
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceExperimental and theoretical study of the reactivity of a series of epoxides with chlorine atoms at 298 K†
Carmen M.
Tovar
 *a,
Alexander
Haack
b,
Ian
Barnes
a,
Iustinian Gabriel
Bejan
*c and
Peter
Wiesen
a
*a,
Alexander
Haack
b,
Ian
Barnes
a,
Iustinian Gabriel
Bejan
*c and
Peter
Wiesen
a
aInstitute for Atmospheric and Environmental Research, University of Wuppertal, 42097 Wuppertal, Germany. E-mail: carmen.tovar_ramos@uni-wuppertal.de
bPhysical and Theoretical Chemistry, University of Wuppertal, 42097 Wuppertal, Germany
c“Alexandru Ioan Cuza” University of Iasi, Faculty of Chemistry and Integrated Center of Environmental Science Studies in the North-Eastern Region – CERNESIM, 700506, Iasi, Romania. E-mail: iustinian.bejan@uaic.ro
First published on 19th February 2021
Abstract
Evaluating the reactivity of epoxides in the gas phase is very important due to their wide distribution in the atmosphere, potential health implications and atmospheric impact. The kinetic rate constants for the oxidation of epoxides have been very little studied until now. From the experimental data obtained in this work has been observed that there is an increase in reactivity towards chlorine atoms as a CH2 group is added to the hydrocarbon chain. The Structure Activity Relationship (SAR) method usually provides a good approximation of the rate constant for a wide series of compounds especially for those without complex structure and multiple organic functions. However, a good determination of the factors included in SAR estimations depends largely on the database of these compounds, which in the case of epoxides is very limited. The SAR estimation method also does not take into account other possible factors that could affect reactivity, such as the geometry of the molecule. The aim of this work is to further evaluate the reactivity of epoxides with chlorine atoms using experimental determinations, theoretical calculations and SAR estimations. For this, rate coefficients have been measured at 298 ± 2 K and 1000 ± 4 mbar pressure of synthetic air in a 1080 l Quartz Reactor (QUAREC) and a 480 l Duran glass reactor for the reaction of chlorine atoms with cyclohexene oxide (CHO), 1,2-epoxyhexane (12EHX), 1,2-epoxybutane (12EB), trans-2,3-epoxybutane (tEB) and cis-2,3-epoxybutane (cEB). Theoretical calculations for the reactions studied are in good agreement with our experimental findings and provide insights about the position of the H atom abstraction and reactivity trends for a series of epoxides. The importance of taking into consideration the geometrical distribution and the ring influence to improve SAR calculations is discussed.
1 Introduction
Epoxides are the simplest known oxygen-containing heterocycles. They are widely distributed in the atmosphere from anthropogenic sources and have important applications in chemical industries due their strong proton affinity.1 They are often used as intermediates in the synthesis of various industrial polymers, and also in electroplating, lithium batteries, polymers and gasoline production.2–5Furthermore, cycloaliphatic epoxides like cyclohexene oxide (CHO), are one of the major types of epoxy resins utilized in coatings industry.6
Epoxides are used as well in fine chemistry, in the production of pharmaceuticals, perfumery, plasticizers and pesticides.7,8 CHO can be enantio-selectively converted into chiral 1,2-aminocyclohexanol or 1,2-diaminocyclohexane, which are both building blocks for the preparation of natural and synthetic biologically active molecules like amino acids.9,10
Whereas, 1,2-epoxyhexane (12EHX) is used as a passivating agent for core–shell aluminium nanoparticles,11 1,2-epoxybutane (12EB) has applications in the chemistry of the modification of carboxylic acid ends groups in swollen polymer particles (e.g. in poly butylene terephthalate, PBT) in supercritical fluids12 and is applied in the production of surfactants and as stabilizer for chlorinated hydrocarbon solvents and gasoline additives.13
Cyclic ethers are also formed in the initial reactions of alkyl radicals with molecular oxygen in combustion and pre-combustion processes that occur at moderate temperatures. They represent a significant part of the oxygenated pollutants found in the exhaust gases of engines.14 Cyclohexene oxide has been detected in the oxidation and auto-ignition of cyclohexene by rapid compression between 600 K to 900 K and 0.7 MPa to 1.4 MPa.15
Recent studies have shown an increasing interest in the atmospheric oxidation processes with chlorine atoms.16
Epoxides contribute in significant amounts to reaction products of the chemical degradation of VOCs in the atmosphere. The formation of cyclic ethers is considered as an important product in the OH radical initiated oxidation of aromatic compounds,17 isoprene18 in the ozonolysis of furans,19 sesquiterpenes20 and biomass derived alcohols.21
Isoprene-derived epoxides can make up a major proportion of the atmospheric SOA in many continental local areas.22,23
A recent study has reported the annual global isoprene emission for about 500 to 750 Tg isoprene (440 to 660 Tg carbon) using MEGAN ranges.24 Extensive studies of SOA formation in the south-eastern United States have identified epoxide intermediates as key species in the formation of isoprene-derived SOA.18,25–30 Global simulations estimate a flux of up to 100 Tg of carbon per year of the dihydroxyepoxides derived from isoprene photooxidation.18
The atmospheric degradation mechanism for many compounds is still unclear, with poor carbon balance.
Prior to evaluating the impact that these compounds have on the atmosphere, it is important to know their reactivity and structural properties, which can be key in the elucidation of reaction mechanisms.
Previous studies suggest that the addition of an ether linkage to an alkane leads to a weakening of the C–H bonds adjacent to the ether site. Whether the enhancement in reactivity is restricted to the C–H bonds located near the ether linkage, or whether the enhancement extends further down the molecule is difficult to ascertain given the data currently available in a series of aliphatic ethers with OH, Cl and NO3.31
Our goal in this work is to gain insight into how the geometrical distribution and ring tension can influence the reactivity of epoxides towards chlorine atoms. Five representative epoxy compounds were selected to develop kinetic calculations to evaluate the reactivity along the hydrocarbon chain. Later, theoretical calculations were developed to deepen the influence of the ring and ether group but considering the geometrical configuration of the molecule, which cannot be properly evaluated with experimental measurements alone.
These insights could improve future SAR calculations and explain the reactivity of epoxide in a more accurate way. These observations will also be useful to feed the atmospheric models for this kind of compounds that have been until now very little studied. To the best of our knowledge, this is the first kinetic and theoretical investigation of these reactions.
2 Methods
2.1 Experimental methods
Experiments have been performed in a 1080/Quartz reactor (6 m) and a 480/Duran glass reactor (3 m) under the following conditions: (298 ± 2) K and (1000 ± 4) mbar total pressure of synthetic air. Fourier Transform Infrared Spectroscopy (FTIR) equipped with a liquid nitrogen cooled mercury–cadmium–telluride detector with a resolution of 1 cm−1 has been employed for reactants monitoring.The 3 m reactor consists of a cylindrical Duran glass vessel (3 m length, 45 cm dia.) closed at both ends by Teflon coated aluminium end flanges. Integrated on the metal flanges are ports for the inlet of reactants into the reactor and for the collection of samples from the reaction mixtures for further analysis. Other accessories, like a mixing fan to ensure homogeneity of the reaction mixtures and a capacitance manometer, are also located on the flanges. Arranged concentrically around the outside of the reactor are 32 super actinic fluorescent lamps (Philips TLA 40 W/05, 300 ≤ λ ≤450 nm, λmax = 360 nm). The vacuum (ca. 10−3 mbar) is maintained by means of a Leybold turbo-molecular pump, model RUVAC WZ 151 (500 m3 h−1), backed by a Leybold double stage rotary fore pump, model D40B (200 m3 h−1). The reactor is equipped with built-in White mirror systems, which were usually operated at total path lengths of 51.6 m for the acquisition of the IR spectra.
Barnes et al.32 provided a detailed description of the 6 m reactor, and in a more recent publication33 the system has been described as follows: the reactor consists of two quartz glass tubes with an inner diameter of 47 cm and a wall thickness of 5 mm. The reactor can be evacuated to 10−3 mbar using a pumping system consisting of a turbo-molecular pump backed by a double stage rotary fore pump. Homogeneous mixing of the reactants in the reactor is ensured by magnetically coupled Teflon mixing fans located inside the reactor. Two types of lamps are available to photo-dissociate the radical/atom precursors: 32 super actinic fluorescent lamps (Philips TL 05/40 W: 320 < λ < 480 nm, λmax = 360 nm) and 32 low-pressure mercury lamps (Philips TUV/40 W, λmax = 254 nm). The lamps are wired in parallel and can thus be switched individually. They are distributed evenly around the photo reactor to ensure homogeneous irradiation within the reactor. A White type multiple-reflection mirror system set at a total optical path length of (484.7 ± 0.8) m is mounted inside the photo reactor. This optical system was used for sensitive monitoring of reactants and products in the IR spectral range 4000–700 cm−1. A Nicolet Nexus FT-IR spectrometer equipped with a KBr beam splitter and a liquid nitrogen cooled mercury–cadmium–telluride (MCT) detector is used to record IR spectra with a spectral resolution of 1 cm−1.
A Globar was used as IR light source. All mirrors are gold coated to increase the reflectivity efficiency. Typically, 60 interferograms were co-added per spectrum over a period of approximately 1 minute and 15–20 such spectra were recorded per experiment.
2.2 Relative rate method
Rate coefficients for the reactions of Cl atoms with epoxides were determined using the relative rate method34 in which the relative disappearance rates of the epoxides compared with different reference compounds were monitored in the presence of Cl atoms.The photolysis of Cl2 with the fluorescent lamps was used to generate Cl atoms:
| Cl2 + hν (320 < λ < 480 nm) → 2Cl | (1) |
Reaction mixtures consisting of a reference organic compound, the sample epoxide and the radical precursor compounds, diluted in synthetic air, were prepared in the reactor and left to mix prior to photolysis for approximately 15 min. Measured amounts of the reagents were flushed into the reactor under reduced pressure by a stream of synthetic air and the reactor was then filled with synthetic air to atmospheric pressure.
Typical photolysis times ranged from 15 to 20 min. In the presence of the chlorine atoms the epoxide under investigation and the reference compound are consumed by the following reactions:
| Cl + Epoxide → Products (kepoxide) | (2) |
| Cl + Reference → Products (kreference) | (3) |
Provided that the reference compound and the epoxides are lost only by reactions (2) and (3), then it can be shown that:
 | (I) |
In order to test for possible loss of the reactants through photolysis and wall deposition, mixtures of the reactants in nitrogen or air in the absence of chlorine were irradiated for 30 min. Photolysis and wall deposition was found to be negligible for all epoxides and the reference compounds. Additionally, various experiments were performed to assess possible loss of the reactants and references via reaction with molecular chlorine. No significant direct reaction of the epoxides with molecular chlorine was observed on leaving the compounds in the dark in the reactor for at least the reaction time duration.
The initial mixing ratios of the oxidant precursor molecular chlorine in the experiments performed in the 6 m reactor, were typically ∼5 ppmv, and ∼42 ppmv in the 3 m reactor. The initial mixing ratios for the epoxides and reference compounds are shown in Table 1 (ESI†).
| Epoxide | Structure | Reference | K epoxide/kreference | k epoxide (cm3 molecule−1 s−1) |
|---|---|---|---|---|
| Cyclohexene oxide |

|
Propylene | 0.803 ± 0.007 | (2.17 ± 0.24) × 10−10 |
| Ethylene | 2.394 ± 0.073 | (2.63 ± 0.72) × 10−10 | ||
| Average | (2.40 ± 0.76) × 10−10 | |||
| 1,2-Epoxyhexane |

|
Propylene | 0.707 ± 0.022 | (1.91 ± 0.22) × 10−10 |
| Ethylene | 1.821 ± 0.042 | (2.00 ± 0.55) × 10−10 | ||
| Methacrolein | 0.539 ± 0.006 | (1.40 ± 0.16) ×10−10 | ||
| Average | (1.77 ± 0.61) × 10−10 | |||
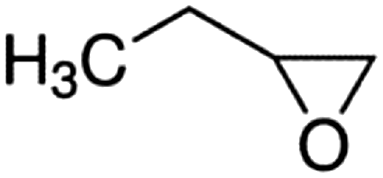
| 1,2-Epoxybutane |
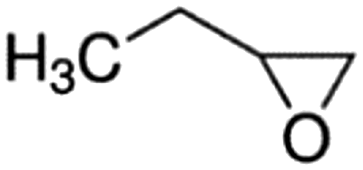
|
Propylene | 0.324 ± 0.013 | (8.65 ± 1.02) × 10−11 |
| Ethylene | 0.746 ± 0.012 | (8.21 ± 2.24) × 10−11 | ||
| Average | (8.43 ± 2.46) × 10−11 | |||
| trans-2,3-Epoxybutane |

|
Propylene | 0.261 ± 0.053 | (7.03 ± 0.79) × 10−11 |
| Ethylene | 0.633 ± 0.009 | (6.96 ± 1.91) × 10−11 | ||
| Average | (6.99 ± 2.06) × 10−11 | |||
| cis-2,3-Epoxybutane |

|
Propylene | 0.247 ± 0.007 | (6.66 ± 0.77) × 10−11 |
| Ethylene | 0.652 ± 0.009 | (7.17 ± 1.96) × 10−11 | ||
| Average | (6.92 ± 2.11) × 10−11 | |||
All epoxy and reference compounds used in this study were obtained from Sigma Aldrich and used without further purification. The stated purities were as follows: cyclohexene oxide 98%; 1,2-epoxyhexane 97%; 1,2-epoxybutane 99%; trans-2,3-epoxybutane 97%; cis-2,3-epoxybutane 97%; propylene 99%; methacrolein 95%; butane 99% and ethylene 99,5%. Synthetic air has been provided by Messer GmbH with a 99.999%.
2.3 Computational methods
All calculations were conducted with the Gaussian16 program package.35 GaussView636 together with a standard text editor were used to create input files and analyse output files. We used density functional theory (DFT) applying the B3-LYP37,38 exchange–correlation function with Grimme's GD3-BJ empirical dispersion corrections39 to account for long range interactions. We chose the def2-TZVPP40 basis set for all atoms because of its generality and good triple-zeta quality. All SCF energies were converged within a threshold of 10−8 Hartree, corresponding to “tight convergence”.All stationary geometries, i.e., minimum as well as transition state structures, were optimized with “tight convergence” criteria (in Hartree Bohr−1: Max force = 1.5 × 10−5, RMS force = 1.0 × 10−5, Max displacement = 6.0 × 10−5 and RMS displacement = 4.0 × 10−5) and normal mode were optimized with tight convergence criteria and normal mode analysis was performed to check the correct number of imaginary frequencies (zero for minima, one for transition states). It was also checked, whether the movement along the imaginary mode in the transition state structures corresponds to the expected reaction path. The harmonic frequencies of the normal modes were used to calculate thermochemical data, i.e. free reaction enthalpies, ΔRG.
Potential energy surface (PES) scans, using the same level of theory (except that the default geometry convergence criteria), were conducted to either find initial guesses for transition states or minimum energy paths for the H-abstraction by the chlorine atom. This was done by gradually changing one coordinate (e.g. elongation or shortening of a bond distance) while allowing all other geometrical parameters to relax. For the H-abstractions, however, some angles were held constant to model, for example, a linear approach of the chlorine atom onto the C–H bond.
3 Results and discussion
Fig. 1 shows the exemplary plots of the rate coefficients of cyclohexene oxide (CHO), 1,2-epoxyhexane (12EHX), 1,2-epoxybutane (12EB), trans-2,3-epoxybutane (tEB) and cis-2,3-epoxybutane (cEB) according to eqn (I), obtained for the gas-phase reaction of chlorine atoms with different reference compounds using 3 m reactor. Very good linear plots with almost zero intercept were obtained for all epoxy compounds, which confirms that no interferences are indicated in the rate constant determination. | ||
| Fig. 1 Rate coefficients for the reaction of chlorine atoms with (a) cyclohexene oxide; (b) 1,2-epoxy hexane; (c) cis-2,3-epoxybutane; (d) trans-2,3-epoxybutane using ethylene, methacrolein and propylene as reference compounds. | ||
The average rate coefficient ratios (kepoxide/kreference) obtained from the slopes of these plots from a minimum of at least two experiments for each compound are given in. Very good agreement between the rate coefficients obtained using two or three different reference hydrocarbons have been observed. Fig. 2 shows the experiments performed for 1,2-epoxybutane in the presence of chlorine atoms using both reactors. It is worth mentioning that the rate coefficients obtained were almost identical.
 | ||
| Fig. 2 Rate coefficients for the reaction of 1,2-epoxybutane towards chlorine atoms with ethylene and propylene as reference compounds (using 6 m and 3 m reactors). | ||
The errors quoted in for the average kepoxide include the 2σ statistical error from the linear regression analyses of the plots with an additional 35% uncertainty contribution in the recommended values of the reaction rate coefficients for the reaction of the reference compounds with chlorine.31
Table 2 compares the experimental rate coefficients with the values obtained by a SAR estimation method and those found in the literature for other epoxy compounds. The third column in Table 2 shows the rate coefficient, kSAR, predicted from the structure–reactivity relationship (SAR) method41 that has been tentatively applied for the epoxides studied by using the factor F(O) = 1.1842,43 and F(O) = 1.45.31 This factor corresponds to the calculated influence of the ether group on the rate constant. In the SAR method, the calculation of the rate constants depends only on the identity of the substituents bonded to these groups.42 As can be seen in Table 2 the SAR predicted values for the rate coefficients are in reasonable agreement with the experimentally determined values of the present study.
| Epoxides | Structure | k SAR | k epoxide (cm3 molecule−1 s−1) | Technique | Ref. |
|---|---|---|---|---|---|
| Calculated using the SAR method41 and substituent factor for the ether group F(O) from:a Ref. 42.b Ref. 31.c Ref. 43. Highlighted values have been used to determine SAR parameters. | |||||
| Cyclohexene oxide |

|
2.09 × 10−10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) a a |
(2.40 ± 0.76) × 10 −10 | RR-FTIR | This work |
| 2.31 × 10−10b |
|||||
| 2.08 × 10−10c |
|||||
| Ethylene oxide |

|
3.10 × 10−12a |
(2.9 ± 0.24) × 10−12 | RR(C2H6) | 49 |
| 4.15 × 10−12b |
(4.6 ± 0.60) × 10−12 | RR(C2H5Cl) | 50 | ||
| 7.25 × 10−10c |
(5.2 ± 0.60) × 10−12 | RR(CH3OCHO) | 50 | ||
| (4.9 ± 0.63) × 10 −12 | Average | 31 | |||
| Propylene oxide |

|
2.89 × 10−11a |
(3.0 ± 0.70) × 10 −11 | RR-FTIR | 43 |
| 2.63 × 10−11b |
|||||
| 2.62 × 10−10c |
|||||
| 1,2-Epoxyhexane |

|
1.98 × 10−10a |
(1.77 ± 0.61) × 10 −10 | RR-FTIR | This work |
| 2.12 × 10−10b |
|||||
| 1.84 × 10−10c |
|||||
| 1,2-Epoxybutane |

|
9.45 × 10−11a |
(8.43 ± 2.46) × 10 −11 | RR-FTIR | This work |
| 9.75 × 10−11b |
|||||
| 9.21 × 10−10c |
|||||
| trans-2,3-Epoxybutane |
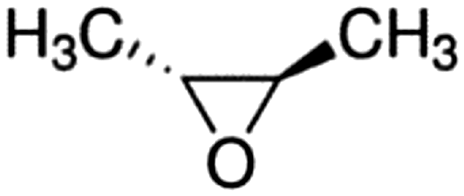
|
5.47 × 10−11a |
(6.99 ± 2.06) × 10 −11 | RR-FTIR | This work |
| 4.84 × 10−11b |
|||||
| 5.25 × 10−10c |
|||||
| cis-2,3-Epoxybutane |

|
5.47 × 10−11a |
(6.92 ± 2.11) × 10 −11 | RR-FTIR | This work |
| 4.84 × 10−11b |
|||||
| 5.25 × 10−11c |
|||||
From the rate coefficients in Table 2 it can be derived that the methyl group (i.e. propylene oxide) added to the cyclic ether moiety (ethylene oxide) increases up to 6 times the reactivity of the epoxy compounds. Furthermore, 2,3-epoxybutane shows a doubled reactivity towards chlorine atoms by comparison with propylene oxide. This enhanced reactivity can be attributed mainly to the additional methyl group attached to the ethylene oxide heterocyclic ring.
Table 3 shows the rate coefficients of the reaction of different ethers and epoxides towards chlorine atoms, where ethers and epoxides of the same number of carbons, increase their reaction rate coefficient as the number of CH2 groups adjacent to ether group increases.


In order to try to elucidate the reason for this reactivity trend in the epoxy compounds we calculated thermochemical data and minimum energy paths on the potential energy surface (PES) for each compound, which give us an important insight about the likelihood of each H abstraction reaction by chlorine atoms. For the PES scans, we picture the potential energy as a function of the difference of the Cl–H and H–C distances, which gives us a coordinate covering the two important geometrical parameters that change during the reaction. Values smaller than zero correspond to a long Cl–H and short H–C bond (reactant channel), values larger than zero correspond to a short Cl–H and long H–C bond (product channel).
For the following discussion, we will consider that for cis-2,3-epoxybutane and trans-2,3-epoxybutane H1 is the hydrogen atom of the CH group directly attached to the ring (position α) and H2 is one of the hydrogen atoms of the CH3 group next to the ring (position β). In the case of the 1,2-epoxybutane, we will use H0 for one of the hydrogen atoms of the CH2 group directly attached to the ring. Similarly, H1 is the hydrogen atom of the CH group directly attached to the ring (position α) and H2 is the H atom abstracted for the CH2 next to the ring (position β) in the aliphatic chain (Fig. 3).
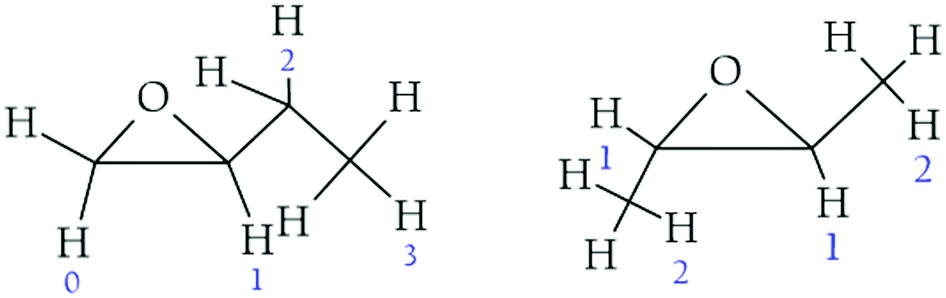 | ||
| Fig. 3 Diagram of the possible hydrogen atoms considered for the abstraction channels. | ||
The H atom abstraction paths for H1 and H2 of cis-epoxybutane and trans-epoxybutane are shown in Fig. 4. Firstly, it can be observed that decreasing the Cl–H distance is accompanied by a monotonically decrease of the potential energy from its asymptotic value and no TS is visible. This is true for all abstraction reactions considered here. This is why the discussion about the rate constants have to focus on the form of the minimum energy paths and the energies of the reactant and product channels.
 | ||
| Fig. 4 Potential energy surface (PES) scans, for the H atom abstraction from cis/trans-2,3-epoxybutane by chlorine atoms. | ||
Comparing the shape of the reaction paths for the H1 and H2 abstractions, it can be noticed that the former has only one minimum lying in the reactant channel and the latter has two, each in the reactant and product channels. Assuming an approach of Cl and the formation of a metastable pre-reactive complex, in the H1 case, it will mostly remain in the reactant channel and in the H2 channel there will be a distribution between the two minima, slightly favouring the product channel one due to its lower energy. If the pre-reactive complex falls apart, this configuration will strongly influence in which direction the reaction occurs. Considering also the energy of both channels leads to the conclusion that for H1 the abstraction reaction is less likely than for H2 because in the latter the product channel is more populated and the energy of the products is lower than of the reactants.‡
Regarding the difference between cis- and trans-epoxybutane the PES scans show no significant difference for both H1 and H2, which is in good agreement with the experimental findings, i.e. that the rate constants are almost identical. This is supported by the Gibbs free reaction enthalpies for the cis and trans isomer, which are −22 kJ mol−1 and −25 kJ mol−1 for H1 and −30 kJ mol−1 and −32 kJ mol−1 for H2, respectively.
The reason for the second minimum and for the lower energy of the products can be explained by the Walsh model.44–46 It is founded in the premise that carbon atoms in ethylene oxide approach the sp2 state, its CH2 units being therefore pseudo-ethylenic in character.47
It has also been shown that such conjugation does not occur if the π orbitals of the unsaturated groups cannot orient themselves with their axes parallel to the plane of the epoxide ring48 (Fig. 5). It follows, therefore, that the orbitals in the epoxide ring, which are responsible for the conjugation, must lie in the plane of the ring (and not above and below as in the benzene case).
 | ||
| Fig. 5 Geometry of trans-2,3-epoxybutane showing the plane of the epoxy ring associated to the conjugation. | ||
Accordingly, with the Walsh configuration, one orbital from each carbon atom overlap with an atomic orbital of oxygen to form the molecular orbital, and two electrons of oxygen move into this orbital. The remaining four electrons populate the delocalized molecular orbitals, which are in the plane of the ring. These latter orbitals are formed by sideways overlap of atomic p orbitals, and their presence accounts for the conjugating ability of the epoxide ring.51
Coulson and Moffit proposed a similar structure. They purposed a structure that was called a “bent-bond”. In this structure the ring atoms do not lie along the lines of greatest electron density of the atomic orbitals from the neighbouring atoms.52,53 If we consider the case of cis/trans-epoxybutane, when the compound has lost one H of the CH3 group, it becomes trigonally (sp2) hybridized, facilitating the possible formation of a double bond with the singly-occupied p-orbital after abstraction of H. Indeed, the optimized geometry shows the CH2 group aligning in such a way that the three hydrogens and two carbons lie within one plane indicating a double bond character of the C–C bond (Fig. 5). This aspect could explain the favoured abstraction of H2 (β-position).
The effect of the side chain can be studied by comparing the minimum energy paths of cis/trans-epoxybutane and 1,2-epoxybutane (Fig. 6). The same effects observed above can be seen for the asymmetric compound: only the H2 abstraction path has a (significant) minimum in the product channel and the product energy is much lower than for the other channels indicating a strong preference toward an abstraction in β-position. In particular, an abstraction of H0 is very unfavourable since in addition to the weak population of the product channel, the energy of the products is even higher than for H1.
 | ||
| Fig. 6 Potential energy surface (PES) scans, for the H atom abstraction from 1,2-epoxybutane by chlorine atoms. | ||
Comparing now the paths for the H2 abstraction, it can be seen that the second minimum (lying in the product channel) is significantly lowered in the asymmetric compound compared to the symmetric ones. The metastable reaction adduct thus strongly populates the product channel increasing the likelihood of the abstraction. This is in agreement with the experimental data, where the rate constant for the 1,2-epoxybutane is significantly larger than for the cis/trans isomers. This effect could be explained by a stabilizing effect of the adjacent CH3 group, which can contribute to the π-interaction through its own p-orbitals to a certain degree (+I effect). The preference of the H2 abstraction is also supported by the thermochemistry of the products. The Gibbs free reaction enthalpies for H0a/b, H1 and H2 are −14 kJ mol−1, −14 kJ mol−1, −22 kJ mol−1 and −50 kJ mol−1, respectively.
We did not calculate energy paths for 1,2-epoxyhexane because of the difficulties due to its flexible nature. However, the increased rate constant (Table 2) compared to 1,2-epoxybutane can be explained by the longer sidechain and thus a stronger +I effect. Additionally, hydrogens in γ, δ,… position can be abstracted as well, increasing the overall rate. The likelihood of these reactions will be discussed below.
Having a cyclic compound, i.e. cyclohexene oxide (Fig. 7), the rate constant increases again compared to 1,2-epoxyhexane, despite having the same number of carbons. From Fig. 8 it becomes clear that, again, H2 abstraction is favoured over H1 abstraction (e and a describe atoms in equatorial and axial position). The side chain again contributes to an energetically low product channel minimum having thus a high population density. Terming H3 the H atoms of the CH2 group in γ-position, it can be seen that they have two minima as well. Additionally, their exit channel is energetically lower than the reactant asymptote, hence, although not as likely as the H2 abstraction, this pathway is also feasible, contributing to the overall rate. Assuming this behaviour being similar in 1,2-epoxyhexane would explain the larger rate constant compared to 1,2-epoxybutane. However, since cyclohexene oxide has four H atoms in β-position compared to only two in 1,2-epoxyhexane, the increased rate from linear to cyclic can be explained as well.
 | ||
| Fig. 7 Geometries of the cis conformer of cyclohexene oxide during the H abstraction reaction in the presence of chlorine atoms, optimized under the B3LYP-GD3BJ/def2-TZVPP. The coordinates of all structures can be found in the ESI.† | ||
 | ||
| Fig. 8 Potential energy surface (PES) scans, for the H atoms abstraction from cyclohexene oxide by chlorine atoms. | ||
We investigated further reaction steps for the decomposition of cyclohexene oxide as can be seen in Fig. 9. Experiments are being developed to analyse the decomposition products as well, giving us the opportunity to test the above discussions by comparing the measured and calculated decomposition products.
 | ||
| Fig. 9 Pathways of H atom abstractions from the cis conformer of cyclohexene oxide (CHO) reacting with chlorine atoms, see Fig. 7 for the explanation of acronyms. | ||
The intramolecular changes now all proceed through TSs and hence their likelihood can be discussed with the concept of these barrier heights. In Fig. 9, all pathways containing an “A” corresponds to opening of the epoxide ring (breaking of a C–O bond) while “B” containing paths correspond to opening of the carbon ring (breaking of a C–C bond). Following the H2 abstraction, the epoxide ring opens through a low TS to form the compound H2-A. Although the compound H1-A is very stable due to the delocalization of the free electron over three atoms (see Fig. 7), we argue that due to kinetic effects regarding first the H abstraction and second the epoxide opening, the H2-A compound is more likely to be observed in experimental data. An H3 abstraction can only be followed by high transition states making progression along this pathway questionable. We thus believe, that the H2-A compound will be the most likely one, probably further decomposing into the H2-AB1 products having a conjugated π system over four atoms (see Fig. 7).
In terms of reactivity trends, the CH2 groups near to the ring are prone to form double bonds after the abstraction of a hydrogen atom and then orient themselves easily with their axes of π orbitals parallel to the plane of the epoxide ring, making the reaction faster. Besides, if we compare the reactivity of cyclohexene oxide with 1,2-epoxyhexane with the same number of C atoms, we can see that kCHO > k12EHX (Table 2). Also, previous studies shows that the electron density in the oxygen atom in ethylene oxide is unusually low compared to that in acyclic and larger cyclic ethers,54,55 that could add to the explanation why the reactivity of cyclohexene oxide is higher.
Conclusions
From the results of this work it can be seen that ethers and epoxides of the same number of carbon atoms increase their reaction rate coefficient as the number of CH2 groups adjacent to ether group increases.According to the Structure–Activity Relationships (SAR) method, the rate coefficients at a primary, secondary and tertiary site on an organic species are modified by substituent factors F(X) corresponding to different functional groups that account for the activating or deactivating effects of neighbouring groups. Comparing the reactivity between aliphatic ethers and epoxides, there is a sharp decrease in the reactivity that could be attributed to the effect of the ring.
Through the theoretical calculations performed in this work for epoxides up to 6 carbon atoms, we present a possible explanation to the reactivity trends observed experimentally and the importance of the selectivity in the reaction of epoxides towards chlorine atoms. The presence of the ring will affect the molecule in a complex manner, and both, ring and ether group have to be considered as a unity to properly evaluate the reactivity of these compounds. Therefore, as explained before, the sites in the molecule are more reactive in β-position when the carbon atoms in ethylene oxide approach the sp2 state and its CH2 units are pseudo-ethylenic in character. That conjugation does not occur if the π orbitals of the unsaturated groups cannot orient themselves with their axes parallel to the plane of the epoxide ring. Such a geometric arrangement in the epoxy molecule would be necessary to allow the abstraction of a hydrogen in the molecule in a preferential way, thus increasing the reactivity of the reaction, as demonstrated here for the molecules under study.
Further investigation about the reaction mechanism products of these compounds will help further improve the knowledge of the nature and reactivity of this type of compounds.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors acknowledge the financial support provided by the European Union's Horizon 2020 research and innovation programme, through the EUROCHAMP-2020 Infrastructure Activity Grant (grant agreement no. 730997). IGB acknowledges the PN-III-P4-ID-PCE-2016-4-0807 and PN-III-P2-2.1-PED2019-4972 UEFISCDI projects. CMT wish to acknowledge to FANTEL for a PhD scholarship.References
- S. Malik, D. Sharma and V. K. Sharma, Topological investigations of mixtures containing cyclic ether and cyclic alkanones, J. Mol. Liq., 2016, 223, 1158–1171 CrossRef CAS.
- K. M. Abraham, J. L. Goldman and D. L. Natwig, Characterization of Ether Electrolytes for Rechargeable Lithium Cells, J. Electrochem. Soc., 1982, 129, 2404 CrossRef CAS.
- C. Pang, F. Ding, W. Sun, J. Liu, M. Hao and Y. Wang, et al., A novel dimethyl sulfoxide/1,3-dioxolane based electrolyte for lithium/carbon fluorides batteries with a high discharge voltage plateau, Electrochim. Acta, 2015, 174, 230–237 CrossRef CAS.
- F. B. Belaribi, N. Abdouche, A. Boussebissi, F. Amireche and G. Boukais-Belaribi, Excess molar enthalpies of binary mixtures of n-octane, isooctane and cyclooctane with morpholine, 1,4-dioxane, piperidine, oxane, N-methyl piperidine and cyclohexane. Experimental results and DISQUAC modelling, J. Mol. Liq., 2015, 212, 650–655 CrossRef CAS.
- S. Altway, M. Taha and M. J. Lee, Liquid + liquid), (solid + liquid), and (solid + liquid + liquid) equilibria of systems containing cyclic ether (tetrahydrofuran or 1,3-dioxolane), water, and a biological buffer MOPS, J. Chem. Thermodyn., 2015, 82, 93–98 CrossRef CAS.
- T. J. Wallington, P. Dagaut and M. J. Kurylo, Correlation between gas-phase and solution-phase reactivities of hydroxyl radicals towards saturated organic compounds, J. Phys. Chem., 1988, 92, 5024–5028 CrossRef CAS.
- S. T. Oyama, Rates, Kinetics, and Mechanisms of Epoxidation: Homogeneous, Heterogeneous, and Biological Routes. in Mechanisms in Homogeneous and Heterogeneous Epoxidation Catalysis, ed. S. T. Oyama, Amsterdam, Elsevier, Chapter 1, 2008. pp. 3–99 Search PubMed.
- G. Grigoropoulou, J. H. Clark and J. A. Elings, Recent developments on the epoxidation of alkenes using hydrogen peroxide as an oxidant, Green Chem., 2003, 5, 1–7 RSC.
- D. J. Ager, I. Prakash and D. R. Schaad, 1,2-Amino Alcohols and Their Heterocyclic Derivatives as Chiral Auxiliaries in Asymmetric Synthesis, Chem. Rev., 1996, 96, 835–876 CrossRef CAS.
- C. A. D. Parrodi and E. Juaristi, Chiral 1,2-Amino Alcohols and 1,2-Diamines Derived from Cyclohexene Oxide: Recent Applications in Asymmetric Synthesis, Synlett, 2006, 2699–2715 CrossRef.
- P. A. Jelliss, S. W. Buckner, S. W. Chung, A. Patel, E. A. Guliants and C. E. Bunker, The use of 1,2-epoxyhexane as a passivating agent for core–shell aluminum nanoparticles with very high active aluminum content, Solid State Sci., 2013, 23, 8–12 CrossRef CAS.
- M. Pagliaro, Supercritical Carbon Dioxide in Polymer Reaction Engineering, in Angewandte Chemie International Edition, ed. M. F. Kemmere and T. Meyer, 2006, vol. 45, pp. 6079–6079 Search PubMed.
- IARC. Re-evaluation of Some Organic Chemicals, Hydrazine and Hydrogen Peroxide (Part 1, Part 2, Part 3).
- I. Auzmendi-Murua and J. W. Bozzelli, Thermochemical Properties and Bond Dissociation Enthalpies of 3- to 5-Member Ring Cyclic Ether Hydroperoxides, Alcohols, and Peroxy Radicals: Cyclic Ether Radical + 3O2 Reaction Thermochemistry. The, J. Phys. Chem. A, 2014, 118, 3147–3167 CrossRef CAS.
- O. Lemaire, M. Ribaucour, M. Carlier and R. Minetti, The production of benzene in the low-temperature oxidation of cyclohexane, cyclohexene, and cyclohexa-1,3-diene, Combust. Flame, 2001, 127, 1971–1980 CrossRef CAS.
- M. R. McGillen, W. P. L. Carter, A. Mellouki, J. J. Orlando, B. Picquet-Varrault and T. J. Wallington, Database for the kinetics of the gas-phase atmospheric reactions of organic compounds, Earth System Sci. Data, 2020, 12, 1203–1216 CrossRef.
- R. H. Schwantes, K. A. Schilling, R. C. McVay, H. Lignell, M. M. Coggon and X. Zhang, et al., Formation of highly oxygenated low-volatility products from cresol oxidation, Atmos. Chem. Phys., 2017, 17(5), 3453–3474 CrossRef CAS.
- F. Paulot, J. D. Crounse, H. G. Kjaergaard, A. Kürten, J. M. St Clair and J. H. Seinfeld, et al., Unexpected epoxide formation in the gas-phase photooxidation of isoprene, Science, 2009, 325(5941), 730–733 CrossRef CAS.
- M. Li, Y. Liu and L. Wang, Gas-phase ozonolysis of furans, methylfurans, and dimethylfurans in the atmosphere, Phys. Chem. Chem. Phys., 2018, 20(38), 24735–24743 RSC.
- S. Richters, H. Herrmann and T. Berndt, Different pathways of the formation of highly oxidized multifunctional organic compounds (HOMs) from the gas-phase ozonolysis of β-caryophyllene, Atmos. Chem. Phys., 2016, 16(15), 9831–9845 CrossRef CAS.
- M. H. Almatarneh and I. A. Elayan, Abu-Saleh AAAA, Altarawneh M, Ariya PA. The gas-phase ozonolysis reaction of methylbutenol: A mechanistic study, Int. J. Quantum Chem., 2019, 119(10), e25888 CrossRef.
- D. R. Worton, J. D. Surratt, B. W. LaFranchi, A. W. H. Chan, Y. Zhao and R. J. Weber, et al., Observational Insights into Aerosol Formation from Isoprene, Environ. Sci. Technol., 2013, 47, 11403–11413 CrossRef CAS.
- W. W. Hu, P. Campuzano-Jost, B. B. Palm, D. A. Day, A. M. Ortega and P. L. Hayes, et al., Characterization of a real-time tracer for isoprene epoxydiols-derived secondary organic aerosol (IEPOX-SOA) from aerosol mass spectrometer measurements, Atmos. Chem. Phys., 2015, 15, 11807–11833 CrossRef CAS.
- A. Guenther, T. Karl, P. Harley, C. Wiedinmyer, P. I. Palmer and C. Geron, Estimates of global terrestrial isoprene emissions using MEGAN (Model of Emissions of Gases and Aerosols from Nature), Atmos. Chem. Phys., 2006, 6, 3181–3210 CrossRef CAS.
- Y. H. Lin, H. Zhang, H. O. T. Pye, Z. Zhang, W. J. Marth and S. Park, et al., Epoxide as a precursor to secondary organic aerosol formation from isoprene photooxidation in the presence of nitrogen oxides, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 6718–6723 CrossRef CAS.
- J. D. Surratt, A. W. H. Chan, N. C. Eddingsaas, M. Chan, C. L. Loza and A. J. Kwan, et al., Reactive intermediates revealed in secondary organic aerosol formation from isoprene, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 6640–6645 CrossRef CAS.
- L. Xu, H. Guo, C. M. Boyd, M. Klein, A. Bougiatioti and K. M. Cerully, et al., Effects of anthropogenic emissions on aerosol formation from isoprene and monoterpenes in the southeastern United States, Proc. Natl. Acad. Sci. U. S. A., 2015, 112, 37–42 CrossRef CAS.
- Y. H. Lin, E. M. Knipping, E. S. Edgerton, S. L. Shaw and J. D. Surratt, Investigating the influences of SO2 and NH3 levels on isoprene-derived secondary organic aerosol formation using conditional sampling approaches, Atmos. Chem. Phys., 2013, 13, 8457–8470 CrossRef.
- S. H. Budisulistiorini, X. Li, S. T. Bairai, J. Renfro, Y. Liu and Y. J. Liu, et al., Examining the effects of anthropogenic emissions on isoprene-derived secondary organic aerosol formation during the 2013 Southern Oxidant and Aerosol Study (SOAS) at the Look Rock, Tennessee ground site, Atmos. Chem. Phys., 2015, 8871–8888 CrossRef CAS.
- S. H. Budisulistiorini, M. R. Canagaratna, P. L. Croteau, W. J. Marth, K. Baumann and E. S. Edgerton, et al., Real-Time Continuous Characterization of Secondary Organic Aerosol Derived from Isoprene Epoxydiols in Downtown Atlanta, Georgia, Using the Aerodyne Aerosol Chemical Speciation Monitor, Environ. Sci. Technol., 2013, 47, 5686–5694 CrossRef CAS.
- J. Calvert, A. Mellouki, J. Orlando, M. Pilling and T. Wallington, Mechanisms of Atmospheric Oxidation of the Oxygenates Oxford, Oxford University Press, 2011 Search PubMed.
- I. Barnes, K. H. Becker and N. Mihalopoulos, An FTIR product study of the photooxidation of dimethyl disulfide, J. Atmos. Chem., 1994, 18, 267–289 CrossRef CAS.
- I. Barnes, P. Wiesen and M. Gallus, Kinetics and mechanism of the OH-radical and Cl-atom oxidation of propylene carbonate, RSC Adv., 2016, 6(100), 98234–98242 RSC.
- B. J. Finlayson-Pitts and J. N. Pitts, Overview of the Chemistry of Polluted and Remote Atmospheres, in Chemistry of the Upper and Lower Atmosphere, ed. B. J. Finlayson-Pitts and J. N. Pitts, Academic Press, San Diego, 2000, ch. 1, pp. 1–14 Search PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb and J. R. Cheeseman, et al., Gaussian_16 Revision A.03, Gaussian Inc., Wallingford CT, 2016 Search PubMed.
- R. Dennington, T. A. Keith and J. M. Millam, GaussView Version 6, 2019 Search PubMed.
- A. D. Becke, Density-functional thermochemistry. III. The role of exact exchange, J. Chem. Phys., 1993, 98, 5648–5652 CrossRef CAS.
- C. Lee, W. Yang and R. G. Parr, Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density, Phys. Rev. B: Condens. Matter Mater. Phys., 1988, 37, 785–789 CrossRef CAS.
- S. Grimme, S. Ehrlich and L. Goerigk, Effect of the damping function in dispersion corrected density functional theory, J. Comput. Chem., 2011, 32, 1456–1465 CrossRef CAS.
- F. Weigend and R. Ahlrichs, Balanced basis sets of split valence, triple zeta valence and quadruple zeta valence quality for H to Rn: design and assessment of accuracy, Phys. Chem. Chem. Phys., 2005, 7, 3297–3305 RSC.
- S. M. Aschmann and R. Atkinson, Rate constants for the gas-phase reactions of alkanes with Cl atoms at 296 ± 2 K, Int. J. Chem. Kinet., 1995, 27, 613–622 CrossRef CAS.
- A. Notario, A. Mellouki and G. L. Bras, Rate constants for the gas-phase reactions of Cl atoms with a series of ethers, Int. J. Chem. Kinet., 2000, 32, 105–110 CrossRef CAS.
- S. Middala, S. Campbell, C. Olea, A. Scruggs and A. S. Hasson, Kinetics and mechanism of the reaction of propylene oxide with chlorine atoms and hydroxy radicals, Int. J. Chem. Kinet., 2011, 43, 507–521 CrossRef CAS.
- A. D. Walsh, The structures of ethylene oxide, cyclopropane, and related molecules, Trans. Faraday Soc., 1949, 45, 179–190 RSC.
- A. D. Walsh, Structures of Ethylene Oxide and Cyclopropane, Nature, 1947, 159(4047), 712–713 CrossRef CAS.
- R. Hoffmann and R. Davidson, Valence orbitals of cyclobutane, J. Am. Chem. Soc., 1971, 93(22), 5699–5705 CrossRef CAS.
- A. Rosowsky, Ethylene oxides, in Chemistry of heterocyclic compounds, ed. A. Weissberger, John Wiley & Sons, Ltd., New York, 1964, p. 1188 Search PubMed.
- N. H. Cromwell and G. V. Hudson, The Steric Requirements for Three-Ring Carbonyl Hyperconjugation1, J. Am. Chem. Soc., 1953, 75, 872–874 CrossRef CAS.
- M. Bartels, K. Hoyermann and U. Lange, An Experimental Study of the Reactions CH3CHO + Cl, C2H4O + Cl, and C2H4O + F in the Gas-Phase, Berichte der Bunsengesellschaft für physikalische Chemie., 1989, 93, 423–427 CrossRef CAS.
- D. Ponomarev, M. D. Hurley and T. J. Wallington, Kinetics of the reactions of fluorine and chlorine atoms with ethylene oxide (oxirane), Int. J. Chem. Kinet., 2002, 34, 122–125 CrossRef CAS.
- R. E. Parker and N. S. Isaacs, Mechanisms Of Epoxide Reactions, Chem. Rev., 1959, 59, 737–799 CrossRef CAS.
- C. A. Coulson and W. E. Moffitt, Strain in Non-Tetrahedral Carbon Atoms, J. Chem. Phys., 1947, 15, 151 CrossRef CAS.
- C. A. Coulson and W. E. I. Moffitt, The properties of certain strained hydrocarbons, London, Edinburgh Dublin Philos. Mag. J. Sci., 1949, 40, 1–35 CrossRef CAS.
- S. Searles and M. Tamres, Hydrogen Bond Formation with Saturated Cyclic Ethers1, J. Am. Chem. Soc., 1951, 73, 3704–3706 CrossRef CAS.
- S. Searles, M. Tamres and E. R. Lippincott, Hydrogen Bonding Ability and Structure of Ethylene Oxides, J. Am. Chem. Soc., 1953, 75, 2775–2777 CrossRef CAS.
- T. Lamberts, G. Fedoseev, J. Kästner, S. Ioppolo and H. Linnartz, Importance of tunneling in H-abstraction reactions by OH radicals – The case of CH4 + OH studied through isotope-substituted analogs, A\&A., 2017, 599, A132 Search PubMed.
- D. E. Heard, Rapid Acceleration of Hydrogen Atom Abstraction Reactions of OH at Very Low Temperatures through Weakly Bound Complexes and Tunneling, Acc. Chem. Res., 2018, 51(11), 2620–2627 CrossRef CAS.
- S. Karmakar, D. Jose and A. Datta, Tunnelling effects in chemistry, Resonance, 2014, 19(2), 160–174 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Cartesian coordinates, geometries and experimental data. See DOI: 10.1039/d0cp06033j |
| ‡ Tunnelling effect could become prominent for reactions at low temperatures.56–58 Here, it is assumed a thermal distribution in these complexes over all minima because the transition state is below the energy of the reactants. |
| This journal is © the Owner Societies 2021 |