experience severe electronic structure problems in this region and the computational procedure described in Section II could not be executed for these stationary points. To guide future work, we provide some details about these ab initio issues. The represented structures are obtained with the MP2 method using the cc-pVDZ and aug-cc-pVDZ basis sets for CH3 HA TS′ (TS′) and CH3 HA TS′′ (TS′′), respectively. The geometry optimizations at CCSD(T)-F12b with the aug-cc-pVDZ and aug-cc-pVTZ basis sets are not converged due to Hartree–Fock (TS′) and CCSD (TS′′) convergence issues. The elongation of the breaking C–H bond is significant (0.154 Å) in the TS′′ geometry, whereas modest in TS′ (0.028 Å). The single-point energy computations can be performed only in the former case (TS′′), because Hartree–Fock does not converge for the latter (TS′). The reason why we do not report only TS′′ is that visualization of the normal mode corresponding to the imaginary frequency shows that this mode does not exactly belong to the C–H bond stretching. Furthermore, Nicovich et al.27 reported a geometry of this transition state obtained at the MP2/cc-pVTZ level of theory, where this C–H bond is 1.134 Å long, like in TS′. Moreover, TS′ provides a lower barrier for HA than TS′′ as seen in Fig. 1. The problems with this CH3 HA TS were also reported by Taylor et al., who found that a TS′-like structure can be obtained at the CCSD(T)/6-31G(d) level, whereas the same method with larger 6-31+G(d,p) and 6-311+G(2df,p) basis sets fails to locate a TS′-like saddle point.25
After the hydrogen-abstractions, the amino-substitution (AS) via the Walden TS has the lowest barrier, 31.74(29.72) kcal mol−1; moreover this channel is only slightly endothermic (ΔE(ΔH0) = 4.76(0.25) kcal mol−1). In spite of this, the AS front-side attack TS (AS TS FS) has the largest barrier height, 52.00(49.55) kcal mol−1, which is higher by 20.26(19.83) kcal mol−1 than that of the corresponding Walden inversion TS (AS TS W). The relative energies of the post-reaction minima are very similar, for example 2.93(−0.97) kcal mol−1 for AS MIN W and 2.43(−1.34) kcal mol−1 for AS MIN FS, in accordance with the similar C–Cl (1.786 Å) and comparably large C–N (3.260/3.254 Å) distances. The barrier of the FS transition state is higher than that of the Walden inversion TS at the methyl hydrogen-substitution (CH3 HS TS) as well. The difference is 6.22(6.08) kcal mol−1 and the relative energy of the CH2ClNH2 + H products is 15.47(10.53) kcal mol−1. The endothermicity of the MS channel, ΔE(ΔH0) = 27.47(22.45) kcal mol−1, is higher than that of the amino-substitution, with 22.71(22.20) kcal mol−1. The most endothermic pathway is the hydrogen-substitution from the amino-group, with ΔE(ΔH0) = 45.56(39.87) kcal mol−1, whose TS (NH2 HS TS) is just slightly above the product level, as seen in Fig. 1.
In the case of the substitution pathways the relative-energy differences between the products and product-like minimum (MIN) complexes are only 0.28−2.32(0.01−1.58) kcal mol−1. In the case of CH3 HA MIN′, CH3 HA MIN′′, and NH2 HA MIN, the product complexes are more stable with De(D0) dissociation energies of 6.09(4.39), 6.57(4.77), and 8.00(6.07) kcal mol−1, respectively. This finding is expected, because the HA MINs are stabilized by dipole–dipole interactions, whereas only dipole-atom and dipole–quadrupole forces occur in the HS and MS complexes, respectively. As seen above, for CH3 HA we have found two minima in the exit channel. CH3 HA MIN′ is along the CH3 HA pathway where the departing HCl fragment hydrogen bonds to the C atom, whereas at the slightly deeper CH3 HA MIN′′ the HCl unit connects to the amino group as shown in Fig. 1 and 2. Adiabatically the CH3 HA MIN′′ complex is the global minimum of the system, whereas classically PREMIN is deeper by 2.44 kcal mol−1.
One may consider an additional product channel of the Cl + CH3NH2 reaction leading to NH3 + CH2Cl. This thermodynamically favorable channel, with ΔE(ΔH0) = −3.70(−8.20) kcal mol−1, must be kinetically hidden, because, unlike the above-discussed six channels, NH3 + CH2Cl requires breaking and forming two bonds instead of one. In the exit channel we have found a CH2Cl⋯NH3 complex with Cs symmetry, where CH2Cl connects to NH3 with a single CH⋯N hydrogen bond. The De(D0) values of the CH2Cl⋯NH3 complex are 3.05(1.91) kcal mol−1 obtained at the CCSD(T)-F12b/aug-cc-pVTZ level of theory and the corresponding classical(vibrationally adiabatic) energies relative to the reactants are −7.40(−10.76) kcal mol−1; thus, PREMIN(CH3 HA MIN′′) remains the global minimum of the system.
Besides the NH3 + CH2Cl channel, the kinetically also hindered NHCl + CH4 formation may be considered, but it turns out to be endothermic with ΔE(ΔH0) = 14.24(9.82) kcal mol−1 (Table 1); thus, this channel is not competitive for the lowest-energy configuration of the title reaction.
| Stationary points | MP2 | CCSD(T)-F12b | δ[T]e | δ[(Q)]f | Δ core | Δ rel | Δ SO | Classicalj | Δ ZPE | Adiabaticl | ||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| aVDZa | aVDZb | aVTZc | aVQZd | |||||||||
| a MP2/aug-cc-pVDZ relative energies obtained at MP2/aug-cc-pVDZ geometries. b CCSD(T)-F12b/aug-cc-pVDZ relative energies obtained at CCSD(T)-F12b/aug-cc-pVDZ geometries. c CCSD(T)-F12b/aug-cc-pVTZ relative energies obtained at CCSD(T)-F12b/aug-cc-pVTZ geometries. d CCSD(T)-F12b/aug-cc-pVQZ relative energies obtained at CCSD(T)-F12b/aug-cc-pVTZ geometries. e CCSDT–CCSD(T) obtained at CCSD(T)-F12b/aug-cc-pVTZ geometries with cc-pVDZ basis set. f CCSDT(Q)–CCSDT obtained at CCSD(T)-F12b/aug-cc-pVTZ geometries with cc-pVDZ basis set. g Core correlation corrections obtained as the differences between all-electron and frozen-core CCSD(T)-F12b/cc-pCVTZ-F12 relative energies at CCSD(T)-F12b/aug-cc-pVTZ geometries. h Scalar relativistic effects obtained as the differences between DK-AE-CCSD(T)/aug-cc-pwCVTZ-DK and AE-CCSD(T)/aug-cc-pwCVTZ relative energies at CCSD(T)-F12b/aug-cc-pVTZ geometries. i Spin–orbit (SO) corrections obtained as the differences between the SO and non-SO ground-state MRCI+Q/aug-cc-pwCVTZ relative energies at CCSD(T)-F12b/aug-cc-pVTZ geometries. j Benchmark classical relative energies obtained as CCSD(T)-F12b/aug-cc-pVQZ relative energies + δ[T] (e) + δ[(Q)] (f) + Δcore (g) + Δrel (h) + ΔSO (i). k Zero-point energy (ZPE) corrections obtained at CCSD(T)-F12b/aug-cc-pVTZ. l Benchmark vibrationally adiabatic relative energies obtained as classical relative energies (j) + ΔZPE (k). m CH3 HA TS′′ relative energies and corrections obtained at MP2/aug-cc-pVDZ geometry. n Obtained with UMP2. | ||||||||||||
| PREMIN | −16.35 | −16.39 | −15.76 | −15.92 | −0.02 | −0.13 | −0.11 | +0.06 | +0.81 | −15.30 | +1.08 | −14.22 |
| CH3 HA TS′′m | 2.33 | 3.49 | 3.99 | 3.92 | −0.13 | −0.14 | +0.00 | +0.07 | +0.82 | 4.55 | −0.65 | 3.90 |
| NH2 HA TS | −0.31n | −4.14 | −3.43 | −3.55 | −0.28 | −0.15 | −0.04 | +0.14 | +0.83 | −3.05 | −4.45 | −7.50 |
| AS TS W | 41.25 | 31.72 | 31.50 | 31.51 | −0.66 | −0.37 | +0.47 | −0.02 | +0.81 | 31.74 | −2.02 | 29.72 |
| AS TS FS | 60.57 | 52.12 | 52.42 | 52.44 | −1.22 | −0.48 | +0.49 | −0.05 | +0.81 | 52.00 | −2.44 | 49.55 |
| MS TS | 40.00 | 33.91 | 34.42 | 34.41 | −0.59 | −0.38 | +0.39 | −0.02 | +0.83 | 34.64 | −2.94 | 31.70 |
| CH3 HS TS W | 38.59 | 35.96 | 36.36 | 36.22 | −0.35 | −0.34 | +0.09 | +0.11 | +0.81 | 36.55 | −4.15 | 32.40 |
| CH3 HS TS FS | 45.30 | 42.70 | 43.13 | 42.85 | −0.56 | −0.40 | −0.03 | +0.09 | +0.83 | 42.77 | −4.29 | 38.48 |
| NH2 HS TS | 50.64 | 46.33 | 47.48 | 47.51 | −0.15 | −0.37 | +0.20 | +0.05 | +0.83 | 48.06 | −4.60 | 43.46 |
| CH3 HA MIN′ | −11.73 | −13.91 | −12.97 | −13.06 | −0.07 | −0.11 | −0.22 | +0.25 | +0.83 | −12.38 | −2.80 | −15.18 |
| CH3 HA MIN′′ | −12.53 | −14.50 | −13.52 | −13.60 | −0.07 | −0.10 | −0.20 | +0.27 | +0.84 | −12.86 | −2.70 | −15.56 |
| NH2 HA MIN | −4.88 | −8.58 | −7.45 | −7.50 | −0.15 | −0.02 | +0.00 | +0.19 | +0.84 | −6.64 | −3.31 | −9.94 |
| AS MIN W | 5.65 | 1.19 | 1.90 | 1.87 | −0.10 | −0.01 | +0.21 | +0.13 | +0.84 | 2.93 | −3.90 | −0.97 |
| AS MIN FS | 5.30 | 0.53 | 1.40 | 1.39 | −0.10 | −0.02 | +0.20 | +0.13 | +0.84 | 2.43 | −3.77 | −1.34 |
| MS MIN | 29.05 | 23.60 | 24.44 | 24.41 | −0.09 | −0.14 | +0.17 | +0.11 | +0.84 | 25.30 | −4.00 | 21.29 |
| CH3 HS MIN W | 11.61 | 13.13 | 14.41 | 14.30 | +0.06 | −0.18 | −0.02 | +0.18 | +0.84 | 15.19 | −4.69 | 10.50 |
| CH3 HS MIN FS | 11.51 | 12.91 | 14.26 | 14.15 | +0.06 | −0.18 | −0.02 | +0.18 | +0.84 | 15.03 | −4.51 | 10.52 |
| NH2 HS MIN | 43.83 | 42.73 | 44.23 | 44.24 | +0.07 | −0.25 | +0.10 | +0.12 | +0.84 | 45.11 | −4.99 | 40.12 |
| CH3NHCl + H | 44.14 | 43.30 | 44.69 | 44.68 | +0.07 | −0.25 | +0.11 | +0.12 | +0.84 | 45.56 | −5.70 | 39.87 |
| NH2Cl + CH3 | 31.60 | 25.94 | 26.65 | 26.55 | −0.07 | −0.13 | +0.17 | +0.11 | +0.84 | 27.47 | −5.02 | 22.45 |
| CH2ClNH2 + H | 11.82 | 13.45 | 14.71 | 14.58 | +0.06 | −0.17 | −0.02 | +0.18 | +0.84 | 15.47 | −4.94 | 10.53 |
| CH3Cl + NH2 | 7.89 | 3.08 | 3.76 | 3.69 | −0.09 | −0.01 | +0.21 | +0.12 | +0.84 | 4.76 | −4.51 | 0.25 |
| HCl + CH3NH | 4.16 | −0.39 | 0.40 | 0.38 | −0.13 | +0.01 | +0.11 | +0.15 | +0.84 | 1.36 | −5.23 | −3.88 |
| HCl + CH2NH2 | −4.42 | −7.64 | −6.99 | −7.09 | −0.07 | −0.06 | −0.15 | +0.25 | +0.84 | −6.29 | −4.50 | −10.79 |
| CH4 + NHCl | 22.56 | 12.59 | 13.57 | 13.53 | −0.32 | −0.09 | +0.26 | +0.02 | +0.83 | 14.24 | −4.42 | 9.82 |
| NH3 + CH2Cl | −0.05 | −5.18 | −4.35 | −4.53 | −0.14 | −0.06 | −0.02 | +0.22 | +0.84 | −3.70 | −4.50 | −8.20 |
 | ||
| Fig. 3 Relative energy and zero-point energy comparisons of stationary points obtained with C1 and Cs point-group symmetry at the MP2/aug-cc-pVDZ and CCSD(T)-F12b/aug-cc-pVDZ levels of theory. | ||
The geometry optimizations are performed at MP2/aug-cc-pVDZ, CCSD(T)-F12b/aug-cc-pVDZ and CCSD(T)-F12b/aug-cc-pVTZ levels of theory. The most important structural parameters of each pathway are collected (Fig. 2). In most cases, the difference between the MP2 and coupled-cluster results occurs in the hundredths of angstrom, while between the aug-cc-pVDZ and aug-cc-pVTZ CCSD(T)-F12b distances the deviations appear in the thousandths of angstrom, showing the excellent basis set convergence of the explicit-correlated CCSD(T)-F12b method.
The benchmark relative energies are obtained using the most accurate CCSD(T)-F12b/aug-cc-pVTZ geometries. To check the geometry effect on the relative energies, we have performed optimizations for the reactant and products at the CCSD(T)-F12b/aug-cc-pVQZ level of theory. We have found that the CCSD(T)-F12b/aug-cc-pVQZ relative energies at the CCSD(T)-F12b/aug-cc-pVQZ and CCSD(T)-F12b/aug-cc-pVTZ geometries agree well within 0.001 kcal mol−1, showing that the geometry effects on the energies are clearly negligible, around the numerical noise level.
The convergence of the relative energies and corresponding auxiliary corrections are shown in Table 1 and presented graphically in Fig. 4 and 5. The CCSD(T)-F12b method is needed to reach the sub-chemical accuracy, because the MP2/aug-cc-pVDZ energies differ from the corresponding CCSD(T)-F12b data by 2.70 kcal mol−1 on average and the maximum deviation is as large as almost 10 kcal mol−1 (AS TS W). The basis convergence of the explicitly-correlated CCSD(T)-F12b method is outstanding, as seen in Fig. 4, which is also confirmed by the fact that the root-mean-square deviation (RMSD) is 0.84 kcal mol−1 for DZ vs. QZ, which decreases to 0.10 kcal mol−1 for the TZ-QZ differences. Furthermore, our previous study showed that the CCSD(T)-F12b method with a QZ basis provides relative energies approaching the standard CCSD(T) QZ/5Z-extrapolation-based complete-basis-set limits within 0.1 kcal mol−1.45 However, the most accurate relative energies, reported in this work, are obtained by considering the energy contributions mentioned below.
 | ||
| Fig. 4 Convergence of the relative energies of the stationary points and products, obtained with MP2/aug-cc-pVDZ and CCSD(T)-F12b with the aug-cc-pVDZ, aug-cc-pVTZ, and aug-cc-pVQZ basis sets. | ||
 | ||
| Fig. 5 Auxiliary energy contributions (eqn (1)–(5)) and zero-point energy corrections for the stationary points and products. | ||
1.1.1. Energy contributions. Based on the values shown in Table 1 and the graphical representation of the corrections (Fig. 5), it is obvious that the harmonic ZPE correction is the most significant. The effects on the relative energies are negative (except for PREMIN) and the mean of the absolute magnitude is 3.89 kcal mol−1 (not considering the CH3 HA TS′′, since the ZPE was computed at MP2 level of theory in this case). The products have always larger zero-point corrections than the corresponding TSs and post-reaction complexes have (the RMSD is 1.37 kcal mol−1).
The post-CCSD(T) correlation corrections are essential to reach “exact” energies, mainly for the transition states, because these structures differ the most from the equilibrium, and hence these geometries have the greatest relevance of the electron correlation. The fact that sub-chemical accuracy cannot be achieved without these corrections is also shown by the finding that in most cases the sum of δ[CCSDT] and δ[CCSDT(Q)] in absolute value is around 1 kcal mol−1 and, for example, nearly 2 kcal mol−1 in the case of AS TS FS.
The core correlation corrections (Δcore) and the scalar relativistic effects (Δrel) are small and usually positive corrections around 0.1–0.2 kcal mol−1 (see Table 1). The largest Δcore values are found for the AS W/FS and MS TSs, i.e., +0.47/0.49 and +0.39 kcal mol−1, respectively, whereas the corresponding Δrel values are small, –(0.02–0.05) kcal mol−1. The largest Δrel effect of +0.27 kcal mol−1 is obtained for CH3 HA MIN′′. In general, the absolute Δcore contributions are larger for saddle points than minima, whereas an opposite trend is seen for Δrel.
It is needed to reckon the increase of the relative energies due to the relativistic spin–orbit interaction in the Cl atom. The 2P non-relativistic ground state (non-SO) of the Cl atom splits into two energy levels, 2P1/2 and 2P3/2. The former level has higher energy by 2ε/3 than non-SO and the latter has lower energy by ε/3. The measured SO splitting of Cl is ε = 2.52 kcal mol−1, and therefore the SO interaction lowers the reactant asymptote by ε/3 = 0.84 kcal mol−1, which is exactly reproduced by our computations. SO corrections for the stationary points are positive in all cases: 0.84 kcal mol−1 for the products and MIN complexes (for CH3 HA MIN′ 0.83 kcal mol−1), while slightly less (0.81–0.83 kcal mol−1) for transition states and PREMIN. These results show that even at PREMIN the SO interaction is almost fully quenched; thus, the SO shift of the reactant level effectively increases the relative energies. In addition, the cases, when the methylamine is approached by the chlorine atom from seven different directions are investigated and the potential curves, relative to the ground non-SO asymptotic limit, are shown in Fig. 6. As seen, due to the interaction with methylamine, six different states are present. The twofold degenerate SO1 and twofold degenerate excited SO2 result from the splitting of the 2P3/2 state and SO3 correlates to 2P1/2. The non-SO ground state (non-SO1) and two non-SO excited states (non-SO2, non-SO3) are the non-relativistic split 2P states. As shown in Fig. 6, the deepest van der Waals wells of SO1 with depths of 9.04, 2.66, and 2.22 kcal mol−1 appear when the methylamine is approached by the chlorine atom with C–N–Cl angle of 108.5° as in PREMIN, perpendicularly to the C–N bond on the side opposite to the hydrogens of the amino-group, and in the case of the attack of the amino-group in line with the C–N bond, respectively. This implies that approach from the methyl-group has a shallower well than from the NH2 as expected, because the latter is a more polar group than the former. Interestingly, as Fig. 6 also shows, the difference between the energy of SO and non-SO ground states does not drop to zero rapidly, the decrease occurs from about 4 to 2 Å inter-fragment distances. In the case of OH + C2H6 and OH + CH4 this effect moves within sharper boundaries.29 At the TS and product regions the SO vs. non-SO energy differences are close to zero, which means that the SO effects on the relative energies are close to ε/3 as mentioned above.
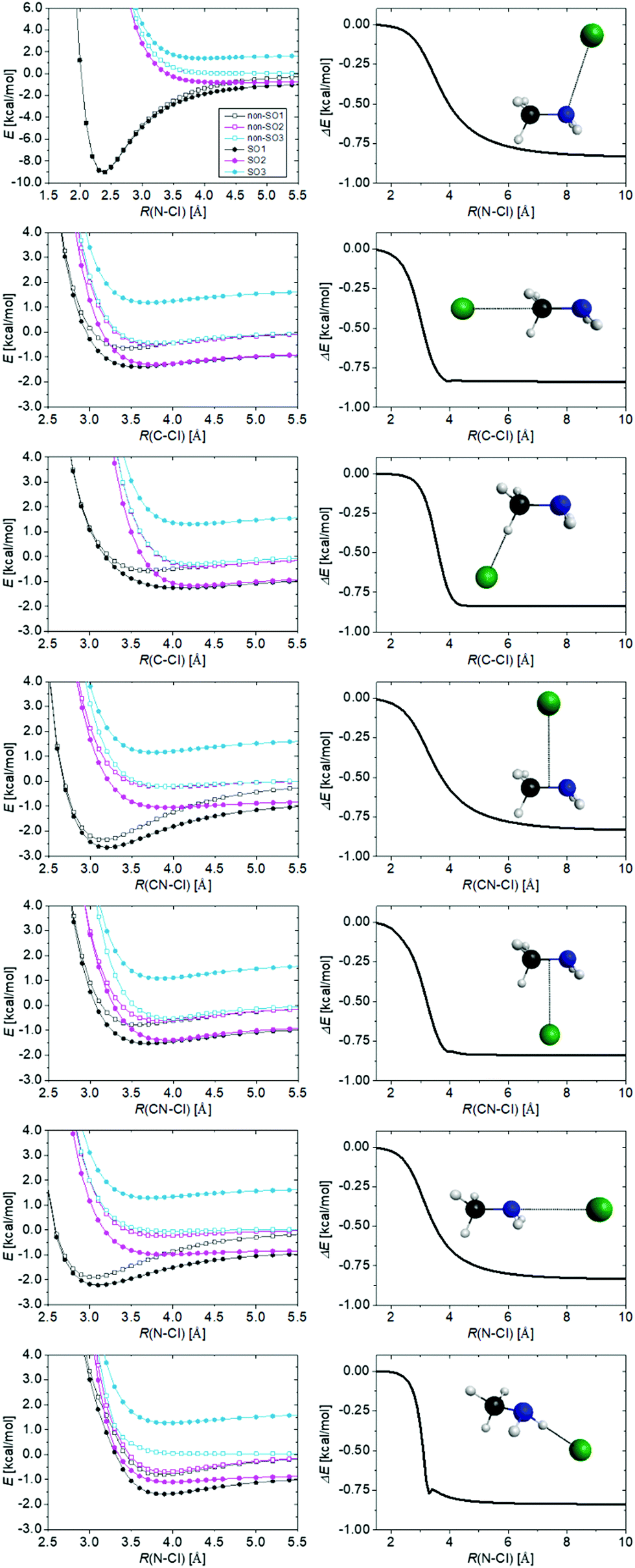 | ||
| Fig. 6 Potential energy curves of the Cl⋯CH3NH2 system obtained at the MRCI+Q/aug-cc-pwCVTZ level of theory for seven different separation directions, while methylamine is kept frozen at its equilibrium geometry. The seven orientations are: the Cl atom approaching CH3NH2 in the same orientation as in the PREMIN complex (first row), approaching the methyl-group (second row), approaching one H atom of the methyl-group (third row), perpendicularly the C–C bond from two directions (fourth and fifth rows), from the amino-group (sixth row) and approaching one H atom of the amino-group (seventh row). The curves in the right panels show the distance-dependence of E(SO1)–E(non-SO1). | ||
| Reaction | This worka | Experimentb |
|---|---|---|
| a Obtained in this work from eqn (7). b Determined from Active Thermochemical Tables (version 1.122p).46 Gaussian error propagation law is applied to determine the uncertainties, based on the uncertainties (corresponding to 95% confidence limits) of the enthalpy of formation data given in ATcT. | ||
| Cl + CH3NH2 → HCl + CH2NH2 | −10.79 | −10.93 ± 0.12 |
| Cl + CH3NH2 → HCl + CH3NH | −3.88 | −4.18 ± 0.13 |
| Cl + CH3NH2 → NH2 + CH3Cl | 0.25 | 0.32 ± 0.08 |
| Cl + CH3NH2 → NH3 + CH2Cl | −8.20 | −7.92 ± 0.22 |
IV. Summary and conclusions
The pathways of the Cl + CH3NH2 multi-channel reaction have been investigated using a high-level composite ab initio method. Besides the previously known exothermic H-abstraction channels leading to HCl + CH2NH2 (−10.79) and HCl + CH3NH (−3.88), amino-, methyl-, and H-substitution pathways, resulting in CH3Cl + NH2 (0.25), NH2Cl + CH3 (22.45), and CH2ClNH2/CH3NHCl + H (10.53/39.87), respectively, are also revealed, showing the benchmark vibrationally adiabatic reaction enthalpies (kcal mol−1) in parentheses. In the four cases, where experimental data are available for comparison, good agreement has been observed between theory and experiment. In the entrance channel there is a CH3NH2⋯Cl complex with substantial De(D0) value of 15.30(14.22) kcal mol−1. The presence of this deep minimum in the entrance channel of the title reaction is in sharp contrast to the Cl + CH4/C2H6 reactions, where the depth of the entrance-channel well is only 0.6–1.0 kcal mol−1.12,16 H-abstraction is clearly/nearly barrierless from the amino/methyl group, whereas the substitution processes have high barriers in the 30–50 kcal mol−1 range and in the amino-, methyl-, and H-substitution increasing energy order. H-substitution is both kinetically and thermodynamically favored from the methyl group. For the methyl-H-substitution and the amino-substitution we have found front-side attack TSs besides the Walden-inversion ones, and the front-side attack pathways have always higher barriers by about 6 and 20 kcal mol−1, respectively. In the exit channels product-like complexes are found, which are the most stable in the case of the H-abstraction channels with about twice as large binding energies than those of the corresponding product complexes of the Cl + CH4/C2H6 reactions. On the basis of the present computations, the Guinness (lowest-energy) structure47 of the Cl + CH3NH2 system corresponds to the CH3NH2⋯Cl and CH2NH2⋯HCl complexes without and with zero-point energies, respectively.Our benchmark energy computations show that the CCSD(T)-F12b/aug-cc-pVQZ results are basis-set converged within 0.1 kcal mol−1, the post-(T) correlation effects are substantial for the TSs where their mean absolute value is 0.41 kcal mol−1, the core and scalar relativistic corrections have small usually positive values around 0.1–0.2 kcal mol−1, and the spin–orbit coupling is almost fully quenched at the TSs, product channels, and even at the CH3NH2⋯Cl pre-reaction complex, and therefore, the SO shift of the Cl atom effectively increases the relative energies by 0.81–0.84 kcal mol−1, and the ZPE effects are the most substantial, often decreasing the relative energies by 3–6 kcal mol−1. Considering the uncertainties of the post-(T) correlation (0.1 kcal mol−1), core correlation (0.1 kcal mol−1), relativistic effects (0.1 kcal mol−1), and harmonic ZPE corrections (0.1 kcal mol−1) as well as basis-set errors (0.1 kcal mol−1), neglected non-Born–Oppenheimer effects (<0.1 kcal mol−1 based on ref. 48) and anharmonicity (∼5% of the ZPE corrections, i.e., 0.1–0.3 kcal mol−1), we estimate 0.2–0.3 and 0.3–0.4 kcal mol−1 uncertainty for the present benchmark classical and vibrationally adiabatic relative energies, respectively.
As a future research direction, one may develop a global analytical potential energy surface for the title reaction, which would allow dynamics investigations and further comparisons with experiments. The present benchmark stationary-point data provide guidance for such dynamics studies and help to assess the accuracy of the potential energy surface. Dynamics simulations can reveal the atomic-level mechanisms of the title reaction and the collision-energy-dependent branching ratios of the different product channels as well as HCl rotational distributions allowing critical comparison with experiment. Furthermore, the new findings of the present study may motivate the research community to consider fundamental reactions as complex, and multi-channel processes.
Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
We thank the National Research, Development and Innovation Office–NKFIH, K-125317, the Ministry of Human Capacities, Hungary grant 20391-3/2018/FEKUSTRAT, and the Momentum (Lendület) Program of the Hungarian Academy of Sciences for financial support.References
- G. C. Schatz and A. Kuppermann, J. Chem. Phys., 1975, 62, 2502 CrossRef.
- G. C. Schatz, M. C. Colton and J. L. Grant, J. Phys. Chem., 1984, 88, 2971 CrossRef CAS.
- H. W. Song, M. H. Yang and H. Guo, J. Chem. Phys., 2016, 145, 131101 CrossRef PubMed.
- Z. H. Kim, A. J. Alexander, H. A. Bechtel and R. N. Zare, J. Chem. Phys., 2001, 115, 179 CrossRef CAS.
- S. Yoon, S. Henton, A. N. Zivkovic and F. F. Crim, J. Chem. Phys., 2002, 116, 10744 CrossRef CAS.
- R. Welsch and U. Manthe, J. Chem. Phys., 2013, 138, 164118 CrossRef PubMed.
- F. Meng, W. Yan and D. Wang, Phys. Chem. Chem. Phys., 2012, 14, 13656 RSC.
- C. Murray, J. K. Pearce, S. Rudić, B. Retail and A. J. Orr-Ewing, J. Phys. Chem. A, 2005, 109, 11093 CrossRef CAS PubMed.
- W. W. Harper, S. A. Nizkorodov and D. J. Nesbitt, J. Chem. Phys., 2000, 113, 3670 CrossRef CAS.
- J. P. Layfield, A. F. Sweeney and D. Troya, J. Phys. Chem. A, 2009, 113, 4294 CrossRef CAS PubMed.
- C. Murray and A. J. Orr-Ewing, Int. Rev. Phys. Chem., 2004, 23, 435 Search PubMed.
- G. Czakó and J. M. Bowman, J. Phys. Chem. A, 2014, 118, 2839 CrossRef PubMed.
- K. Liu, J. Chem. Phys., 2015, 142, 080901 CrossRef PubMed.
- B. Fu, X. Shan, D. H. Zhang and D. C. Clary, Chem. Soc. Rev., 2017, 46, 7625 RSC.
- J. Espinosa-Garcia, J. C. Corchado, M. Garcia-Chamorro and C. Rangel, Phys. Chem. Chem. Phys., 2018, 20, 19860 RSC.
- D. Papp, B. Gruber and G. Czakó, Phys. Chem. Chem. Phys., 2019, 21, 396 RSC.
- D. Papp, V. Tajti, T. Győri and G. Czakó, J. Phys. Chem. Lett., 2020, 11, 4762 CrossRef CAS PubMed.
- D. Papp and G. Czakó, J. Chem. Phys., 2020, 153, 064305 CrossRef CAS.
- A. W. Ray, J. Agarwal, B. B. Shen, H. F. Schaefer III and R. E. Continetti, Phys. Chem. Chem. Phys., 2016, 18, 30612 RSC.
- M. L. Weichman, J. A. DeVine, M. C. Babin, J. Li, L. Guo, J. Ma, H. Guo and D. M. Neumark, Nat. Chem., 2017, 9, 950 CrossRef CAS PubMed.
- D. Lu, J. Li and H. Guo, Chem. Sci., 2019, 10, 7994 RSC.
- D. Lu, J. Li and H. Guo, CCS Chem., 2020, 2, 882 CrossRef CAS.
- D. Lu and J. Li, Theor. Chem. Acc., 2020, 139, 157 Search PubMed.
- S. Rudić, C. Murray, J. N. Harvey and A. J. Orr-Ewing, Phys. Chem. Chem. Phys., 2003, 5, 1205 RSC.
- M. S. Taylor, S. A. Ivanic, G. P. F. Wood, C. J. Easton, G. B. Bacskay and L. Radom, J. Phys. Chem. A, 2009, 113, 11817 CrossRef CAS PubMed.
- H. Feng, W. Sun, Y. Xie and H. F. Schaefer III, ChemPhysChem, 2013, 14, 896 CrossRef CAS PubMed.
- J. M. Nicovich, S. Mazumder, P. L. Laine, P. H. Wine, Y. Tang, A. J. C. Bunkan and C. J. Nielsen, Phys. Chem. Chem. Phys., 2015, 17, 911 RSC.
- L. Krotos and G. Czakó, J. Phys. Chem. A, 2017, 121, 9415 CrossRef CAS PubMed.
- B. Gruber and G. Czakó, Phys. Chem. Chem. Phys., 2020, 22, 14560 RSC.
- R. D. Amos, J. S. Andrews, N. C. Handy and P. J. Knowles, Chem. Phys. Lett., 1991, 185, 256 CrossRef CAS.
- T. H. Dunning, Jr., J. Chem. Phys., 1989, 90, 1007 CrossRef.
- G. Knizia, T. B. Adler and H.-J. Werner, J. Chem. Phys., 2009, 130, 054104 CrossRef PubMed.
- H.-J. Werner, P. J. Knowles, G. Knizia, F. R. Manby and M. Schütz and others, Molpro, version 2015.1, a package of ab initio programs, see http://www.molpro.net.
- K. Raghavachari, G. W. Trucks, J. A. Pople and M. Head-Gordon, Chem. Phys. Lett., 1989, 157, 479 CrossRef CAS.
- J. Noga and R. J. Bartlett, J. Chem. Phys., 1987, 86, 7041 CrossRef CAS.
- M. Kállay and J. Gauss, J. Chem. Phys., 2005, 123, 214105 CrossRef PubMed.
- J. G. Hill, S. Mazumder and K. A. Peterson, J. Chem. Phys., 2010, 132, 054108 CrossRef PubMed.
- M. Douglas and N. M. Kroll, Ann. Phys., 1974, 82, 89 CAS.
- W. A. de Jong, R. J. Harrison and D. A. Dixon, J. Chem. Phys., 2001, 114, 48 CrossRef CAS.
- A. Berning, M. Schweizer, H.-J. Werner, P. J. Knowles and P. Palmieri, Mol. Phys., 2000, 98, 1823 CrossRef CAS.
- S. R. Langhoff and E. R. Davidson, Int. J. Quantum Chem., 1974, 8, 61 CrossRef CAS.
- K. R. Shamasundar, G. Knizia and H.-J. Werner, J. Chem. Phys., 2011, 135, 054101 CrossRef CAS PubMed.
- K. A. Peterson and T. H. Dunning, Jr., J. Chem. Phys., 2002, 117, 10548 CrossRef CAS.
- MRCC, a quantum chemical program suite written by M. Kállay, Z. Rolik, I. Ladjánszki, L. Szegedy, B. Ladóczki, J. Csontos and B. Kornis, See also Z. Rolik and M. Kállay, J. Chem. Phys., 2011, 135, 104111 CrossRef PubMed , as well as: http://www.mrcc.hu.
- G. Czakó, I. Szabó and H. Telekes, J. Phys. Chem. A, 2014, 118, 646 CrossRef PubMed.
- B. Ruscic and D. H. Bross, Active Thermochemical Tables (ATcT) values based on ver. 1.122p of the Thermochemical Network (2020); available at http://ATcT.anl.gov.
- M. A. Suhm, Angew. Chem., Int. Ed., 2014, 53, 1714 CrossRef CAS PubMed.
- G. Czakó, B. C. Shepler, B. J. Braams and J. M. Bowman, J. Chem. Phys., 2009, 130, 084301 CrossRef PubMed.
| This journal is © the Owner Societies 2021 |