
Hydrogen bond donor functionalized poly(ionic liquid)s for efficient synergistic conversion of CO2 to cyclic carbonates†
Haibin
Gou
ab,
Xifei
Ma
ab,
Qian
Su
 a,
Lei
Liu
a,
Ting
Ying
a,
Wei
Qian
ab,
Li
Dong
a and
Weiguo
Cheng
*a
a,
Lei
Liu
a,
Ting
Ying
a,
Wei
Qian
ab,
Li
Dong
a and
Weiguo
Cheng
*a
aCAS Key Laboratory of Green Process and Engineering, State Key Laboratory of Multiphase Complex Systems, Beijing Key Laboratory of Ionic Liquids Clean Process, Institute of Process Engineering, Chinese Academy of Sciences, Beijing 100190, China. E-mail: wgcheng@ipe.ac.cn; Fax: +86 10 8262 7080; Tel: +86 10 8262 7080
bUniversity of Chinese Academy of Sciences, School of Chemical Engineering, Beijing 100049, China
First published on 17th December 2020
Abstract
The development of metal-free, high effective and recyclable catalysts plays a pivotal role in transforming CO2 into high value-added products such as cyclic carbonates. In this paper, we introduced the hydrogen bond donor (HBD) groups into poly(ionic liquid)s via free radical polymerization, which successfully combined the HBD and ionic liquids (ILs) into one heterogeneous catalyst. The HBD could synergistically activate epoxides with hydroxyl functionalized ionic liquids and efficiently catalyze the cycloaddition of CO2 into cyclic carbonates. The yield of propylene carbonate (PC) reached 94% (at 105 °C, 2 MPa CO2, 3 h), which far exceeded poly(ionic liquid)s without HBDs functionalization (PC yield 72%), and even approached bulk ionic liquids (PC yield 95%). Moreover, HBD-functionalized poly(ionic liquid)s (HPILs) exhibited excellent recyclability after five runs and afforded wide substrate scope. According to the experimental results, 1H NMR spectra and density functional theory (DFT) calculations showed 2-hydroxyethyl methacrylate (HEMA) and the hydroxyl of ILs would form strong H-bonds with epoxides contributing to the ring-opening process of epoxides, and a possible HBD and nucleophilic anion synergistically catalytic mechanism was proposed. The method herein paved a brand new way for green technology and utilization of poly(ionic liquid)s.
Introduction
Carbon dioxide, the dominant culprit of greenhouse effect and climate change, is a type of abundant and renewable C1 feedstock for the synthesis of high value-added chemicals as well.1–6 Therefore, it is extremely urgent to find effective and rational approaches to capture and chemically immobilize the abundant CO2. Typically, the fixation of CO2 into epoxides implements the recyclable and valuable utilization, conforming to the atom economy and green chemistry1 (Scheme 1). In addition, the products, cyclic carbonates, are important commercial products for synthesizing durable polycarbonates, polar solvents, and green electrolytes for batteries.1,2,4 However, the thermodynamic stability and kinetic inertia of CO2, which has a specific linear molecular structure, prevents this reaction from occurring sufficiently. Introducing an efficient catalyst, which could active CO2 and epoxides, is mandatory to facilitate the cycloaddition reaction.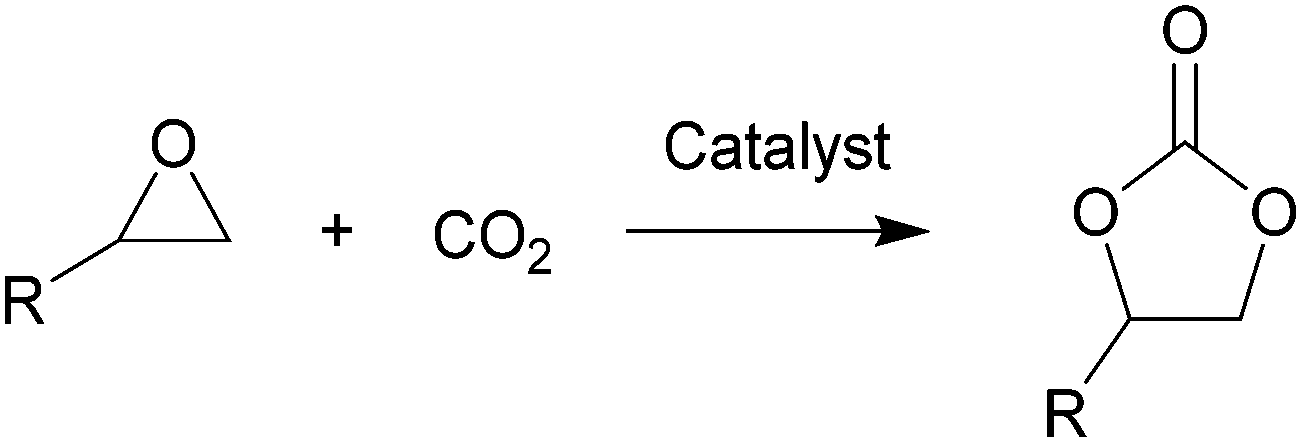 | ||
| Scheme 1 CO2 cycloaddition reaction. | ||
Hitherto, many catalysts have been developed for the cycloaddition reaction, such as ammonium salts,7–9 phosphonium salts,10 alkali metal salts,11,12 and organic catalysts.13–17 These homogeneous catalysts with higher activity are complicatedly separated from the reaction system. However, the heterogeneous catalysts, including transition metal complexes,18,19 metal–organic framework (MOFs),20 covalent organic frameworks,21,22 are easily separated from the products, but disadvantaged by the limitation of interphase mass transition. Therefore, the development of catalysts with high-activity and easy separation becomes the focal point in recent researches.
Ionic liquids (ILs) showed excellent chemical–physical properties, such as low vapor pressure, low flammability, and electrochemical stability. For example, the acyclic and cyclic alkyl and ether-functionalized sulfonium ILs, with low melting pint, low viscosity and good conductivity, could be used as potential electrolytes for electrochemical applications.23 Furthermore, some ILs could be applied as catalysts.24,25 Although the ILs with halogen ion exhibited excellent catalytic performance in the cycloaddition reaction,26–29 they were usually too complicated to separate from these products. Nonetheless, the supported ILs showed their advantages of superior stability and reusability but hindered by relatively low reactivity and easy depletion of active sites in the supports.30,31 Moreover, poly(ionic liquid)s (PILs) are usually built up by IL units, which contain the properties derived from the ILs and polymers such as flexibility, high thermal stability and easy preparation.32–35 Typically, the structures and properties of PILs can flexibly be regulated by changing the constituents or pendants, which are suitable for the catalysis.30,36,37 Therefore, the PILs could be used as a type of efficient catalysts for the cycloaddition reaction. Owing to the high specific surface area and efficient active sites, the PILs are promising candidates for CO2 capture and chemical conversion.38–43 Han et al.44 first synthesized the cross-linked polymer with supported ionic liquids by radical polymerization, which performed efficient activity in the CO2 cycloaddition reaction. According to the previous research, the Brønsted acid sites45,46 and Lewis basic sites47 can activate and accelerate the ring-opening process of epoxides, which facilitate the smooth conduction of the cycloaddition reaction.
Therefore, designing PILs that contain high-density nucleophile electrophile sites is a rational approach to attain high catalytic activity.48 Wang et al.37 developed a type of amino acid anion paired mesoporous PILs, which effectively catalyze the CO2 conversion without co-catalysts and metal-/halide-free in an ambient condition. Xie et al.38 synthesized a series of sponge-like hypercrosslinked imidazolium-based mesoporous PILs (PVIm-6-SCD) that could simultaneously facilitate CO2 capture and conversion. Next, Li et al.36 synthesized a series of heterogeneous PILs catalysts via thiol–ene click reaction under ultraviolet light. The thiourea acted as an electrophilic center and ammonium bromide as nucleophilic sites in the PILs could synergistically catalyze CO2 conversion. The two hydrogen atoms of thiourea in the PILs could activate the epoxides by forming strong hydrogen bonds (H-bonds).
Nevertheless, these PILs generally exhibited lower reactivity than bulk ionic liquids, thus increasing the consumption of ionic liquids. Chen et al.43 synthesized the crystalline viologen-based porous PILs via the Menshutkin reaction. They summarized that the synergistic effect of the enriched Br− anion and available H-bonds coming from intrinsic water molecules leads to the remarkable catalytic performance. However, our group has previously reported that the synergistic effect of H-bonds with the halogen anion could activate the oxygen of C–O, resulting in the ring-opening of epoxides.49–51
Furthermore, extra HBDs could form H-bonds with the epoxides except for the hydroxyl-functionalized ILs. Han et al.52 found that the hydroxyl of diols and cellulose could form H-bonds with PO to activate the epoxide and accelerate the reaction. Sun et al.53 studied and simulated the ethylene glycol (EG)/HEBimBr binary catalysts in the CO2 cycloaddition reaction, where EG as HBDs could form H bonds with epoxides. Our preliminary research found that adding a little amount of H2O or alcohol in this reaction system could enhance the conversion of epoxides.49,51,54 However, these HBDs usually acted as co-catalysts to form a homogeneous binary system or as additional agents. It needed additional purifying process to obtain the pure cyclic carbonates products.
Herein, we prepared a series of HBD-functionalized poly(ionic liquid)s (HPILs) that were synthesized through the free radical polymerization of hydroxyl-functionalized ILs monomers, HBDs monomers and cross-linkers as shown in Scheme 2. Through 1H NMR, FT-IR, and element analysis, the structure of the HPILs was confirmed. All of the prepared HPILs acting as recyclable heterogeneous catalysts were evaluated for cycloaddition reaction in solvent-free and additive-free conditions. Interestingly, the HPILs exhibited remarkable catalytic performance than those without HBD-functionalized PILs under the same reaction conditions. High stability and excellent recyclability were tested under optimum conditions. To confirm the superior catalytic performance, various analogue HPILs were prepared and catalytically evaluated. By characterization analysis and DFT simulation calculations, a possible hydrogen bond donor and nucleophilic anion synergistic catalytic mechanism was proposed. This work may provide a novel strategy for synthesizing HBD-functionalized poly(ionic liquid)s and improving the reactivity of poly(ionic liquid)s.
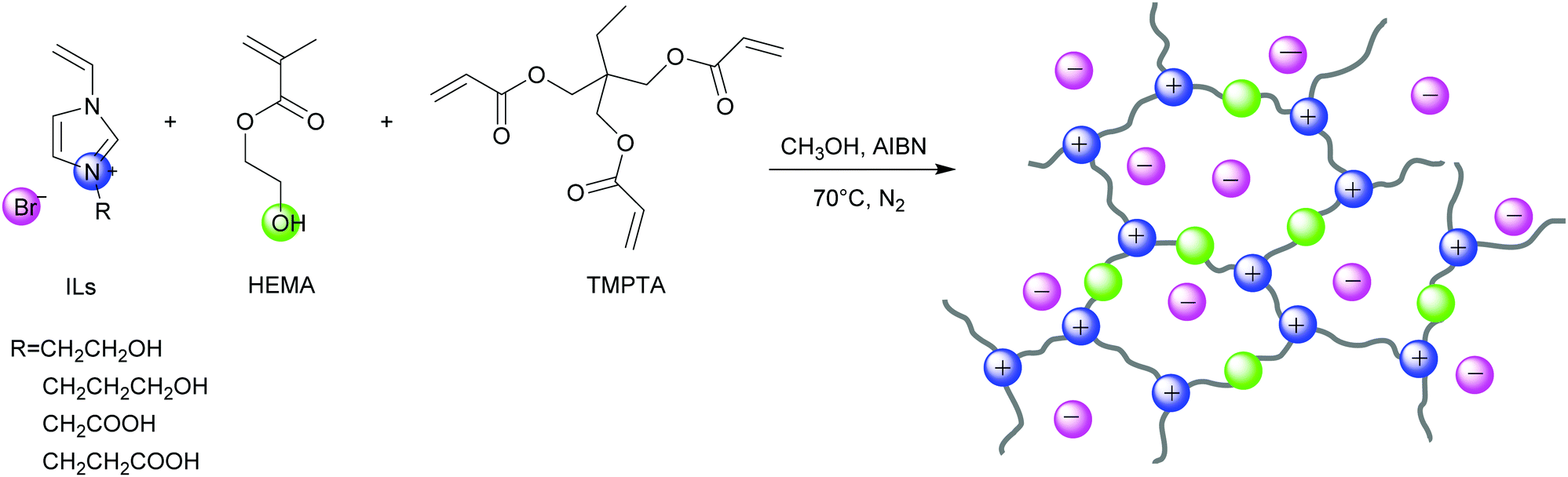 | ||
| Scheme 2 The synthesis of HPILs. | ||
Experimental
Materials
All the chemical materials were purchased directly without any other treatment. 1-Vinylimidazole (99%, Macklin), 2-bromoethanol (95%, Aladdin), 3-bromo-1-propanol (97%, Macklin), bromoacetic acid (AR, 98% (GC), Aladdin), 3-bromopropionic acid (98%, Aladdin),2,2′-Azobls(2-methylproplonirle) (AIBN, 99%, recrystallized, Aladdin), 2-hydroxyethyl methacrylate (HEMA, 96%, Rhawn), trimethylolpropane triacrylate (TMPTA, 95%, Rhawn), anhydrous methanol, anhydrous ethanol, ethyl acetate (Tianjin Zhiyuan), and acetonitrile (Xilong Scientific).
Synthesis of ionic liquids monomers
3-Hydroxypropyl-1-vinylimidazolium bromide (VHPImBr) was synthesized in the same method. The 1H NMR spectrum is shown in Fig. S2 (ESI†).
Similarly, 3-carboxyethyl-1-vinylimidazolium bromide (VCEImBr) was prepared in the same way. The 1H NMR spectrum is shown in Fig. S4 (ESI†).
Synthesis of poly(ionic liquid)s
The poly(ionic liquid)s were synthesized through free radical polymerization of ILs monomer and HEMA at different molar ratios using TMPTA as the cross-linker and AIBN as the initiator. s-PIL-1, HPIL-1, HPIL-2, HPIL-3 and HPIL-4 were synthesized based on VCEImBr. The molar ratio of n(VCEImBr)![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :n(TMPTA) in s-PIL-1 was 6:1, and the molar ratios of n(VCEImBr):n(TMPTA):n(HMEA) in HPIL-1, HPIL-2, HPIL-3 and HPIL-4 were 6:1:3, 6:1:6, 6:1:9 and 6:1:12, respectively.
:n(TMPTA) in s-PIL-1 was 6:1, and the molar ratios of n(VCEImBr):n(TMPTA):n(HMEA) in HPIL-1, HPIL-2, HPIL-3 and HPIL-4 were 6:1:3, 6:1:6, 6:1:9 and 6:1:12, respectively.
Similarly, s-PIL-2, HPIL-5, HPIL-6, HPIL-7, HPIL-8, HPIL-9 and HPIL-10 were synthesized based on VHPImBr. The molar ratio of n(VHPImBr):n(TMPTA) in s-PIL-2 was 6:1. Moreover, the molar ratios of n(VHPImBr):n(TMPTA):n(HEMA) in HPILs were 6:1:3, 6:1:6, 6:1:9, 6:1:12, 6:0.5:9 and 6:2:9, respectively. The detail molar ratio of the feed materials could be seen in the ESI,† (Table S1). s-PILs could not be dissolved in any conventional solvents. Because HPILs had a poor solubility in conventional solvents, the molecular weight of these PILs could not be accurately tested.
Typically, the molar ratio of n(VCEImBr):n(TMPTA):n(HMEA) was 6:1:6 in HPIL-2. VCEImBr (1.5 g), TMPTA (0.3 g), HEMA (0.79 g), and AIBN (50 mg) were dissolved in 30 mL methanol, and then the Schlenk line was used to replace the air with N2. Subsequently, the mixture solution was stirred at 70 °C for 24 h. Then, the product was precipitated from ethanol, washed and dried in the vacuum at 50 °C for 12 h.
Likewise, the other PILs were synthesized in the same way only changing the ratio of ILs, TMPTA and HEMA. The detailed polymerization process will be discussed in the ESI.† The 1H NMR spectra of HPILs are shown in Fig. S5 and S6 (ESI†).
CO2 cycloaddition with epoxides
The cycloaddition of CO2 with epoxides was carried out in a high-pressure reactor (15 mL) equipped with a temperature detector. Typically, propylene oxide (PO) (0.83 g, 14.3 mmol) and VHPImBr (50 mg, 1.5 mol% of PO) were stirred at the desired temperature, pressure, and time. After the reaction was completed, the reactor was cooled to room temperature in ice water, and then the product was isolated from the mixtures by centrifugation. The internal standard n-dodecane (0.5 g) was added to the liquid phase. Finally, the selectivity and yield were analyzed by a gas chromatograph (Agilent 7890B) equipped with a flame ionization detector (FID) and a capillary column (HP-5, 30 m × 0.25 mm × 0.25 μm). As for other epoxides, the cycloaddition reactions were conducted in a similar method.Reusability of the catalysts
Recycling performance of these catalysts in the CO2 cycloaddition of epoxides was evaluated in a five-run test. After each run, the catalyst was separated by centrifugation and washed with anhydrous ethanol, dried in vacuum, and then charged into the next run.Characterization
1H nuclear magnetic resonance (NMR) spectra were recorded by a Bruker AVANCE III 600 MHz spectrometer in deuterated water (D2O) and CDCl3 using tetramethylsilane (TMS) as an internal standard. Element Analysis (EA) was measured on a Vario EL Cube elemental analyzer. Fourier transform infrared (FT-IR) spectra were detected on Thermo Nicolet 380 with anhydrous KBr as a standard. Scanning electron microscopy (SEM) images were acquired with JEM6700E, JEOL. Thermal gravimetric analysis (TGA) experiments were carried out on a DTG-60H thermal analyzer (Shimadzu, Japan) under atmosphere, and the heating process was set as 10 °C min−1 in the range of 25–500 °C. Mettler-Toledo differential scanning calorimeter (DSC) instrument was used to estimate the glass transition temperatures of all PILs and set a heating rate of 10 °C min−1 with N2 purging (50 mL min−1); the result was obtained from the second heating process. X-ray photoelectron spectroscopy (XPS) was performed on an X-ray photoelectron spectroscope (ESCALAB250Xi). The conversion and selectivity of propene oxide were analyzed via GC system (Agilent 7890B) using an FID detector. The density functional theory (DFT) calculations were carried out using the Gaussian 09 program.DFT simulation calculations
The density functional theory (DFT) calculations were carried out using the Gaussian 09 program.55 The systems of VHPImBr with PO, VHPImBr and HMEA with PO were calculated under the same conditions. The geometric optimization of the selected chemical structures was achieved under the B3LYP56 hybrid functional and the 6-31G(d,p)57–61 basis set. The single point energy of the optimized structure was obtained at the level of M06-2X58 and 6-311++G(d,p).59–61 To simply and fully understand the role of HEMA in the HPILs, we selected the VHPImBr with/without HEMA monomers and PO molecule as the calculation models.62Results and discussion
Characterization
The ILs and PILs were synthesized by steps described in the Experimental section and their corresponding structures are demonstrated in Scheme 3. s-PILs were synthesized through free radical polymerization of ILs and TMPTA. Similarly, the HPILs, with ILs as active centres, HEMA as HBDs, and TMPTA as a cross-linker, were obtained by the same approach.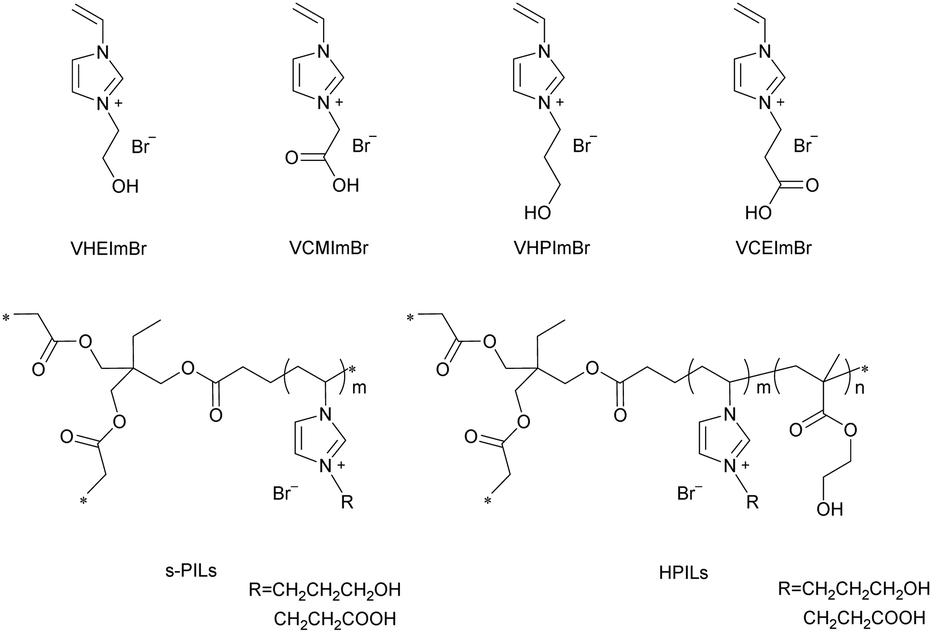 | ||
| Scheme 3 Structures of the synthesized ionic liquids, s-PILs and HPILs. | ||
The structural integrity and chemical composition of the catalysts were confirmed by 1H NMR, FT-IR, and XPS. 1H NMR showed that the PILs were successfully prepared with the disappearance of the H atom in the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C double bond, and each component of the HPILs could be found in the 1H NMR spectra (see Fig. S5 and S6, ESI†). In addition, the VHPImBr characteristic peaks (δ = 9.2 ppm and δ = 7.7 ppm) could be found in Fig. S5 (ESI†). The characteristic peaks of δ = 4.2 ppm and δ = 3.9 ppm belonged to HEMA. Similarly, the characteristic peaks of VCEImBr and HMEA could be found in Fig. S6 (ESI†). In addition, the ILs characteristic peaks decreased with the HEMA content increasing while the HEMA's peaks increased. The peaks of TMPTA was overlapped with them. The element analysis (EA) (see Table S2, ESI†) shows that the N content, which was only contained in ILs monomers indicated the ILs content in the PILs. So, the PILs catalysts dosage in catalytic performance evaluation was based on the EA results.
C double bond, and each component of the HPILs could be found in the 1H NMR spectra (see Fig. S5 and S6, ESI†). In addition, the VHPImBr characteristic peaks (δ = 9.2 ppm and δ = 7.7 ppm) could be found in Fig. S5 (ESI†). The characteristic peaks of δ = 4.2 ppm and δ = 3.9 ppm belonged to HEMA. Similarly, the characteristic peaks of VCEImBr and HMEA could be found in Fig. S6 (ESI†). In addition, the ILs characteristic peaks decreased with the HEMA content increasing while the HEMA's peaks increased. The peaks of TMPTA was overlapped with them. The element analysis (EA) (see Table S2, ESI†) shows that the N content, which was only contained in ILs monomers indicated the ILs content in the PILs. So, the PILs catalysts dosage in catalytic performance evaluation was based on the EA results.
The FT-IR spectra of the IL monomer, s-PILs, and HPILs are demonstrated in Fig. 1 and Fig. S7 (ESI†). The PILs displayed a typical broad peak corresponding to –OH group stretching vibration at around 3720–3030 cm−1. The peaks at 1560 cm−1 and 1074 cm−1 were assigned to the stretching vibration of imidazole.40,63 Moreover, the strong peak at 1720 cm−1 was related to the stretching vibration of CO,64 which only existed in HEMA and TMPTA. The peak around 2930 cm−1 was assigned to the stretch vibration of –CH2– in the PILs’ carbon chain skeleton.
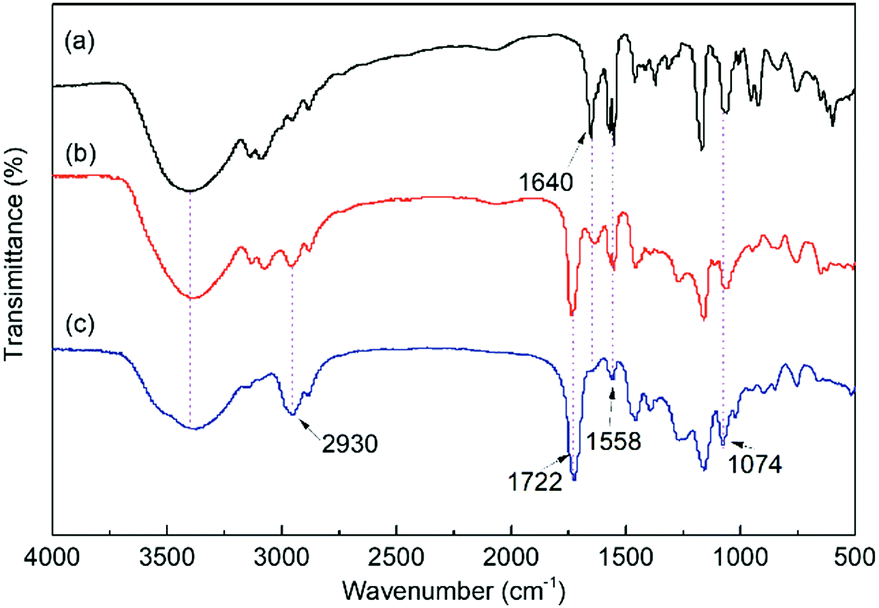 | ||
| Fig. 1 The FT-IR spectra of (a) VHPImBr, (b) s-PIL-2 and (c) HPIL-7. | ||
The characterization of XPS spectra revealed that, in the N 1s XPS spectrum of VHPImBr, the two peaks derived from the imidazolium group were at binding energies (BEs) of 399.5 eV and 401.3 eV, which shifted to 399.1 eV and 401.4 eV, respectively, for HPIL-7 (Fig. 2a). The binding energy of Br 3d was 67.4 eV and 68.5 eV for VHPImBr, 67.5 eV and 68.4 eV for HPIL-7, respectively (Fig. 2b). It displayed no significant change of the Br chemical state, indicating the Br− anion holding similar ability to combine with epoxide groups. The polymerization process did not affect the ability of HPIL-7 and VHPImBr combining with the epoxide groups. Consequently, it could be deduced that HPIL-7 had a similar combination ability with the bulk ILs (VHPImBr).
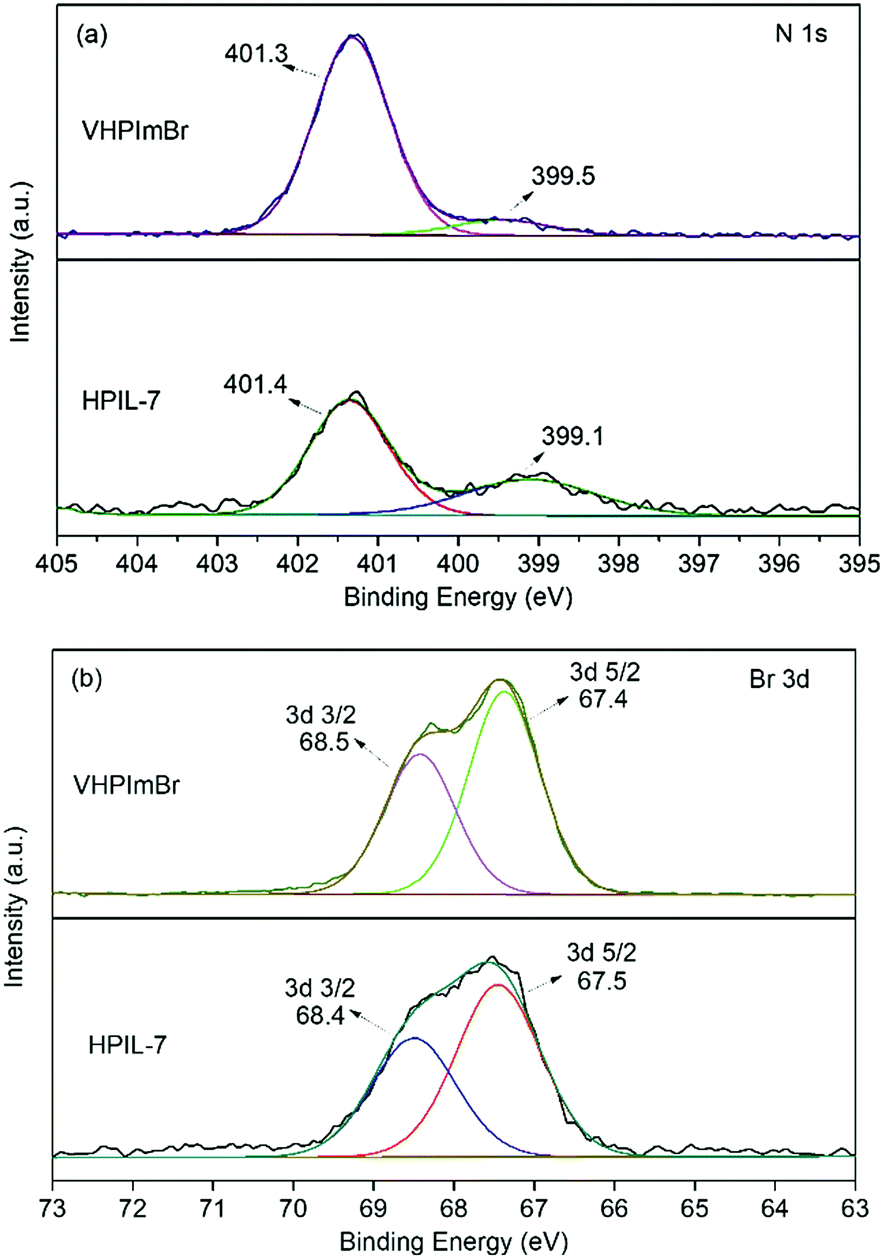 | ||
| Fig. 2 XPS spectra of N 1s and Br 3d for VHPImBr and HPIL-7. | ||
To test the stability of PILs under reaction temperature, all catalysts were investigated by thermo-gravimetric analysis (TGA). As shown in Fig. S8 and S9 (ESI†), the TGA results demonstrated all PILs started to decompose above 200 °C, which was much higher than the reaction temperature. Moreover, the weight loss of the catalysts at about 200 °C was ascribed to the degradation of HPILs. Obviously, the introduction of HEMA had little effect on the thermal stability of PILs (Fig. S8 and S9, ESI†). All PILs showed excellent thermal stability, and DSC was used to test the glass transition phase of VHPImBr and HPIL-7. The structure had a distinct change after the VHPImBr polymerized with HEMA (Fig. S10 and S11, ESI†).
Besides, the morphology and element distribution of s-PIL-2 and HPIL-7 were characterized by SEM. The SEM images (Fig. 3a–d) showed that the PILs possessed a rough surface, which induced the higher surface area of HPIL-7 and facilitated the catalytic performance. Compared with s-PIL-2, HPIL-7 showed a more irregular surface with many small humps and displayed a complex layer structure with ditches and cracks (c and d). These structures could expose as much as possible accessible active sites during the reaction process. The corresponding element mapping images showed that C, O, N, and Br were homogeneously distributed in HPIL-7 (see Fig. S12, ESI†).
 | ||
| Fig. 3 SEM images of (a and b) s-PIL-2, (c and d) HPIL-7, and (e and f) used HPIL-7. | ||
Catalytic ability
Taking PO, for example, we first chose four hydroxyl/carboxyl-functionalized imidazolium ionic liquids to investigate the catalytic activity. The conversion and selectivity of PO were detected by GC system under certain identical reaction conditions. The loading of PILs was according to the results of element analysis whose ionic liquids content was equal to the bulk ionic liquids. All results were summarized in Table 1, due to the synergistic effect of hydroxyl and the Br− anion,49 the ILs (entry 1 to entry 4) showed good PC selectivity (>99%) and yield in the cycloaddition reaction.| Entry | Catalysts | Sel.b (%) | Yieldb (%) |
|---|---|---|---|
| Reaction conditions:a PO (0.83 g, 14.3 mmol), catalysts (1.5 mol% of PO, the dosage was according to the EA analysis, see in ESI), 105 °C, 2 MPa CO2, 3 h.b Based on GC analysis.c Temperature = 120 °C, 1.5 MPa, t = 3 h.d The catalyst were used for 5 times. | |||
| 1 | VHEImBr | 100 | 92 |
| 2 | VHPImBr | 100 | 95 |
| 3 | VCMImBr | 100 | 36 |
| 4 | VCEImBr | 100 | 86 |
| 5 | s-PIL-1 | >99 | 60 |
| 6 | s-PIL-2 | >99 | 72 |
| 7 | HPIL-1 | >99 | 84 |
| 8 | HPIL-2 | >99 | 87 |
| 9 | HPIL-3 | >99 | 90 |
| 10 | HPIL-4 | >99 | 94 |
| 11 | HPIL-5 | >99 | 91 |
| 12 | HPIL-6 | >99 | 93 |
| 13 | HPIL-7 | >99 | 94 |
| 14 | HPIL-8 | >99 | 95 |
| 15 | HPIL-9 | >99 | 92 |
| 16 | HPIL-10 | >99 | 90 |
| 17c | HPIL-7 | >99 | 99 |
| 18cd | HPIL-7 | >99 | 97 |
Comparing entry 1 with 2 (or entry 3 with 4), the reactivity of ILs monomers was related to the carbon chain length attached to the hydroxyl (or carboxyl), the PC yield reached 95% for VHPImBr which was higher than VHEImBr, and hydroxyl-functionalized ILs were better than carboxylated ILs. Therefore, VHPImBr and VCEImBr were selected as model ILs monomers to prepare self-polymerized PILs (s-PILs) and HPILs through free radical polymerization. s-PIL-1, HPIL-1, HPIL-2, HPIL-3 and HPIL-4 were synthesized based on VCEImBr, and s-PIL-2, HPIL-5, HPIL-6, HPIL-7, HPIL-8, HPIL-9 and HPIL-10 were synthesized based on VHPImBr. The detailed synthesis methods could be seen in the ESI.†
In Table 1, entries 5 and 6 showed s-PIL-1 and s-PIL-2 exhibited good PC selectivity (>99%), but the PC yields were only 60% and 72% at 105 °C, CO2 2 MPa, 3 h, respectively. Moreover, HBD-functionalized HPILs exhibited a better catalytic activity than the s-PILs without HBDs (Table 1, entry 5–16). From entries 11–14 and 7–10, we could see that the HPILs based on VHPImBr exhibited better catalytic performance than those based on VCEImBr. Although HPIL-8 showed the best catalytic activity, it required a higher dosage than others. Therefore, we selected HPIL-7 (entry 13) as the best catalyst, which had the similar catalytic performance with HPIL-8 (entry 14). The ratio of VHPImBr and HEMA in HPIL-7 was 1:1.5 (based on the feed material ratio in Table S1, ESI†). Besides, we tested the catalytic performance between VHPImBr/HEMA mixtures. As shown in Table S3 (ESI†), the PC yield increased with the HEMA content both in VHPImBr/HEMA mixtures and HPILs. When VHPImBr/HEMA molar ratio was higher than 1:1.5, the PC yield insignificantly increased.
When HEMA was introduced into HPILs, the catalysts would be enriched in hydroxyl, which could form H bonds with PO molecules. From entries 7–11 and 11–14, we could see that the reactivity of HPILs increased with the HBDs content because the HEMA group acted as an extra hydrogen bond donor in HPILs and could synergistically activate the PO with bromide anion in the catalytic process. Besides, the content of cross-linker could affect the reactivity of HPILs (entry 15 and 16), which explained the proper amount of TMPTA could improve the performance of HPILs. To summarize, Table 1 showed that the introduction of HEMA in HPILs could enhance the reactivity of HPILs, which was much higher than s-PILs, even close to that of IL monomers.
Subsequently, the influence of reaction conditions, such as temperature, CO2 pressure, and reaction time, were investigated in the following experiments. Fig. 4a shows that the reaction temperature could significantly affect the reactivity of HPIL-7, and the PC yield obviously increased from 68% to 99%. At 110 °C and CO2 1.5 MPa, increase in the reaction time, PC yield increased from 90% to 97.6% (Fig. 4b). Compared with the temperature and reaction time, the CO2 pressure had a negligible influence on the HPIL-7 catalytic reactivity as shown in Fig. 4c. Then, we got the optimal conditions for the reaction (120 °C, CO2 1.5 MPa, 3 h), the PC yield could reach 99% (Table 1, entry 17).
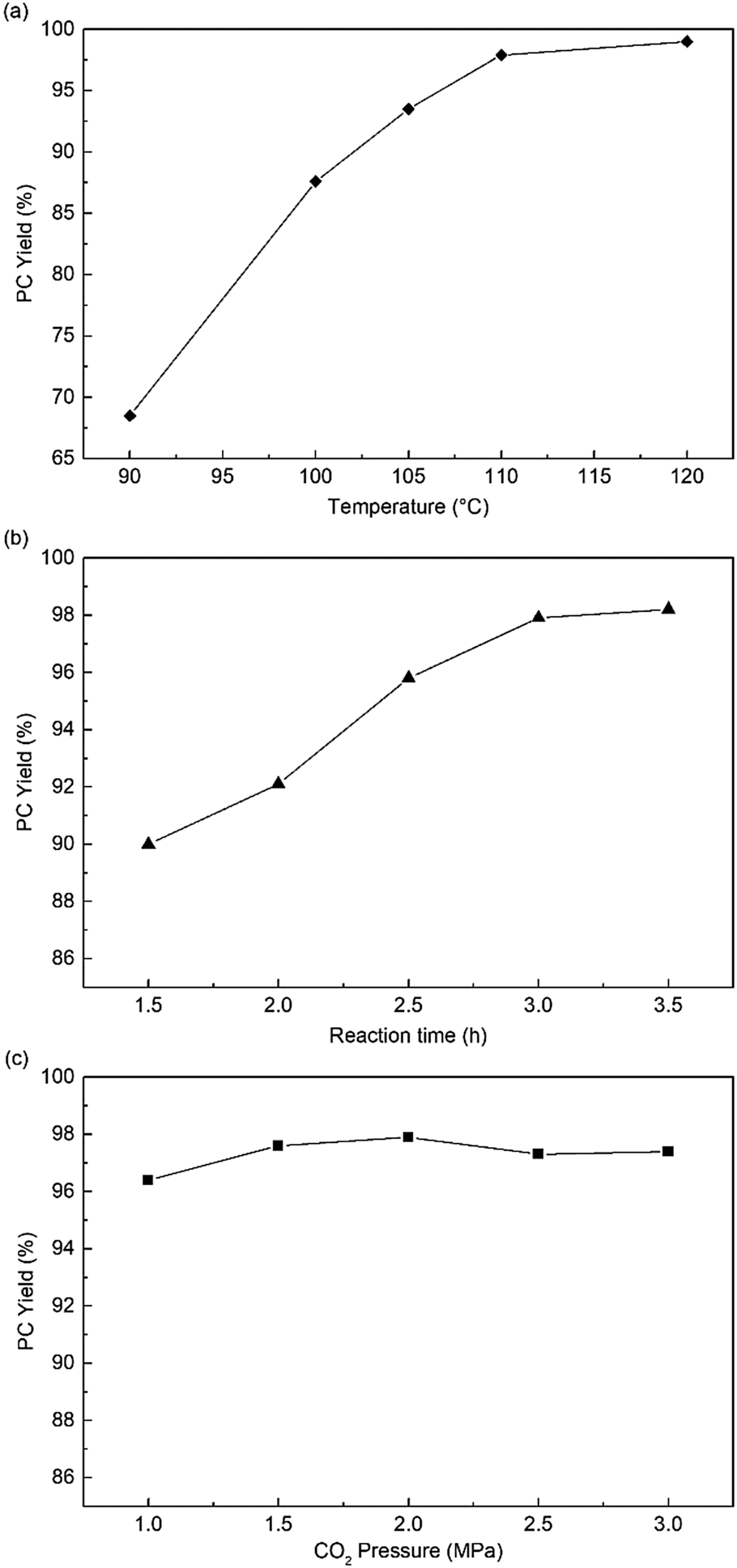 | ||
| Fig. 4 The effects of different reaction conditions to PC yield. Reaction conditions: PO (0.83 g, 14.3 mmol), HPIL-7 (180 mg, 1.5 mol%, based on the EA results), (a) 2 MPa CO2, 3 h. (b) 110 °C, 1.5 MPa (c) 110 °C, 3 h. | ||
Recyclability and stability
HPIL-7 exhibited the best catalytic reactivity under the optimum condition (120 °C, CO2 1.5 MPa, 3 h). To investigate the recyclability and stability of HPIL-7, we washed used HPIL-7 with ethanol several times and dried in vacuum, and then used it in next running. HPIL-7 showed excellent recyclability and stability after it was used five times, and the selectivity of PC remained at >98% and PC yield remained at 97% (Fig. 5). After five runs, the reactivity of HPIL-7 only slightly decreased. Although the VHPImBr exhibited higher conversion than the corresponding HPILs, it was limited by the complicated separation process. However, the HPILs could be easily separated by simple filtration. In this concept, we successfully combined the HBDs and ionic liquids into heterogeneous catalysts and simplified the separation process. Moreover, the introduction of HBDs significantly improved the performance of s-PILs.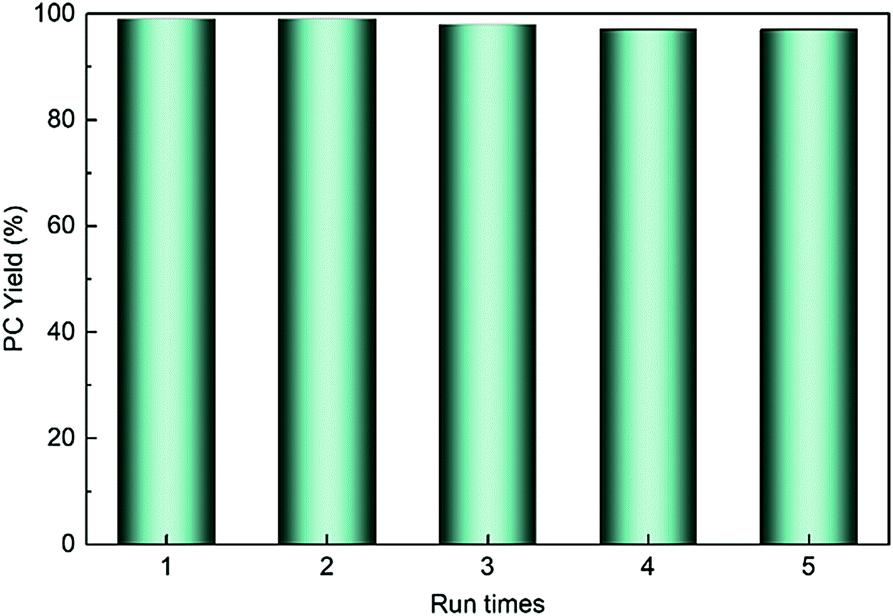 | ||
| Fig. 5 HPIL-7 reused test. | ||
After that, we characterized the used catalyst and found that the structure and composition were basically unchanged. It could be seen in Fig. 6 that the FT-IR spectra of fresh and used HPIL-7 had no significant change. Also, the SEM images (Fig. 3e and f) showed that the morphology of HPIL-7 had no obvious change, which explained the excellent recyclability and stability.
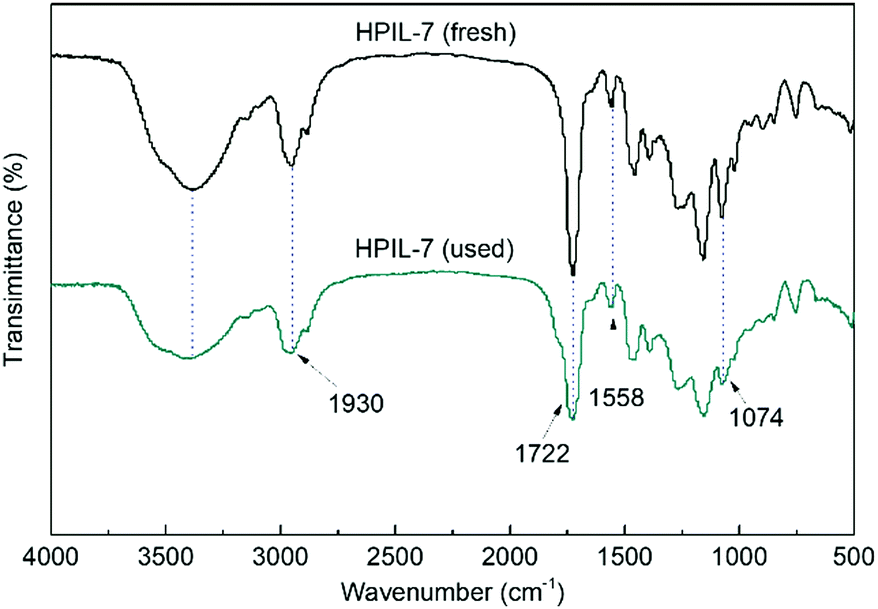 | ||
| Fig. 6 The FT-IR spectra of fresh and used HPIL-7. | ||
Substrate scope of epoxides
In addition, for investigating the potential and general applicability of HPIL-7, the cycloaddition of epoxides to different terminal epoxides was carried out and summarized in Table 2. HPIL-7 could effectively catalyze various epoxides into corresponding cyclic carbonates with good conversion and selectivity (>98%). However, for the epoxides with large substituent groups, the catalytic performance dramatically decreased. Due to the steric hindrance from epoxides, the nucleophilic group (Br− anion) could not effectively attack the cyclohexene oxide, resulting in less conversion (48%) even under a longer time reaction (Table 2, entry 6). Nonetheless, it was worth mentioning that HPIL-7 was efficient for the epoxides converting under a relatively mild condition and reflecting the outstanding generality.
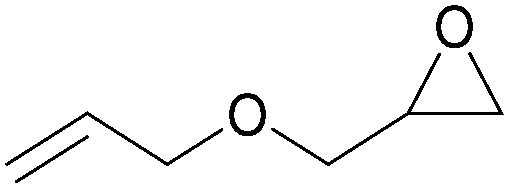
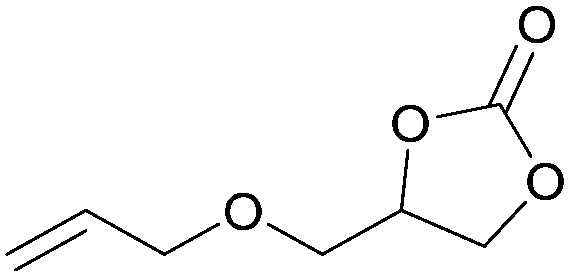
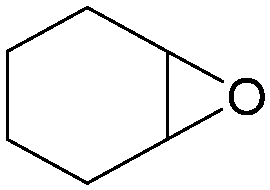
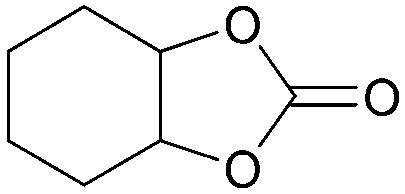
DFT simulation calculations
To deeply understand the mechanism of HEMA in the reaction system and investigate the ability of ILs monomers and HEMA molecule to combine with PO molecule,62 two systems were constructed and optimized. The geometric optimization of the selected chemical structure was achieved under the B3LYP hybrid functional and the 6-31G(d,p) basis set. The single point energy of the optimized structure was obtained at the level of M06-2X and 6-311++G(d,p). As shown in Fig. 7a, the simulation model was depicted that the VHPImBr could form a strong H-bond with PO. The H atom of the hydroxyl in VHPImBr and the O atom of PO ring formed a strong H-bond with a bond length of 1.83 Å, and the interaction energy of −4.25 kcal mol−1 between VHPImBr and PO. When HEMA was introduced into the simulation system, both the VHPImBr and HEMA could form strong H-bonds with PO. Moreover, the H atom of the hydroxyl of VHPImBr and HEMA simultaneously formed a strong H-bond with PO, of which the length were 1.87 Å and 1.98 Å, respectively. The interaction energy was −10.82 kcal mol−1 between the system composed by VHPImBr with HEMA and PO in Fig. 7b. The DFT results preliminarily indicate the synergistic effect of VHPImBr and HEMA in the ring-opening process of PO.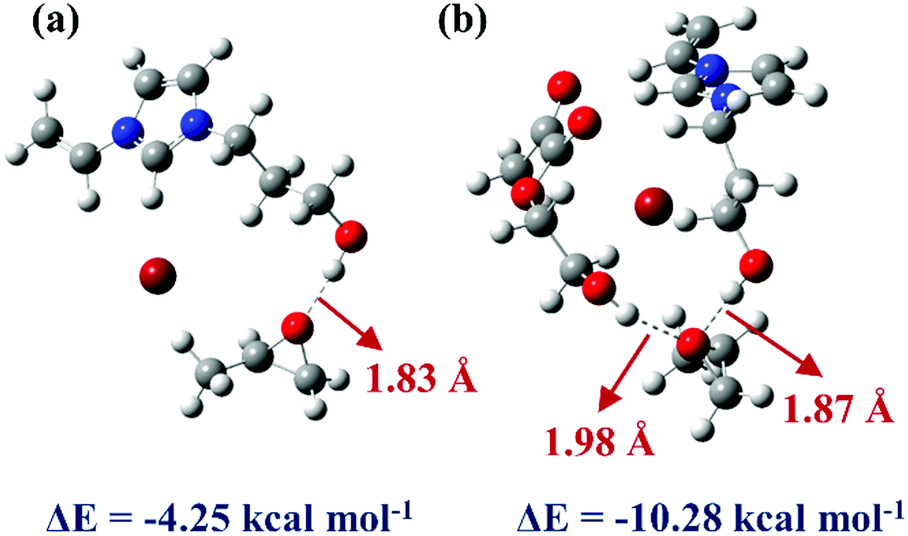 | ||
| Fig. 7 The M06-2X/6-311++G(d,p) optimized geometries of PO and catalysts. (a) VHPImBr and PO. (b) VHPImBr, HEMA and PO. | ||
Namely, the HBD-functionalized HPILs with more negative interaction energy were easier to activate PO than that of s-PILs, which is in accordance with the experimental results. Nonetheless, in the reaction system, both VHPImBr and HEMA could interact with PO by forming H-bond and improve the utilization of nucleophilic reagent (Br−), thus explaining why the introduction of HBDs could enhance the catalytic performance of HPILs (Table 1, entries 5, 6 and 7–16). Furthermore, the introduction of HEMA could make the active sites dispersed in HPILs, thus improving the utilization of ILs.
Possible reaction mechanism
In addition, the 1H NMR spectra of the mixture of VHPImBr, HEMA and PO indicated that the HBDs could significantly influence the C–O bond of PO through a strong H-bond. The 1H NMR spectra of HEMA with and without PO were determined and presented in Fig. 8. This showed that HEMA could form a strong H-bond with PO, and the peak of H atom in –OH significantly downshifted from 2.12 ppm to 2.50 ppm.52 Thus, HEMA could activate the PO by forming H-bond, like H2O and EG.53,54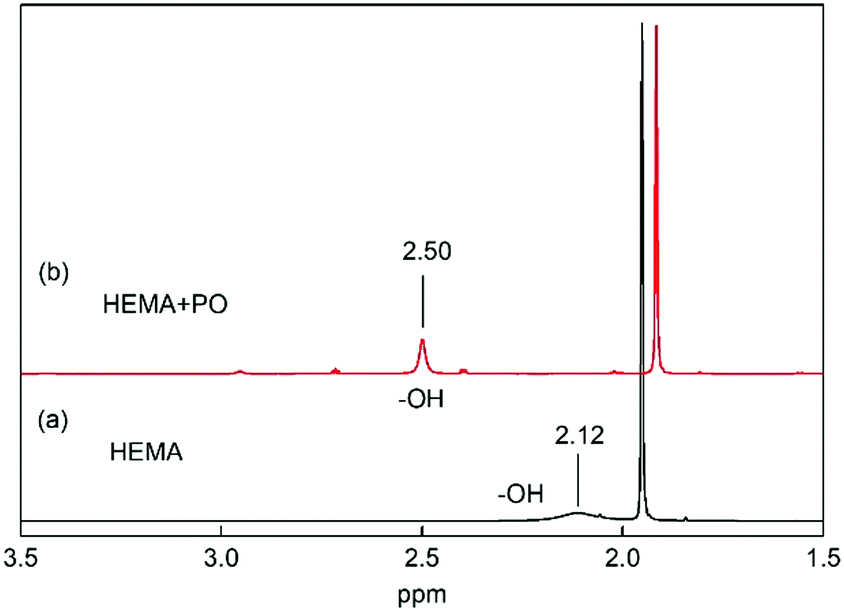 | ||
| Fig. 8 1H NMR spectra of HEMA with and without PO in CDCl3. (a) HEMA 10 μL, CDCl3 0.5 mL. (b) PO 30 μL, HEMA 10 μL, CDCl3 0.5 mL. | ||
Therefore, we proposed a possible mechanism in Scheme 4 based on the experiments, 1H NMR spectra, and DFT calculations. First, the hydroxyl of VHPImBr could induce the polarization of C–O bond through forming H-bond with PO,49 and HEMA could form H-bond with the PO simultaneously to enhance the polarization process of C–O bond. Subsequently, the Br− anion attacked the less sterically hindered carbon atom of PO, and the epoxide ring was successfully opened. Next, CO2 attacked the oxygen anion forming the alkylcarbonate anion, which would be transformed into PC and the catalyst regenerated. Hence, the heterogeneous HPILs catalyst with hydrogen bond donors, cation and anion of ionic liquids smoothly promoted the cycloaddition reaction.
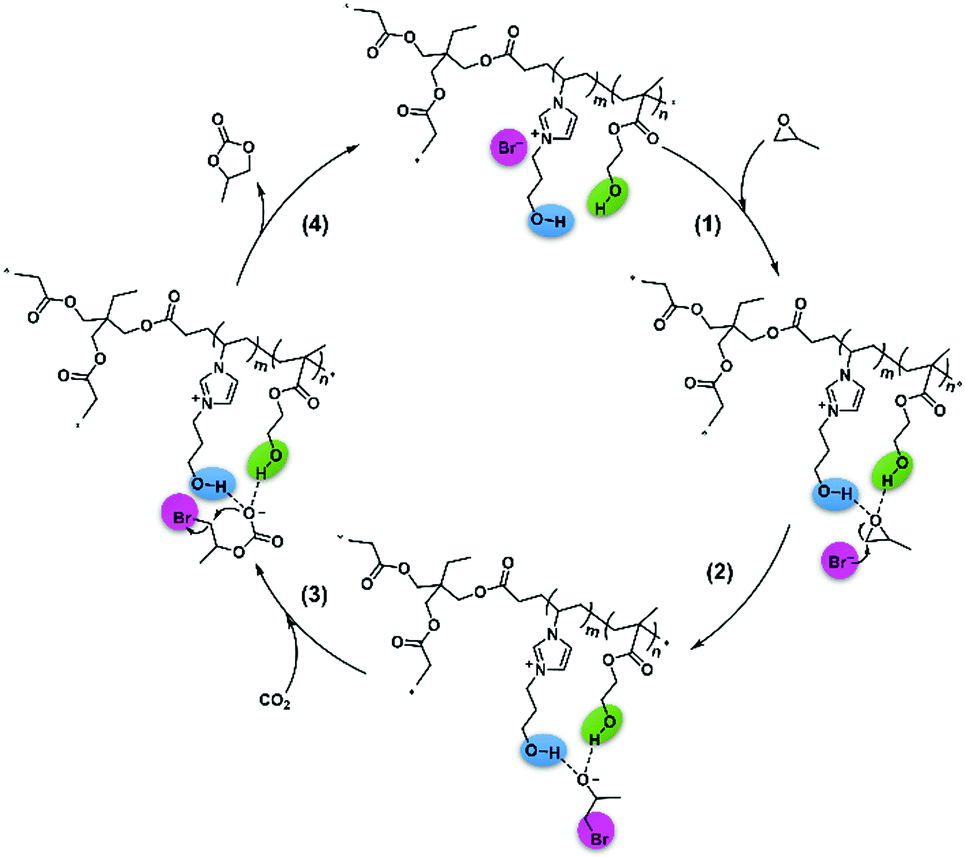 | ||
| Scheme 4 Proposed possible mechanism for CO2 cycloaddition with PO catalyzed by HPIL-7. | ||
Conclusions
In summary, we successfully implemented the hydrogen bond donors and ionic liquids into one heterogeneous catalyst through free radical polymerization. Impressively, HPIL-7 exhibited remarkable catalytic performance in the cycloaddition reaction of CO2 with epoxides. The yield of propylene carbonate (PC) reached 94%, which far exceeded the s-PIL (PC yield 72%), even approached to bulk ionic liquids (PC yield 95%). In addition, the HPIL-7 could be easily recycled from the reaction system and could be reused five times without obvious loss of activity. Further, the 1H NMR spectra and DFT calculations demonstrated that both hydroxyl of ILs and HBDs could form strong H-bonds with PO, which favour the activation of PO and accelerate the ring-opening process. A probable reaction mechanism based on the synergistic effect of HBD and nucleophilic anion in HPILs was proposed. This work reported herein provided a new way to build HBD-functionalized heterogeneous catalysts for practical conversion of CO2 into high value-added chemicals.Author contributions
Haibin Gou: Conceptualization, Data curation, Formal Analysis, Investigation, Methodology, Validation, Visualization, Writing – original draft, Writing – review & editing. Xifei Ma: Software, Writing – review & editing. Qian Su: Conceptualization, Formal Analysis, Writing – review & editing. Lei Liu: Software, Writing – review & editing. Ting Ying: Writing – review & editing. Wei Qian: Writing – review & editing. Li Dong: Funding acquisition, Project administration. Weiguo Cheng: Conceptualization, Funding acquisition, Methodology, Resources.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We sincerely acknowledge the financial support from the Innovation Academy for Green Manufacture, Chinese Academy of Sciences (No. IAGM-2019-A01), the “Transformational Technologies for Clean Energy and Demonstration”, Strategic Priority Research Program of the Chinese Academy of Sciences, Grant No. XDA 21030500 and National Natural Science Foundation of China (21890763 and 21908226)References
- T. Sakakura, J. C. Choi and H. Yasuda, Chem. Rev., 2007, 107, 2365–2387 CrossRef CAS.
- M. Mikkelsen, M. Jørgensen and F. C. Krebs, Energy Environ. Sci., 2010, 3, 43–81 RSC.
- Q. Liu, L. Wu, R. Jackstell and M. Beller, Nat. Commun., 2015, 6, 5933 CrossRef.
- S. Dabral and T. Schaub, Adv. Synth. Catal., 2019, 361, 223–246 CrossRef CAS.
- W. Sun, M. Wang, Y. Zhang, W. Ding, F. Huo, L. Wei and H. He, Green Energy Environ., 2020, 5, 183–194 CrossRef.
- Z. Dai, L. Ansaloni and L. Deng, Green Energy Environ., 2016, 1, 102–128 CrossRef.
- S. Foltran, R. Mereau and T. Tassaing, Catal. Sci. Technol., 2014, 4, 1585–1597 RSC.
- Y. Zhao, C. Yao, G. Chen and Q. Yuan, Green Chem., 2013, 15, 446–452 RSC.
- J. Q. Wang, K. Dong, W. G. Cheng, J. Sun and S. J. Zhang, Catal. Sci. Technol., 2012, 2, 1480–1484 RSC.
- Y. P. Patil, P. J. Tambade, S. R. Jagap and B. M. Bhanage, J. Mol. Catal. A: Chem., 2008, 289, 14–21 CrossRef CAS.
- N. Kihara, N. Hara and T. Endo, J. Org. Chem., 1993, 58, 6198–6202 CrossRef CAS.
- L. Guo, C. Wang, X. Luo, G. Cui and H. Li, Chem. Commun., 2010, 46, 5960–5962 RSC.
- X. Wu, C. Chen, Z. Guo, M. North and A. C. Whitwood, ACS Catal., 2019, 9, 1895–1906 CrossRef CAS.
- N. Liu, Y. Xie, C. Wang, S. Li, D. Wei, M. Li and B. Dai, ACS Catal., 2018, 9945–9957 CrossRef CAS.
- T. T. Liu, J. Liang, Y. B. Huang and R. Cao, Chem. Commun., 2016, 52, 13288–13291 RSC.
- C. A. Montoya, C. F. Gómez, A. B. Paninho, A. V. M. Nunes, K. T. Mahmudov, V. Najdanovic Visak, L. M. D. R. S. Martins, M. F. C. Guedes da Silva, A. J. L. Pombeiro and M. Nunes da Ponte, J. Catal., 2016, 335, 135–140 CrossRef CAS.
- J. J. Chen, Y. C. Xu, Z. L. Gan, X. Peng and X. Y. Yi, Eur. J. Inorg. Chem., 2019, 1733–1739 CrossRef CAS.
- R. R. Shaikh, S. Pornpraprom and V. D’Elia, ACS Catal., 2017, 8, 419–450 CrossRef.
- F. Della Monica, B. Maity, T. Pehl, A. Buonerba, A. D. Nisi, M. Monari, A. Grassi, B. Rieger, L. Cavallo and C. Capacchione, ACS Catal., 2018, 8, 6882–6893 CrossRef CAS.
- J. Zhu, P. M. Usov, W. Xu, P. J. Celis-Salazar, S. Lin, M. C. Kessinger, C. Landaverde Alvarado, M. Cai, A. M. May, C. Slebodnick, D. Zhu, S. D. Senanayake and A. J. Morris, J. Am. Chem. Soc., 2018, 140, 993–1003 CrossRef CAS.
- H. Li, X. Feng, P. Shao, J. Chen, C. Li, S. Jayakumar and Q. Yang, J. Mater. Chem. A, 2019, 7, 5482–5492 RSC.
- F. Yang, Y. Li, T. Zhang, Z. Zhao, G. Xing and L. Chen, Chem. – Eur. J., 2020, 26, 4510–4514 CrossRef CAS.
- S. Murphy, F. Ivol, A. R. Neale, P. Goodrich, F. Ghamouss, C. Hardacre and J. Jacquemin, ChemPhysChem, 2018, 19, 3226–3236 CrossRef CAS.
- J. Tharun, G. Mathai, R. Roshan, A. C. Kathalikkattil, K. Bomi and D. W. Park, Phys. Chem. Chem. Phys., 2013, 15, 9029–9033 RSC.
- H. Sabet-Sarvestani, M. Izadyar, H. Eshghi and N. Norozi-Shad, Phys. Chem. Chem. Phys., 2020, 22, 223–237 RSC.
- T. Y. Shi, J. Q. Wang, J. Sun, M. H. Wang, W. G. Cheng and S. J. Zhang, RSC Adv., 2013, 3, 3726 RSC.
- W. G. Cheng, F. Xu, J. Sun, K. Dong, C. K. Ma and S. J. Zhang, Synth. Commun., 2016, 46, 497–508 CrossRef CAS.
- W. Li, W. Cheng, X. Yang, Q. Su, L. Dong, P. Zhang, Y. Yi, B. Li and S. Zhang, Chin. J. Chem., 2018, 36, 293–298 CrossRef CAS.
- X. Meng, Z. Ju, S. Zhang, X. Liang, N. von Solms, X. Zhang and X. Zhang, Green Chem., 2019, 21, 3456–3463 RSC.
- T. Ying, X. Tan, Q. Su, W. Cheng, L. Dong and S. Zhang, Green Chem., 2019, 21, 2352–2361 RSC.
- Y. Xiong, J. Liu, Y. Wang, H. Wang and R. Wang, Angew. Chem., Int. Ed., 2012, 51, 9114–9118 CrossRef CAS.
- M. J. Earle and K. R. Seddon, Pure Appl. Chem., 2000, 72, 1391–1398 CAS.
- N. V. Plechkova and K. R. Seddon, Chem. Soc. Rev., 2008, 37, 123–150 RSC.
- H. P. Steinrück and P. Wasserscheid, Catal. Lett., 2014, 145, 380–397 CrossRef.
- X. Wang, S. Ma, B. Chen, J. Zhang, Y. Zhang and G. Gao, Green Energy Environ., 2020, 5, 138–146 CrossRef.
- T. F. Dong, Y. J. Zheng, G. W. Yang, Y. Y. Zhang, B. Li and G. P. Wu, ChemSusChem, 2020, 13, 4121–4127 CrossRef CAS.
- Y. Zhou, W. Zhang, L. Ma, Y. Zhou and J. Wang, ACS Sustainable Chem. Eng., 2019, 7, 9387–9398 CrossRef CAS.
- Y. Xie, J. Liang, Y. Fu, M. Huang, X. Xu, H. Wang, S. Tu and J. Li, J. Mater. Chem. A, 2018, 6, 6660–6666 RSC.
- J. Cao, W. Shan, Q. Wang, X. Ling, G. Li, Y. Lyu, Y. Zhou and J. Wang, ACS Appl. Mater. Interfaces, 2019, 11, 6031–6041 CrossRef CAS.
- H. Song, Y. Wang, M. Xiao, L. Liu, Y. Liu, X. Liu and H. Gai, ACS Sustainable Chem. Eng., 2019, 7, 9489–9497 CrossRef CAS.
- Y. Zhang, K. Liu, L. Wu, H. Zhong, N. Luo, Y. Zhu, M. Tong, Z. Long and G. Chen, ACS Sustainable Chem. Eng., 2019, 7, 16907–16916 CrossRef CAS.
- G. Chen, Y. Zhang, J. Xu, X. Liu, K. Liu, M. Tong and Z. Long, Chem. Eng. J., 2020, 381, 122765 CrossRef CAS.
- Y. Zhang, K. Zhang, L. Wu, K. Liu, R. Huang, Z. Long, M. Tong and G. Chen, RSC Adv., 2020, 10, 3606–3614 RSC.
- Y. Xie, Z. Zhang, T. Jiang, J. He, B. Han, T. Wu and K. Ding, Angew. Chem., Int. Ed., 2007, 46, 7255–7258 CrossRef CAS.
- L. F. Ma, Z. Z. Shi, F. F. Li, J. Zhang and L. Y. Wang, New J. Chem., 2015, 39, 810–812 RSC.
- B. A. Vara, T. J. Struble, W. Wang, M. C. Dobish and J. N. Johnston, J. Am. Chem. Soc., 2015, 137, 7302–7305 CrossRef CAS.
- Y. Jiang, J. Li, P. Jiang, Y. Li and Y. Leng, Catal. Commun., 2018, 111, 1–5 CrossRef CAS.
- Y. Xie, J. Liang, Y. Fu, J. Lin, H. Wang, S. Tu and J. Li, J. CO2 Util., 2019, 32, 281–289 CrossRef CAS.
- J. Sun, S. Zhang, W. Cheng and J. Ren, Tetrahedron Lett., 2008, 49, 3588–3591 CrossRef CAS.
- J. Q. Wang, W. G. Cheng, J. Sun, T. Y. Shi, X. P. Zhang and S. J. Zhang, RSC Adv., 2014, 4, 2360–2367 RSC.
- W. Cheng, B. Xiao, J. Sun, K. Dong, P. Zhang, S. Zhang and F. T. T. Ng, Tetrahedron Lett., 2015, 56, 1416–1419 CrossRef CAS.
- S. G. Liang, H. Z. Liu, T. Jiang, J. L. Song, G. Y. Yang and B. X. Han, Chem. Commun., 2011, 47, 2131–2133 RSC.
- M. Liu, K. Gao, L. Liang, F. Wang, L. Shi, L. Sheng and J. Sun, Phys. Chem. Chem. Phys., 2015, 17, 5959–5965 RSC.
- J. Sun, J. Ren, S. Zhang and W. Cheng, Tetrahedron Lett., 2009, 50, 423–426 CrossRef CAS.
- M. J. Frisch, G. W. Trucks and H. B. Schlegel, et al., Gaussian 09, Revision D.01, Gaussian, Inc., Wallingford CT, 2013 Search PubMed.
- A. D. Becke, J. Chem. Phys., 1993, 98, 5648–5652 CrossRef CAS.
- W. J. Hehre, R. Ditchfield and J. A. Pople, J. Chem. Phys., 1972, 56, 2257 CrossRef CAS.
- Y. Zhao and D. G. Truhlar, Theor. Chem. Acc., 2008, 120, 215–241 Search PubMed.
- R. Krishnan, J. S. Binkley, R. Seeger and J. A. Pople, J. Chem. Phys., 1980, 72, 650–654 CrossRef CAS.
- T. Clark, J. Chandrasekhar, G. Spitznagel and P. Schleyer, J. Comput. Chem., 1983, 4, 294–301 CrossRef CAS.
- M. J. Frisch, J. A. Pople and J. S. Binkley, J. Chem. Phys., 1984, 80, 3265–3269 CrossRef CAS.
- Z. Guo, Q. Jiang, Y. Shi, J. Li, X. Yang, W. Hou, Y. Zhou and J. Wang, ACS Catal., 2017, 7, 6770–6780 CrossRef CAS.
- T. Biswas and V. Mahalingam, Sustainable Energy Fuels, 2019, 3, 935–941 RSC.
- Y. Zuo, Z. Jiao, L. Ma, P. Song, R. Wang and Y. Xiong, Polymer, 2016, 98, 287–293 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0cp06041k |
| This journal is © the Owner Societies 2021 |