
Proton conduction in hydronium solvate ionic liquids affected by ligand shape†
Kio
Kawata
a,
Atsushi
Kitada
 *a,
Naoki
Tsuchida
a,
Masayuki
Saimura
b,
Takashi
Nagata
b,
Masato
Katahira
b,
Kazuhiro
Fukami
a and
Kuniaki
Murase
a
*a,
Naoki
Tsuchida
a,
Masayuki
Saimura
b,
Takashi
Nagata
b,
Masato
Katahira
b,
Kazuhiro
Fukami
a and
Kuniaki
Murase
a
aDepartment of Materials Science and Engineering, Kyoto University, Sakyo-ku, Kyoto 606-8501, Japan. E-mail: kitada.atsushi.3r@kyoto-u.ac.jp; Fax: +81-75-753-5463; Tel: +81-75-753-5475
bInstitute of Advanced Energy, Kyoto University, Gokasho, Uji, Kyoto 611-0011, Japan
First published on 8th December 2020
Abstract
We investigated the ligand dependence of the proton conduction of hydronium solvate ionic liquids (ILs), consisting of a hydronium ion (H3O+), polyether ligands, and a bis[(trifluoromethyl)sulfonyl]amide anion (Tf2N−; Tf = CF3SO2). The ligands were changed from previously reported 18-crown-6 (18C6) to other cyclic or acyclic polyethers, namely, dicyclohexano-18-crown-6 (Dh18C6), benzo-18-crown-6 (B18C6) and pentaethylene glycol dimethyl ether (G5). Pulsed-field gradient spin echo nuclear magnetic resonance results revealed that the protons of H3O+ move faster than those of cyclic 18C6-based ligands but as fast as those of acyclic G5 ligands. Based on these results and density functional theory calculations, we propose that the coordination of a cyclic ether ligand to the H3O+ ion is essential for fast proton conduction in hydronium solvate ILs. Our results attract special interest for many electro- and bio-chemical applications such as electrolyte systems for fuel cells and artificial ion channels for biological cells.
Introduction
When two or more substances are mixed, the properties of the mixture can drastically change from those of pure substance. Moreover, even a slight structural change in one of the components can significantly affect the properties, which donate the designability of functions. The neutralization of Brønsted or Lewis acids and bases often results in ionization, e.g. dissociation, protonation, solvation, and complexation. Sometimes, the acid–base mixtures can be classified as ionic liquids (ILs), which are defined as salts that melt below 100 °C. ILs show various characteristics, such as high ionic conductivity, high chemical and thermal stability, and solubility. Consequently, ILs have received significant attention as media for chemical, biochemical, and electrochemical systems.1–6ILs can be classified into four types: solvate ILs, protic ILs, aprotic ILs, and inorganic ILs.7 We previously reported a hydronium (H3O+) solvate IL [H3O+·18C6]Tf2N (18C6 = 18-crown-6; Tf2N = bis[(trifluoromethyl)sulfonyl]amide, Tf = SO2CF3) as the first example of a molten salt of the [H3O+·ligand] complex,8 while other groups had reported solid state [H3O+·ligand] complexes using common anions (e.g. ClO4−, SbF6−, PF6−, TfO−, BF4−, and FeCl4−).9–12 The hydronium solvate IL intersects the solvate and protic types of ILs, where a protic H3O+ ion (i.e., solute) is solvated by the 18C6 ligand (i.e., solvent) to form a [H3O+·18C6] complex cation (i.e., solvate) and Tf2N− as the counter anion. Its strong Brønsted acidity among ILs (Hammett acidity H0 = −4.48) indicates that it may be used as an acid catalyst and in fuel cells.6,13–20
Moreover, the proton conduction of hydronium solvate ILs has been of special interest. Pulsed-field gradient spin echo nuclear magnetic resonance (PGSE-NMR) measurements revealed that the protons of H3O+ move faster than those of 18C6 ligands in [H3O+·18C6]Tf2N.21 Among solvate ILs and protic ILs,22–25 this was the first observation of ligand exchange conduction without free neutral molecules. Furthermore, unlike common solvate ILs and protic ILs, this cooperative proton relay is suppressed and H3O+ is found to move as slow as 18C6 ligands when diluted using equimolar 18C6 solvent.26 The proton conduction of the hydronium-based electrolytes would be interesting from the viewpoint of recently proposed hydronium ion batteries27,28 as well as fuel cells.6,13–15
In these studies, however, a key factor of the fast proton conduction has not been revealed. To understand the anomalous proton conduction, it is important to study analogous compounds with different ligands. Since 18C6 is a plausible ligand of H3O+ and a good acceptor that binds the H3O+ cation selectively owing to its cavity size,9–12,29 other candidate ligands are also cyclic or acyclic polyethers with six ether oxygens akin to 18C6.
In this work, three novel hydronium solvate ILs analogous to [H3O+·18C6]Tf2N are synthesized and the proton conduction of these hydronium solvate ILs is studied. A set of 1H PGSE-NMR measurements was performed to determine the self-diffusion coefficients of H3O+ and the ligands of the hydronium solvate ILs. The obtained results are also compared with density functional theory (DFT) calculations to reveal the distinguishable effect of ligand shape associated with cyclic or acyclic ligands on the proton conduction of hydronium solvate ILs.
Experimental
Materials
Dicyclohexano-18-crown-6 (Dh18C6; Tokyo Chemical Industry Co., Ltd, >98.0% purity), benzo-18-crown-6 (B18C6; Tokyo Chemical Industry Co., Ltd, >96.0% purity), pentaethylene glycol dimethyl ether (G5; Nippon Nyukazai Co., 99.0% purity), and bis[(trifluoromethyl)sulfonyl]imide (HTf2N; Kanto Chemical, 99% purity) were used without further purification. Ultrapure water was prepared using a Merck Milli-Q Reference A system.Synthesis of hydronium solvate ILs
We synthesized [H3O+·Dh18C6]Tf2N and [H3O+·B18C6]Tf2N in an Ar-filled glovebox using the methods reported in ref. 8 and 26, respectively. On the other hand, [H3O+·G5]Tf2N was prepared in an Ar-filled glovebox as follows. Water was mixed with an equimolar amount of G5 in a septum-sealed vial at room temperature (RT), and then this mixture was added dropwise to HTf2N at −10 °C.Characterization of hydronium solvate ILs
Melting points (Tm) and glass transition temperatures (Tg) were determined using a differential scanning calorimeter (DSC; Rigaku, DSC8231) at a heating rate of 5 °C min−1. Proton nuclear magnetic resonance (1H NMR) spectra of [H3O+·Dh18C6]Tf2N at 60 °C, [H3O+·B18C6]Tf2N at 75 °C, and [H3O+·G5]Tf2N at 25 °C were obtained at 600 MHz using a JNM-ECA600 FT NMR spectrometer (JEOL Ltd). The use of a double NMR tube, purchased from Shigemi Corp. (Catalog No. SC-002), prevented the sample from mixing with the external standard. Traces of dimethyl sulfoxide-d6 (Cambridge Isotope Laboratories, Inc.) were placed in the outer tube (5.2 mm ∅) and compounds were added to the internal tube (5.0 mm ∅). Thermal gravimetric analysis (TGA) was conducted for [H3O+·Dh18C6]Tf2N, [H3O+·B18C6]Tf2N, and [H3O+·G5]Tf2N at a heating rate of 5 °C min−1, using a TG-DTA8122 (Rigaku) instrument in a dry air atmosphere. Aluminum pans were used for the measurements. For each measurement, a 10 mg sample was placed on the pan and Al2O3 was used as a standard.The ionic conductivity (σ) of the electrolyte was determined by electrochemical impedance spectroscopy for [H3O+·Dh18C6]Tf2N from 50 °C to 90 °C, [H3O+·B18C6]Tf2N from 60 °C to 85 °C, and [H3O+·G5]Tf2N from 5 °C to 45 °C. A Bio-Logic Science Instruments SAS, VSP-300, was used with stainless steel electrodes. The cell constant was calibrated with 0.1 and 1 mol dm−3 KCl aqueous solutions. The measurement was conducted in a thermostatic chamber (Espec Co., SU-222). Viscosity (η) measurements were performed for each sample in the same temperature range as conductivity measurements using a viscometer (Kyoto Electronics Manufacturing Co., Ltd, EMS-1000). Using the measured values of weight and volume, densities (ρ) for each compound were calculated.
Proton PGSE-NMR measurements were also performed for [H3O+·Dh18C6]Tf2N (at 70 °C), [H3O+·B18C6]Tf2N (at 75 °C), and [H3O+·G5]Tf2N (at 25 °C) by the same equipment used for NMR measurement. The self-diffusion coefficients of H3O+ and ligands (Dh18C6, B18C6, and G5) in these compounds were measured using a simple Hahn spin echo sequence and analyzed using the Stejskal equation:
| ln(I/I0) = −D(γgδ)2(Δ − δ/3), |
The Gaussian 16 program31 was used for the ab initio molecular orbital calculations. The basis sets implemented in the Gaussian program were used. The geometry of 18C6, G5, Dh18C6, and B18C6 complexes with H3O+ was fully optimized at the B3LYP/6-311+G** level. Additionally, to investigate the degree of orientation for two cation complexes, the geometry of two [H3O+·(ligand)] (ligand = 18C6 and G5) was also fully optimized at the B3LYP/6-311+G** level under a tetrahydrofuran atmosphere, because the dielectric permittivity is similar to that of common ILs.32
Results and discussion
Characterization of hydronium solvate ILs
Both an equimolar mixture made of Dh18C6, HTf2N, and H2O, i.e., [H3O+·Dh18C6]Tf2N, and that made of G5, HTf2N, and H2O, i.e., [H3O+·G5]Tf2N, were yellowish liquids, while that made of B18C6, HTf2N, and H2O, i.e., [H3O+·B18C6]Tf2N, was a white solid at RT. The DSC curves for these compounds are shown in Fig. 1, and their melting points Tm or glass transition points Tg are listed in Table 1. All samples have melting points below 100 °C, satisfying the fourth criterion of solvate ILs.8,33 In Fig. 1, no peaks are seen near the melting points of pure ligands or that of HTf2N. Therefore, these compounds show no physicochemical properties based on both pure ligands and precursor salts, satisfying the third criterion of solvate ILs.8,33 | ||
| Fig. 1 DSC curves for (a) [H3O+·Dh18C6]Tf2N, (b) [H3O+·B18C6]Tf2N, (c) [H3O+·G5]Tf2N, and (d) [H3O+·18C6]Tf2N (ref. 8) at a heating rate of 5 °C min−1. | ||
| Ideal composition and formula weight (g mol−1) | T m or Tg (°C) | σ (mS cm−1) | η (mPa s) | ρ (g cm−3) |
|---|---|---|---|---|
| a Data for [H3O+·18C6]Tf2N are from ref. 8. | ||||
| [H3O+ Dh18C6]Tf2N (671.67) | T g −24 | 0.58 at 70 °C | 155.1 at 70 °C | 1.31 at RT |
| [H3O+·B18C6]Tf2N (611.53) | T m 52–54 | 0.85 at 75 °C | 106.8 at 75 °C | 1.33 at 60 °C |
| [H3O+·G5]Tf2N (565.51) | T g −90 | 2.22 at 25 °C | 50.2 at 25 °C | 1.45 at 25 °C |
| [H3O+·18C6]Tf2Na (563.48) | T m 66–68 | 2.36 at 75 °C | 42.9 at 75 °C | 1.32 at 70 °C |
Table 2 shows data of the elemental analysis for H, C, N, F, and S content of each mixture, which was performed at the Center for Organic Elemental Microanalysis, Kyoto University. Oxygen atoms cannot be analyzed in the principle of the elemental analysis. The data were consistent with the suggested formulations [H3O+·Dh18C6]Tf2N, [H3O+·B18C6]Tf2N, and [H3O+·G5]Tf2N. The H2O contents, as analyzed by Karl–Fischer coulometric titration of [H3O+·Dh18C6]Tf2N, [H3O+·B18C6]Tf2N, and [H3O+·G5]Tf2N, were 2.61, 3.03, and 3.20 wt%, which are in good agreement with the calculated values for each sample (2.68 wt% for [H3O+·Dh18C6]Tf2N, 2.94 wt% for [H3O+·B18C6]Tf2N, and 3.18 wt% for [H3O+·G5]Tf2N). These quantitative analyses support that each complex had a ligand to HTf2N to H2O molar ratio of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1:1, suggesting that all the mixtures in this study form a solvate compound between an ion and a ligand in a certain stoichiometric ratio. This satisfies the first criterion of solvate ILs.8,33
:1:1, suggesting that all the mixtures in this study form a solvate compound between an ion and a ligand in a certain stoichiometric ratio. This satisfies the first criterion of solvate ILs.8,33
| Compound | H (%) | C (%) | N (%) | F (%) | S (%) | |
|---|---|---|---|---|---|---|
| [H3O+·Dh18C6]Tf2N | Experimental | 5.77 | 38.68 | 2.13 | 17.21 | 9.70 |
| Calculated | 5.85 | 39.34 | 2.09 | 16.97 | 9.55 | |
| [H3O+·B18C6]Tf2N | Experimental | 4.64 | 35.51 | 2.42 | 18.52 | 10.31 |
| Calculated | 4.45 | 35.35 | 2.29 | 18.64 | 10.49 | |
| [H3O+·G5]Tf2N | Experimental | 5.33 | 29.52 | 2.45 | 20.13 | 11.29 |
| Calculated | 5.17 | 29.74 | 2.48 | 20.16 | 11.34 | |
Fig. 2 exhibits the schematic structure of the cations and the 1H NMR spectra for the hydronium solvate ILs. The assignment of NMR signals for H3O+, Dh18C6, B18C6, and G5 is also displayed in each spectrum. The signals from H3O+ in [H3O+·Dh18C6]Tf2N, [H3O+·B18C6]Tf2N, and [H3O+·G5]Tf2N appear at 11.03, 10.86, and 11.58 ppm, respectively. As reported previously,8 in [H3O+·18C6]Tf2N, where unprotonated water is excluded and H3O+ is solvated by 18C6 ligands, the chemical shift of the H3O+ NMR signal is 10.85 ppm. As also discussed in our previous report,21 one 1H NMR singlet is observed at 8.18 ppm in an equimolar mixture of H2O and HTf2N (HTf2N·H2O), where its degree of dissociation is low and unprotonated H2O exists to some extent. Every H3O+ signal in the compounds of this study is comparable to that of [H3O+·18C6]Tf2N. Thus, these signals correspond to H3O+ solvated by a ligand, satisfying the second criterion of solvate ILs.8,33 Furthermore, we can consider all NMR signals of each ligand to be from bound molecules, hence there are no physicochemical properties based on a pure ligand. This supports the DSC results.
 | ||
| Fig. 2 (a–c) Schematic cation structure of (a) [H3O+·Dh18C6], (b) [H3O+·B18C6], and (c) [H3O+·G5]; (d–f) 1H NMR spectra; and (g–i) enlarged plots for (d and g) [H3O+·Dh18C6]Tf2N at 60 °C, (e and h) [H3O+·B18C6]Tf2N at 75 °C, and (f and i) [H3O+·G5]Tf2N at 25 °C. | ||
In TGA data (Fig. 3) with a heating rate of 5 °C min−1, [H3O+·Dh18C6]Tf2N, [H3O+·B18C6]Tf2N, and [H3O+·G5]Tf2N start volatilizing at a much higher temperature than HTf2N·H2O does, as is the case for [H3O+·18C6]Tf2N.8 More precisely, 5 wt% loss is detected at around 150 °C for [H3O+·Dh18C6]Tf2N and [H3O+·B18C6]Tf2N, similar to the behavior of [H3O+·18C6]Tf2N, while for [H3O+·G5]Tf2N it occurs at 110 °C. The vapor pressures of [H3O+·Dh18C6]Tf2N, [H3O+·B18C6]Tf2N, and [H3O+·G5]Tf2N are negligible at temperatures lower than 100 °C, satisfying the fifth criterion of solvate ILs.8,33
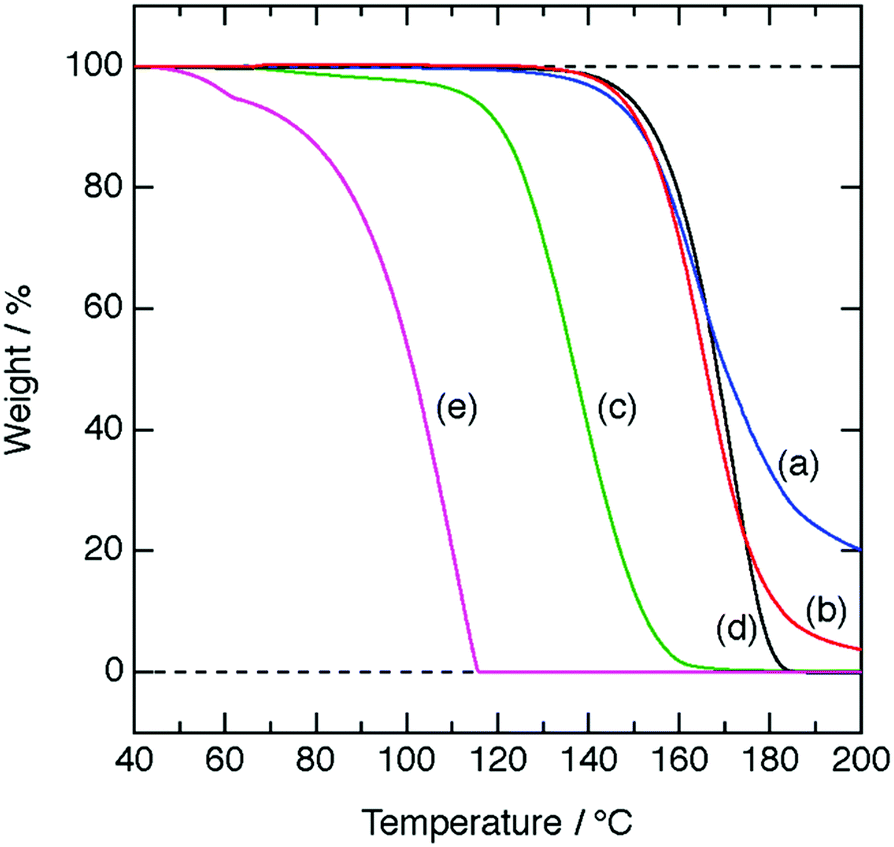 | ||
| Fig. 3 TGA curves at a heating rate of 5 °C min−1; (a) [H3O+·Dh18C6]Tf2N, (b) [H3O+·B18C6]Tf2N, (c) [H3O+·G5]Tf2N, (d) [H3O+·18C6]Tf2N, and (e) HTf2N·H2O (a–c: this work, e: ref. 8, f: ref. 26). | ||
The DSC, 1H NMR results, and elemental analysis suggest that compounds synthesized in this work consist of H3O+ solvated by ligands (Dh18C6, B18C6, and G5) and Tf2N−. Additionally, TGA clarifies that [H3O+·Dh18C6]Tf2N, [H3O+·B18C6]Tf2N, and [H3O+·G5]Tf2N have low volatility at 100 °C. Therefore, [H3O+·Dh18C6]Tf2N, [H3O+·B18C6]Tf2N, and [H3O+·G5]Tf2N are classified as solvate ILs according to the criteria,33 as well as the original hydronium solvate IL [H3O+·18C6]Tf2N.8
The physical properties (ionic conductivity σ, viscosity η, and density ρ) of [H3O+·Dh18C6]Tf2N, [H3O+·B18C6]Tf2N, [H3O+·G5]Tf2N, and [H3O+·18C6]Tf2N are summarized in Table 1. In addition, ionic conductivities and viscosities for each hydronium solvate IL are listed in Tables S1–S3 (ESI†). A plot of molar conductivity (Λ/S cm2 mol−1) vs. fluidity (η−1/Poise−1; 1 Poise = 0.1 Pa s), i.e., the Walden plot, is displayed in Fig. 4. The plot reveals that all compounds synthesized in this work are categorized as “good ILs” as well as [H3O+·18C6]Tf2N and common ILs.8,33–35 The activation energies for ionic conductivity and viscosity estimated from Arrhenius plots are listed in Table 3. Similar to [H3O+·18C6]Tf2N,8 the activation energy of all the compounds in this work for ionic conductivity is somewhat smaller than that for viscosity.
 | ||
| Fig. 4 Walden plots for [H3O+·Dh18C6]Tf2N, [H3O+·B18C6]Tf2N, [H3O+·G5]Tf2N, and [H3O+·18C6]Tf2N (ref. 8). | ||
Proton conduction of hydronium solvate ILs
The PGSE-NMR results in Fig. 5 show plots of echo signal attenuation based on the Stejskal equation for H3O+ (black circles) and ligand (red circles) of [H3O+·Dh18C6]Tf2N at 70 °C, [H3O+·B18C6]Tf2N at 75 °C, [H3O+·G5]Tf2N at 25 °C, and [H3O+·18C6]Tf2N at 75 °C.21 As shown, the plots are linear. According to the Stejskal equation, the gradients of the fitted lines are proportional to the diffusion coefficients. Table 4 lists the estimated values of the self-diffusion coefficients for [H3O+·Dh18C6]Tf2N, [H3O+·B18C6]Tf2N, [H3O+·G5]Tf2N, and previously reported [H3O+·18C6]Tf2N.21 As in [H3O+·18C6]Tf2N, the diffusion coefficients of H3O+ are larger than those of the ligands (Dh18C6 and B18C6) in [H3O+·Dh18C6]Tf2N and [H3O+·B18C6]Tf2N, respectively. For [H3O+·G5]Tf2N, in contrast, the coefficients of H3O+ and G5 are very similar, as in common Li-glyme based solvate ILs where the diffusion coefficient ratio of glyme to Li falls in the range of 0.9–1.1.35 Consequently, the protons of H3O+ move faster than ligands in [H3O+·Dh18C6]Tf2N and [H3O+·B18C6]Tf2N as in the case of [H3O+·18C6]Tf2N, while they move as fast as ligands in [H3O+·G5]Tf2N. That is to say, the fast proton conduction is observed in all cyclic-ligand-based hydronium solvate ILs, while it was not observed in the acyclic-ligand-based one. Note that in [H3O+·Dh18C6]Tf2N the H2O content analyzed by Karl–Fischer titration (2.61 wt%) is somewhat smaller than the calculated value (2.68 wt%) as above. Therefore, it is not the case that a little extra water in hydronium solvate ILs assists the cooperative proton relay. It is also notable that the activation energy for ionic conductivity is smaller than that for viscosity, not only in [H3O+·Dh18C6]Tf2N and [H3O+·B18C6]Tf2N but also in [H3O+·G5]Tf2N.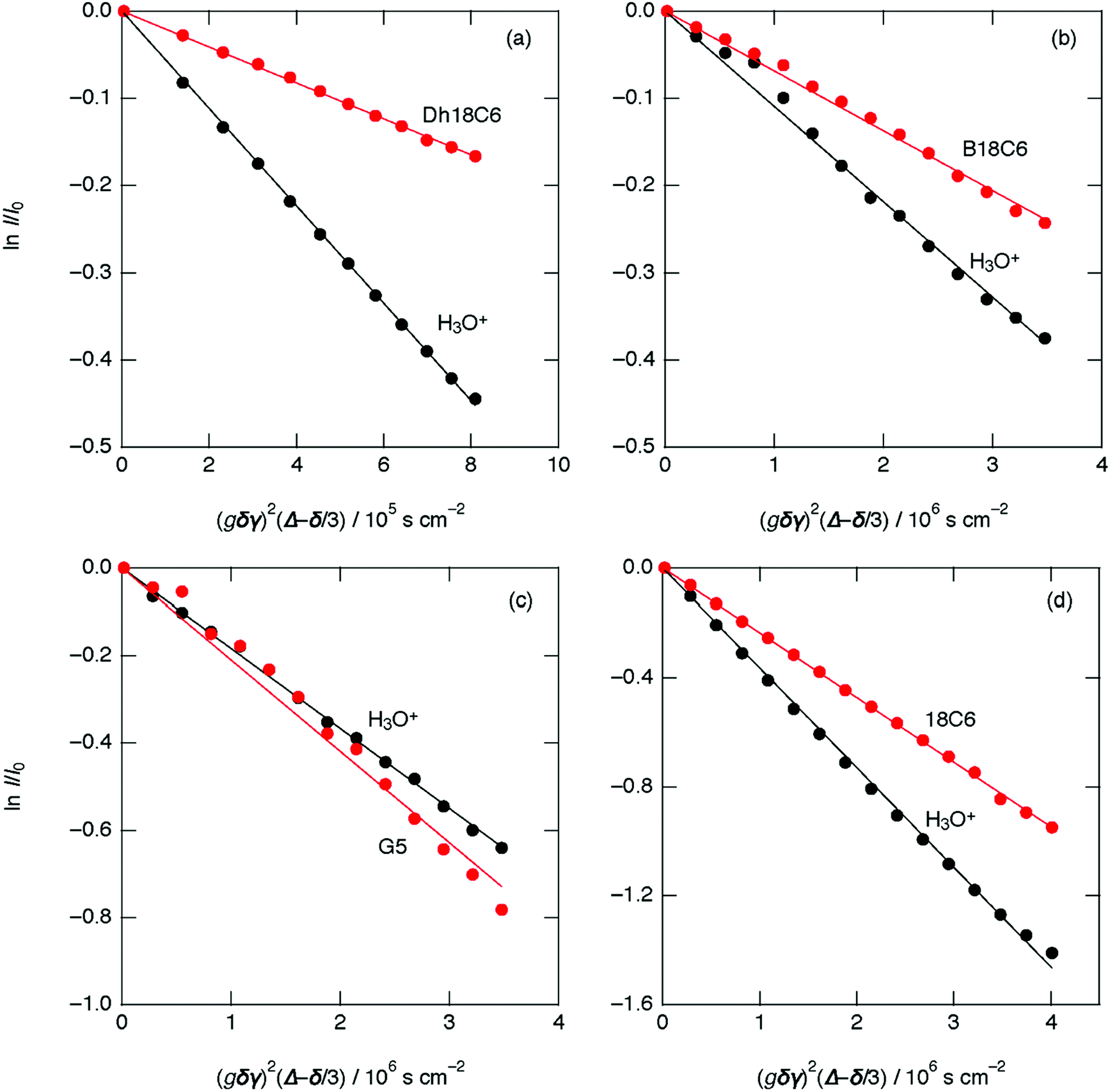 | ||
| Fig. 5 Plots and best-fit lines of echo signal attenuation based on the Stejskal equation for (a) [H3O+·Dh18C6]Tf2N at 70 °C, (b) [H3O+·B18C6]Tf2N at 75 °C, (c) [H3O+·G5]Tf2N at 25 °C, and (d) [H3O+·18C6]Tf2N at 75 °C (ref. 21): ligand (red circles) and H3O+ (black circles). | ||
To discuss the difference of proton conduction between cyclic-ether-based hydronium solvate ILs and acyclic one from a structural viewpoint, graphical representations of the equilibrium geometry of the most stable conformers of [H3O+·18C6] and [H3O+·G5] are displayed in Fig. 6a and b, respectively. In the former case, every other oxygen atom of 18C6 coordinates to the H3O+, forming a highly symmetric coordination via three equivalent hydrogen bonds, which agrees with some pioneering works,36–38 and so do those of both Dh18C6 and B18C6 (Fig. S1 and S2, ESI†). In the latter case, G5 does not wrap around the H3O+ cation completely, and the first to fourth of the six oxygen atoms of G5 participate in coordination to the H3O+. A [H3O+·G5] complex does not form an 18C6-like coordination geometry but instead forms an asymmetric structure. These discussions are supported by the fact that [H3O+·G5]Tf2N undergoes glass transition at −90 °C while [H3O+·18C6]Tf2N crystallizes at 66–68 °C, although the molar weights of the two compounds are almost the same. Consequently, the degree of structural freedom of G5 is larger than that of the 18C6-based ligand because G5 is an acyclic, open-chain ligand.
 | ||
| Fig. 6 Graphical representation of the equilibrium geometry of the most stable conformers of (a) 18C6 complexes with H3O+ (B3LYP/6-311+G** level) and (b) G5 complexes with H3O+ (B3LYP/6-311+G** level): oxygens in red, carbons in gray, and hydrogens in white. | ||
In common protic ILs, the reorientation of a proton acceptor is required for the fast proton transfer.39 Although the proton transfer mechanism differs between hydronium solvate ILs and common protic ILs—in that the former does not include excess neutral molecules that can help proton relay—we consider that ligand reorientation is also needed for the fast proton conduction in hydronium solvate ILs. In fact, the optimized structure of two solvate cations, which is used to investigate their degree of orientation, clearly shows that the pairs of [H3O+·18C6] face each other (Fig. 7a), while two [H3O+·G5] do not (Fig. 7b). In the system like a biological ion channel, where ions pass through the specific path, ligands are needed to line up in parallel. A solid state system where crown ether stacks are known as an artificial analogue of such an ion channel.40 In the hydronium solvate ILs, where solvate cations and anions move relative to each other, the solvate cations will not stay and stack at specific area. Nonetheless, whether the ligands face each other frequently or not, i.e. whether the ligand reorientation occurs frequently, is important for the proton conduction. That is, the results shown in Fig. 7 strongly indicate the large difference in the probabilities of ligand reorientation for the crown ether and the glyme solvates. In [H3O+·G5]Tf2N, fast proton conduction has not been found, evidencing that states in which two [H3O+·G5] face each other are infrequent. Certainly, the open G5 ligand is more flexible than the closed 18C6-based ligands. However, G5 cannot maintain the specific states of ligand orientation required for the fast proton conduction due to its high flexibility, which may suppress fast proton conduction. In contrast, the ligand reorientation occurs frequently for the 18C6-based ligands owing to its rigidity. As a result, conduction paths are available for a cooperative proton relay and fast proton conduction takes place.
 | ||
| Fig. 7 Graphical representation of the equilibrium geometry of the most stable conformers of (a) two [H3O+·18C6] cations and (b) two [H3O+·G5] cations (B3LYP/6-311+G** level): oxygens in red, carbons in gray, and hydrogens in white. | ||
Here we further discuss why such different proton conduction is observed in the cyclic- and acyclic-ligand-based hydronium solvate ILs. Crown ethers show amphiphilicity because of the hydrophobic –CH2CH2– groups and the hydrophilic inherent ether oxygens. The ether oxygens are well situated to coordinate with a cation located at the interior of the ring, whereas the exterior of the ring is hydrophobic. The cyclic molecules can divide the hydrophilic area and hydrophobic area due to its closed nature. Therefore, the fast proton conduction appears in cyclic-ligand-based hydronium solvate ILs. In particular, Dh18C6 shows more exterior hydrophobicity than 18C6 owing to two cyclohexane moieties, resulting in a larger ratio of diffusion coefficients of H3O+ and ligand in [H3O+·Dh18C6]Tf2N than in [H3O+·18C6]Tf2N. Apparently, B18C6 also shows exterior hydrophobicity compared to 18C6. The benzene ring, however, is known not only as a hydrophobic solute but also as a hydrogen bond acceptor.41 As a result, the ratio of diffusion coefficients of H3O+ and ligand is almost the same in [H3O+·B18C6]Tf2N and in [H3O+·18C6]Tf2N. On the other hand, glyme molecules can also show both hydrophobicity and hydrophilicity. The two properties, however, cannot completely be interior/exterior for such open-chain and flexible molecules. Thus, it is the closed nature of crown ether or “partitioned amphiphilicity” that gives conduction paths for the cooperative proton relay. In [H3O+·G5]Tf2N, such conduction paths for cooperative proton relay are not available. It may be notable that hydronium solvate ILs may relate to the ionics in biological ion channels. By utilizing crown ethers, artificial ion channels have widely been studied,40,42–47 where the hydrophilic ion channel in a hydrophobic lipid bilayer is mimicked. Although these studies focus on ion selectivity and ion transport, as far as we know, the effect of the partitioned amphiphilicity on the ionics by comparing the ligand structure has not been reported.
Consequently, the proton conduction of cyclic-ligand-based hydronium solvate ILs and that of the acyclic counterpart are different. The DFT calculations show that the rigidity of 18C6 allows two [H3O+·18C6] cations to be parallel, implying small ligand reorientation, while two [H3O+·G5] cations are not. Therefore, the fast proton conduction only appears in cyclic-ligand-based hydronium solvate ILs. Additionally, since the conduction carrier is protons of H3O+ or H3O+ itself in hydronium solvate ILs, the hydrophilic area of the ligand plays a role as a proton acceptor. The hydrophilic and hydrophobic areas of the cyclic ligand can be divided owing to its closed nature, while those of the acyclic one cannot because it is an open chain. Therefore, we propose that the topology of the ethereal ligands is a clue for the proton conduction of hydronium solvate ILs.
Conclusions
The dependence of ligand shape on the proton conduction in hydronium solvate ILs was studied. While the protons of H3O+ move faster than the cyclic 18C6-based ligands, such fast proton conduction was not observed in the case of acyclic G5 ligands. Consequently, whether the H3O+ ion is coordinated by cyclic or acyclic ligands—in other words, whether H3O+ wears a “crown” or “tiara”—is a key factor for proton conduction in hydronium solvate ILs, and the “coronation” or the crown-ether coordination matters. These findings should reflect the topological difference of the ethereal ligands. For further studies, molecular dynamics simulations for these hydronium solvate ILs, which would detect temporal ion channels in liquids, are of special interest. Our findings can provide guidelines to design new electrolyte systems for fuel cells and artificial ion channels for biological cells.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors acknowledge Prof. Yasuhiro Umebayashi (Niigata University) for his support with the DFT calculations. This work was supported financially by Grants-in-Aid for Scientific Research (S) (No. 20H05663: K. M.), Scientific Research (B) (No. 19H02490: A. K.) and Grant-in-Aid for Challenging Research (Exploratory) (No. 19K22056: A. K.) from the Japan Society for the Promotion of Science. A. K. also thanks the Joint Usage/Research Program on Zero-Emission Energy Research, Institute of Advanced Energy, Kyoto University (ZE31A-1 and ZE2020A-09).References
- T. Welton, Chem. Rev., 1999, 99, 2071–2083 CrossRef CAS PubMed.
- K. Fujita, K. Murata, M. Masuda, N. Nakamura and H. Ohno, RSC Adv., 2012, 2, 4018–4030 RSC.
- T. L. Greaves and C. J. Drummond, Chem. Rev., 2015, 115, 11379–11448 CrossRef CAS PubMed.
- P. A. Hunt, C. R. Ashworth and R. P. Matthews, Chem. Soc. Rev., 2015, 44, 1257–1288 RSC.
- M. Armand, F. Endres, D. R. MacFarlane, H. Ohno and B. Scrosati, Nat. Mater., 2009, 8, 621–629 CrossRef CAS PubMed.
- A. S. Amarasekara, Chem. Rev., 2016, 116, 6133–6183 CrossRef CAS PubMed.
- C. A. Angell, Y. Ansari and Z. Zhao, Faraday Discuss., 2012, 154, 9–27 RSC.
- A. Kitada, S. Takeoka, K. Kintsu, K. Fukami, M. Saimura, T. Nagata, M. Katahira and K. Murase, J. Electrochem. Soc., 2018, 165, H121–H127 CrossRef CAS.
- G. S. Heo and R. A. Bartsch, J. Org. Chem., 1982, 47, 3557–3559 CrossRef CAS.
- R. Chênevert, A. Rodrigue, P. Beauchesne and R. Savoie, Can. J. Chem., 1984, 62, 2293–2298 CrossRef.
- R. Chênevert and A. Rodrique, J. Chem. Educ., 1984, 61, 465–466 CrossRef.
- R. Chênevert, D. Chamberland, M. Simard and F. Brisse, Can. J. Chem., 1989, 67, 32–36 CrossRef.
- T. Yasuda and M. Watanabe, MRS Bull., 2013, 38, 560–566 CrossRef CAS.
- J. Gao, G. Wang, Z. Wang, Y. Wang, J. Liu, W. Liu and Z. Zou, J. Mater. Chem. A, 2014, 2, 19275–19281 RSC.
- C. Ke, J. Li, X. Li, Z. Shao and B. Yi, RSC Adv., 2012, 2, 8953–8956 RSC.
- C. Chiappe and S. Rajamani, Eur. J. Org. Chem., 2011, 5517–5539 CrossRef CAS.
- R. Skoda-Földes, Molecules, 2014, 19, 8840–8884 CrossRef PubMed.
- Z. Duan, Y. Gu, J. Zhang, L. Zhu and Y. Deng, J. Mol. Catal. A: Chem., 2006, 250, 163–168 CrossRef CAS.
- Y. L. Geng, L. Y. Hu, X. Q. Zhao, H. L. An and Y. J. Wang, Chin. J. Chem. Eng., 2009, 17, 756–760 CrossRef CAS.
- H. Xing, T. Wang, Z. Zhou and Y. Dai, J. Mol. Catal. A: Chem., 2007, 264, 53–59 CrossRef CAS.
- A. Kitada, K. Kintsu, S. Takeoka, K. Fukami, M. Saimura, T. Nagata, M. Katahira and K. Murase, J. Electrochem. Soc., 2018, 165, H496–H499 CrossRef CAS.
- K. Yoshida, M. Nakamura, Y. Kazue, N. Tachikawa, S. Tsuzuki, S. Seki, K. Dokko and M. Watanabe, J. Am. Chem. Soc., 2011, 133, 13121–13129 CrossRef CAS PubMed.
- M. A. B. H. Susan, A. Noda, S. Mitsushima and M. Watanabe, Chem. Commun., 2003, 938–939 RSC.
- A. Noda, M. A. B. H. Susan, K. Kudo, S. Mitsushima, K. Hayamizu and M. Watanabe, J. Phys. Chem. B, 2003, 107, 4024–4033 CrossRef CAS.
- H. Doi, X. Song, B. Minofar, R. Kanzaki, T. Takamuku and Y. Umebayashi, Chem. – Eur. J., 2013, 19, 11522–11526 CrossRef CAS PubMed.
- K. Kawata, A. Kitada, N. Tsuchida, M. Saimura, T. Nagata, M. Katahira, K. Fukami and K. Murase, J. Electrochem. Soc., 2020, 167, 046508 CrossRef CAS.
- X. Wang, C. Bommier, Z. Jian, Z. Li, R. S. Chandrabose, I. A. Rodríguez-Pérez, P. A. Greaney and X. Ji, Angew. Chem., Int. Ed., 2017, 56, 2909–2913 CrossRef CAS PubMed.
- Y. Zhu, X. Yang and X. Zhang, Angew. Chem., Int. Ed., 2017, 56, 6378–6380 CrossRef CAS PubMed.
- P. C. Junk, New J. Chem., 2008, 32, 762–773 RSC.
- E. O. Stejskal and J. E. Tanner, J. Chem. Phys., 1965, 42, 288–292 CrossRef CAS.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. V. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery, Jr., J. E. Peralta, F. Ogliaro, M. J. Bearpark, J. J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, T. A. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman and D. J. Fox, Gaussian 16, Revision A.03, Gaussian Inc., Wallingford CT, 2016 Search PubMed.
- C. Daguenent, P. J. Dyson, I. Krossing, A. Oleinikova, J. Slattery, C. Wakai and H. Weingärtner, J. Phys. Chem. B, 2006, 110, 12682–12688 CrossRef PubMed.
- T. Mandai, K. Yoshida, K. Ueno, K. Dokko and M. Watanabe, Phys. Chem. Chem. Phys., 2014, 16, 8761–8772 RSC.
- A. Kitada, D. Ishikawa, K. Fukami and K. Murase, J. Electrochem. Soc., 2017, 164, H5119–H5123 CrossRef CAS.
- K. Ueno, K. Yoshida, M. Tsuchiya, N. Tachikawa, K. Dokko and M. Watanabe, J. Phys. Chem. B, 2012, 116, 11323–11331 CrossRef CAS PubMed.
- M. Bühl and G. Wipff, J. Am. Chem. Soc., 2002, 124, 4473–4480 CrossRef PubMed.
- A. Varnek, G. Wipff, A. Famulari, M. Raimondi, T. Vorob’eva and E. Stoyanov, J. Chem. Soc., Perkin Trans. 2, 2002, 887–893 RSC.
- M. Bühl, R. Ludwig, R. Schurhammer and G. Wipff, J. Phys. Chem. A, 2004, 108, 11463–11468 CrossRef.
- M. L. Hoarfrost, M. Tyagi, R. A. Segalman and J. A. Reimer, J. Phys. Chem. B, 2012, 116, 8201–8209 CrossRef CAS PubMed.
- K. M. Fromm and R. D. Bergougnant, Solid State Sci., 2007, 9, 580–587 CrossRef CAS.
- T. R. Raschke and M. Levitt, J. Phys. Chem. B, 2004, 108, 13492–13500 CrossRef CAS.
- V. E. Carmichael, P. J. Dutton, T. M. Fyles, T. D. James, J. A. Swan and M. Zojaji, J. Am. Chem. Soc., 1989, 111, 767–769 CrossRef CAS.
- M. F. M. Roks and R. J. M. Nolte, Macromolecules, 1992, 25, 5398–5407 CrossRef CAS.
- A. Cazacu, C. Tong, A. van der Lee, T. M. Fyles and M. Barboiu, J. Am. Chem. Soc., 2006, 128, 9541–9548 CrossRef CAS PubMed.
- T. Liu, C. Bao, H. Wang, L. Fei, R. Yang, Y. Long and L. Zhu, New J. Chem., 2014, 38, 3507–3513 RSC.
- T. M. Fyles, T. D. James and K. C. Kaye, J. Am. Chem. Soc., 1993, 115, 12315–12321 CrossRef CAS.
- Z. Sun, M. Barboiu, Y.-M. Legrand, E. Petit and A. Rotaru, Angew. Chem., 2015, 127, 14681–14685 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available: Lists of ionic conductivities and viscosities for each hydronium solvate IL. Graphical representation of the equilibrium geometry of the most stable conformers of two [H3O+·Dh18C6] cations and two [H3O+·B18C6] cations (B3LYP/6-311+G** level). See DOI: 10.1039/d0cp05025c |
| This journal is © the Owner Societies 2021 |