
Proline isomerization effects in the amyloidogenic protein β2-microglobulin†
Maria Celeste
Maschio
 abc,
Jacopo
Fregoni
ad,
Carla
Molteni
*c and
Stefano
Corni
*ad
abc,
Jacopo
Fregoni
ad,
Carla
Molteni
*c and
Stefano
Corni
*ad
aCNR-Nano S3, via Campi 215/a, Modena, Italy
bDepartment of Physics, University of Modena and Reggio Emilia, Via Campi 213/a, 41125 Modena, Italy
cDepartment of Physics, King's College London, Strand, London WC2R 2LS, UK. E-mail: carla.molteni@kcl.ac.uk
dDepartment of Chemical Sciences, University of Padova, Via Marzolo 1, Padova, Italy. E-mail: stefano.corni@unipd.it
First published on 21st December 2020
Abstract
The protein β2-microglobulin (β2-m) can aggregate in insoluble amyloid fibrils, which deposit in the skeletal muscle system of patients undergoing long-term haemodialysis. The molecular mechanisms of such amyloidogenesis are still not fully understood. A potential, although debated, triggering factor is the cis to trans isomerization of a specific proline (Pro32) in β2-m. Here we investigate this process in the native protein and in the aggregation-prone mutant D76N by means of molecular dynamics and the enhanced sampling method metadynamics. Our simulations, including the estimation of the free energy difference between the cis and trans isomers, are in good agreement with in vitro experiments and highlight the importance of the hydrogen bond and hydrophobic interaction network around the critical Pro32 in stabilizing and de-stabilizing the two isomers.
Among amino acids, proline has unique properties due to the cyclic structure of its side chain: it behaves exclusively as proton acceptor in hydrogen bonds and its torsional rigidity leads to disruption of both α-helices and β-sheets in proteins.1,2 For this reason, proline is usually not buried in protein cores but located at the apex of loops and other flexible portions of biomolecules.3 Furthermore, even if the trans geometry is the thermodynamically preferred one, proline can adopt the cis isomer of the peptydil–prolyl imide bond more frequently than other amino acids, with ∼5–6% of such bonds in the cis isomer in proteins native states. Anyhow, the transition from trans to cis conformer can be considered a rate-limiting step in the folding of the protein.4 The interconversion between the two isomers is limited by a high energy barrier experimentally estimated between ∼70 kJ mol−1 and ∼90 kJ mol−1 for proline and derivatives, depending on the system.5,6
The proline isomerization switch can be thought as an effective trigger for conformational changes and has been proposed e.g. for the gating of ligand-gated ion channels7,8 and in other regulatory mechanisms.9 Proline isomerization plays also an important role in amyloidogenical diseases, for example by influencing prion proteins10 and α-synuclein in Parkinson's disease.11 Likewise, it has been suggested as a triggering factor in the amyloid aggregation of β2-microglobulin (β2-m), a protein known to form insoluble fibrils which deposit in the skeletal muscle system of patients undergoing long-term haemodialysis.12 The structure of β2-m is shown in Fig. 1. β2-m amyloids seem to be induced by several environmental factors: by collagen and glycosaminoglycans at neutral pH13,14 or by the presence of Cu2+ metal ions.15,16 Aggregation can also be facilitated by low pH environments17 and contact with fibrillogenical β2-m variants.18
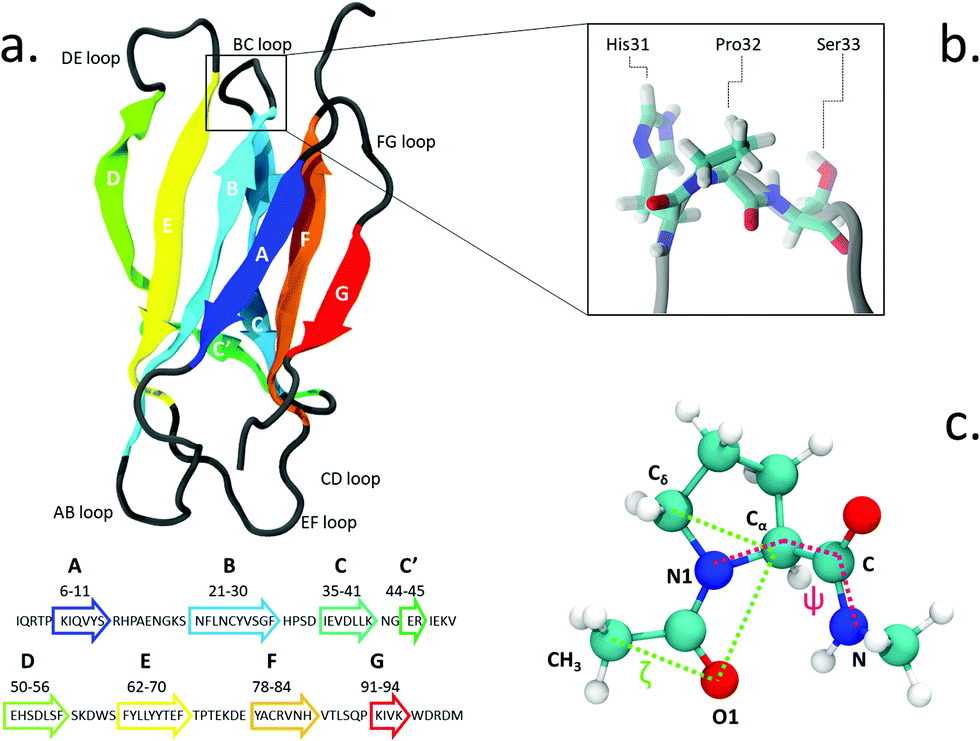 | ||
| Fig. 1 (a) The structure and sequence (in one-letter code amino acids) of β2-m; (b) zoomed view of Pro32 and its side residues His31 and Ser33 in the BC loop; (c) proline dipeptide, with the torsional angles ζ (green) and ψ (magenta) highlighted. | ||
The isomerization of the His31-Pro32 peptidyl-prolyl is an event in the β2-m amyloidogenic process whose importance and role are still debated. In fact, analyses of the wild-type (WT) crystal structure reveal that Pro32, located in the water exposed BC loop (Fig. 1b), is found in the infrequent cis geometry, while the other four prolines within the protein are in the usual trans configuration. On the contrary, within in vivo fibrils, the His31-Pro32 peptide bond adopts the trans configuration. Based on such evidence, cis-Pro32 has been suggested as crucial for maintaining β2-m solubility and hindering aggregation, which may be facilitated by isomerization to trans. Several studies have subsequently focused on the criticality of this peptidyl-prolyl bond19–21 that is relevant to the aggregation of β2-m. Yet, this contributes to increase the complexity in unveiling the mechanism leading to diseases. In fact, Eichner and Radford highlighted the isomerization toward the trans state as a key factor for amyloidogenesis: in an experiment correlating different species with fibril elongation, they identified a folding intermediate state trapped in a non-native trans-prolyl conformation as a precursor of fibril elongation.22 On the other hand Esposito et al., considering the emerging scenario from several studies,23–26 suggested that the cis-to-trans isomerization is a necessary but not sufficient condition for amyloid formation.27
Beside the environmental factors and specific amino acid rearrangements mentioned above, changes in protein sequence can also promote the amyloidogenic behavior. In fact, ΔN6β2-m, the form devoid of the six N-terminal residues, has a high tendency to aggregate and form fibrils at neutral pH conditions;28 furthermore, it can prime the aggregation of wild-type β2-m.29 The highly unstable D76Nβ2-m (with an aspartate substituted by an asparagine at position 76)30 shows a strongly amyloidogenic behavior: it produces fibrils without seeding when placed in vitro in its native form in physiological conditions and it provokes a systemic amyloidosis with deposits in internal organs.31 To complement and help interpret experiments, which cannot easily access energetic and molecular details, computational methods play an important role in elucidating the mechanism of Pro32 isomerization in β2-m.
Fogolari et al.32 used the adaptive biasing force method to simulate His31-Pro32 isomerization in WT β2-m, using the CHARMM v.2733 force-field with the CMAP correction,34 to probe the stabilizing and destabilizing conditions for β2-m. In particular, they examined the impact of the β2-m scaffold on the proline free energy surface. Starting the dynamics with the Pro32 in cis, the free energy difference between the cis and trans isomers was estimated 45 kJ mol−1 with a cis → trans barrier 93.3 kJ mol−1 high. Regarding the conformational change related to the Pro32 isomerization, they found that the cis isomer is thermodynamically stabilized by a fluctuating hydrogen bond network surrounding the critical residue Pro32. Such framework induces large changes in the FG loop and N-terminal RMSD (Root Mean Square Deviation), resulting in a more stable cis isomer.
Stober and Abrams35 simulated the WT β2-m at neutral and acidic conditions by performing “on-the-fly string method” calculations with the CHARM22 force-field. Tracking the variation of a large number of distances characterizing the protein structure, they monitored the evolution of the native system toward stable amyloidogenic state within different histidine protonations – hence different acidic conditions. In the neutral case, the free energy difference among the cis and trans isomers was estimated around 52.9 kJ mol−1, while the barrier height was calculated as 62.3 kJ mol−1. The main results described the stabilization of the neutral cis isomer through the Pro32–His84 hydrogen bond and of the protonated trans isomer through the His84–Asp34 interaction.
In the present work, we explore the Pro32 isomerization with the aid of the enhanced sampling method metadynamics36 and perform simulations of a monomer of β2-m in water for the wild-type and the D76N variant. Using extended full atomistic calculations, we want to shed light on the Pro32 isomerization debate, simulating, at the same physiological conditions, the two most important variants of such amyloidogenic protein. We highlight the difference in the relative stability of the trans and cis isomers for the the wild-type and the D76N mutants. We compare the free energy landscape to several experiments on the two protein variants, definitely relating the amyloidogenical behaviour to a major stability of the trans isomer of Pro32. We relate a significant stabilization of trans-Pro32 for the more amyloidogenic D76N variant to the rearrangemente of the hydrogen bond network. We also observe a further stabilization of trans-Pro32 due to the effect of the His84 protonation proposed by Stober and Abrams35 for the D76N mutant. Finally, we analyze the effect of Pro32 isomerization on the secondary structure. Through the analysis presented in the current work, we strongly confirm the link between the amyloidogenical behaviour and the Pro32 isomerization, highlighting how such factors play a key role in making the D76N mutation so strongly amyloidogenical.
1 Methods
We use the enhanced sampling method metadynamics,37 where the sampling of the configurational space as a function of a few selected slowly varying collective variables (CVs) is enhanced by pushing the system to visit unexplored regions and to avoid the formerly visited ones. This is achieved through a history-dependent bias potential, which is built as a sum of Gaussians deposited on-the-fly during the trajectory in the CV-space. The selected CVs should be effective in describing the process of interest, identifying the relevant free energy minima. At convergence, the sum of Gaussians defining the history-dependent potential allows to reconstruct the underlying free energy landscape as a function of the chosen CVs.38 In particular, we employ well-tempered metadynamics, a specific flavor of metadynamics in which the deposition of the bias potential is adaptively decreased.39Previous works in the literature suggest two optimal torsional angles to drive proline isomerization:4,8,40,41 the improper dihedral angle ζlit (defined by the atoms Cα–Cδ–O1–CH3 in Fig. 1, with values of 180° for the trans and 0° for the cis isomer) and the torsional angle ψ (defined by the atoms N1–Cα–C–N). ζlit accounts for the cis–trans isomerization and the pyramidalization of the imide nitrogen N1 (that is the distortion of a trigonal planar geometry toward a tetrahedral geometry), while ψ controls the C-terminal amide orientation. In the present work, the actual definition of ζ, shown in (Fig. 1c), i.e. Cδ–Cα–O1–CH3, results in the value of 180° for the cis conformer and 0° for the trans conformer.
We apply well-tempered metadynamics to Pro32 isomerization within β2-microglobulin, simulated as a monomer in water in presence of two Na+ counterions for the wild-type and one Na+ counterion for D76N to achieve electroneutrality of the simulation box. Specifically, we simulate two variants: the wild-type, based on Esposito et al.'s NMR structure (PDB entry: 1JNJ),42 and the amyloidogenic D76N β2-m, based on the X-ray structure by Ricagno et al. (PDB entry: 4FXL).30 The protonation states of the hystidines at neutral pH (all de-protonated for both WT and D76N) have been previously assigned on the basis of electrostatic pKa calculations.27,30,31 In a previous study, Stober and Abrams simulating the wild-type at acidic pH, pointed out the specific role of the protonation of His84 as a source of increased amyloidogenicity.35 To investigate whether His84 protonation has a similar effect in D76N, we run well-tempered metadynamics simulations on the D76N variant by protonating only the His84 residue. We remark that at low pH also His31 would be protonated, but we want to keep the focus on the His84 protonation only.
All calculations are performed with GROMACS 2018.243 and the OPLS-AA force field,44 while the Plumed 2.0 plugin45 is used for metadynamics.46 The MD simulations are run for both cis and trans isomer of wild-type and D76N. Each of these systems is first equilibrated in explicit solvent (using the SPC-E model for water47) with molecular dynamics (MD), in a periodically repeated box of volume 8.20 nm × 8.20 nm × 8.20 nm. Using the protein force-field (OPLS-AA) and the water model (SPC-E) preserves the stability of the experimental NMR structures. Such protocol has been previously tested for β2-m in different situations.48–51 OPLS-AA is known to produce larger torsional barriers than CHARMM and AMBER.8,40,52,53 Despite the overestimation, the OPLS-AA force field is expected to qualitatively compare the free proline dipeptide isomerization with the same process in complex environments like proteins.
A stochastic velocity rescaling thermostat54 and the Parrinello–Rahman barostat55 are applied during the MD production simulation – which lasts 500 ns – to keep the temperature at 300 K and the pressure at 1 atm respectively. The cutoff for non-bonded interactions is 10 Å, and long range electrostatics are handled through Particle Mesh Ewald summation. The bond lengths are constrained with the LINCS algorithm56 and a time step of 2 fs for numerical integration of the equations of motions is used. With the same computational protocol as the MD, metadynamics calculations are carried out to estimate the free energy landscape of the proline isomerization process. To approach convergence, proline dipeptide in water is run for 40 ns, while the two protein systems are simulated for about 800 ns. Within the well-tempered metadynamics regime, the initial Gaussian height is 0.8 kJ mol−1 and the width is 5°. The reference temperature is 300 K and the bias factor is set to 15. A Gaussian is deposited every 400 fs.
Hydrogen bonds (h-bonds) are defined using a distance cutoff of 3.5 Å between donor (D) and acceptor (A), and a cutoff for the angle H–D–A of 30 degrees.
2 Results and discussions
2.1 Free energy surfaces (FESs) for proline dipeptide, WT and D76N
While solvated proline dipeptide is a relatively straightforward system to simulate, a proline embedded in a protein is subjected to more complex interactions with nearby amino acids and solvent molecules. The free energy surfaces (FESs) of the solvated dipeptide as well as of Pro32 within wild-type β2-m and D76N are mapped as a function of the two torsional angles ζ and ψ in Fig. 2. The reference case is the proline dipeptide isomerization in water. Consistently with previous studies,8,40 the proline dipeptide FES exhibits four symmetric minima as shown in the Fig. 2a. The difference in free energy between the minima and the barriers along ζ varies in the range between 60° and 100° along the ψ coordinate. This behaviour suggests that the proline dipeptide isomerization along ζ is altered by the ψ torsion. The FESs of the Pro32 isomerization in the two protein variants (wild-type and the D76N mutation of the β2-m) show substantial differences with respect to that of proline dipeptide in water (Fig. 2). Both the FESs are less symmetric, with an asymmetric barrier shape and a change in the structure of the cis and trans minima. In particular, we observe that the trans minimum corresponding to ψ ∼ 350° (T2 in the dipeptide panel), is strongly destabilised (disappearing in practice) in both WT and D76N. Conversely, each variant presents a different landscape for the two cis minima that for proline deptide are located at ψ ∼ 140° (C1) and at ψ ∼ 350° (C2). For the WT, the most stable cis minimum (C1) is greatly stabilized and sits at about −20 kJ mol−1 compared to T1, entailing an inversion of stability of the isomers with respect to proline dipeptide. C2 is much less stabilized, and its free energy remains comparable to T1. The situation for D76N seems intermediate between proline dipeptide and WT: C1 is stabilized enough to become comparable/slightly more stable than T1, but it is not as deep as for WT. | ||
| Fig. 2 Upper panels: FESs of the isomerization of proline dipeptide in water (left), of Pro32 in wild-type β2-m (center) and of Pro32 in D76N (right), as a function of the torsional angles ζ and ψ. Lower panels: the corresponding potentials of mean force (PMFs) as a function of ζ only. While the FESs are not averaged, the projection on ζ is averaged by discarding the initial equilibration time (20 ns for proline dipeptide, 100 ns for wild-type and D76N β2-m). The profiles along ζ are integrated by following the procedure outlined in ref. 57. Further details are presented in Fig. S2 of the ESI.† The absolute minimum is set to zero for each system. The errors, also computed as in ref. 57, are shown as thin red lines. | ||
Another signature of the broken symmetry in the protein (WT and D76N) FES is the shape of the barriers. For proline dipeptide the two barriers separating trans from cis isomers (one at ζ ∼ 90° and the other at ζ ∼ 270°) are identical. This is not the case for WT and D76N. For the latter, it is particularly noticeable the lower (by about 30 kJ mol−1) path available through ζ ∼ 90°.
The estimation of the free energy differences between the cis and the trans minima can be evaluated through the potential of mean force (PMF) along ζ, that can be obtained by integrating out ψ from the bidimensional free energy surface.57 We note that the PMF profile does not correspond to a minimum free energy path calculation, but it is intended to provide an estimate of the thermodynamic quantities for each point of the configurational space explored during the dynamics. The calculation of the PMF is performed by taking the free energy profiles along the trajectory after an initial equilibration time (20 ns for proline dipeptide, 100 ns for wild-type and D76N β2-m) and by aligning them as detailed in ref. 57. In the following, a discussion of the one-dimensional profiles along ζ obtained for the simulated systems (lower panels in Fig. 2) and a comparison between our results and the available computational works in the literature (Table 1) are presented.
| Ref. | Method/technique | Force field | ΔF (kJ mol−1) t → c | Barrier (kJ mol−1) t → c | ||
|---|---|---|---|---|---|---|
| Proline | Theor. | This work | Well-T metadynamics | OPLS-AA | 4.7 ± 0.5 | 87.2 ± 0.4 |
| Melis40 | Metadynamics | AMBER2003 | 4.6 | 65.5 | ||
| Crnjar8 | Metadynamics | AMBERff14SB | 4.2 | 65.0 | ||
| Exp. | Beausoleil58 | NMR in D2O | — | 2.5 | — | |
| Taylor59 | NMR in D2O | — | 2.5 | — | ||
| Cheng6 | 13C NMR | — | — | 79.5–87.8 | ||
| Wild type | Theor. | This work | Well-T Metadynamics | OPLS-AA | −23.6 ± 9.3 | 89.3 ± 8.5 |
| Fogolari32 | Adaptive Biasing Force | CHARMM | −12 to −45 | 48.3 | ||
| Stober35 | Finite Temperature string MD | CHARMM22 | −52.9 | 62.3 | ||
| Exp. | Mangione31 | DSC | — | −7.5 | 72.4 | |
| D76N | Theor. | This work | Well-T Metadynamics | OPLS-AA | −3.7 ± 7.6 | 78.9 ± 8.7 |
| Exp. | Mangione31 | DSC | — | −2.7 | 68.6 | |
2.2 Comparison with experimental data and previous calculations
NMR experiments58,59 reported a free energy difference ΔF between the trans and cis conformers for the proline dipeptide (same molecule simulated in the present work) around 2.4 kJ mol−1, favoring the trans isomer. From our simulations, in the solvated proline dipeptide, the relative free energy difference between the isomers is 4.7 ± 0.5 kJ mol−1, resulting in a more stable trans isomer. While the quantitative agreement between experiment and theory is certainly satisfactory, our setup seems to slightly overstabilize the trans conformer by 2–3 kJ mol−1 Our result is also in agreement with previous computational investigation obtained with different force fields (see Table 1).8,40 We can also compare the free energy barrier separating the two isomers: our calculated value appears on the higher end of available experimental estimates (in agreement with the anticipated behavior of the OPLS-AA force field), with previously calculated values8,40 at the lower end. Anyway, the agreement between the simulated results and experimental data is certainly satisfactory. This sets the stage for the more challenging comparison of simulation and experiments for the proteins.Specific data for β2-m come from the experimental work by Mangione et al.,31 who investigated the isomerization of Pro32 in wild-type and D76N β2-m: there, they measured the stability of each isomer and the barrier separating them. From their results, the free energy difference between the two isomers (ΔFt→c) is equal to −7.5 kJ mol−1 and −3.7 kJ mol−1 for wild-type and D76N, respectively. Although the free energy difference is small in D76N, the cis isomer is found to be more stable than the trans as in WT.
The metadynamics simulations of the two protein variants provide values of the relative stability ΔFt→c equal to −23.6 ± 9.3 kJ mol−1 for WT, correctly identifying cis as the most stable isomer. The fluctuations of ΔFt→c occur in a range where the cis isomer is always more stable than the trans, even when the error is considered. The higher stability of cis compared to trans is in agreement with experimental findings, although quantitatively our computed data seems to overstabilize the trans isomer by ∼15 kJ mol−1. Previous simulations also suffer from overestimation of the cis stability: Fogolari et al. provided ΔFt→c values comparable to ours, slightly higher on average (−12 to −45 kJ mol−1);32 Stober and Abrams got even more stable values (−52.9 kJ mol−1) for the cis isomer. Starting the simulations from the cis experimental structure (the trans conformer is too unstable to be amenable of full structural characterization) may introduce a bias in the overall conformation of the protein that is not fully recovered even by extensive sampling. Identifying and then including in the metadynamics additional independent slow degrees of freedom, would be a possible strategy to check this point. However we could not clearly single out any such additional CVs, whose addition would anyway be computationally demanding.
For the D76N case, we compute a ΔFt→c of −3.7 ± 7.6 kJ mol−1. Here, the free energy difference ΔFt→c fluctuates around zero and the stability of cis is comparable to that of trans. Despite the large uncertainty, the result is in the same range of the −2.7 kJ mol−1 value reported experimentally.31 More importantly, the relative cis–trans stability trends represented by ΔFt→c in passing from proline dipeptide to D76N to WT is properly reproduced, justifying a molecular level analysis of the present simulations to understand the microscopic determinants of such trends (see Section 2.3). Likewise the lowering of the trans → cis barriers going from WT to D76N (89.3 ± 8.5 kJ mol−1 to 78.9 ± 8.7 kJ mol−1) follows the experimental trend reported by Mangione et al.31 Together with the trans → cis barriers which are lower in D76N than in WT, the destabilization of the cis isomer in D76N compared to WT results in a lower barrier for the cis → trans isomerization of Pro32 in D76N than in WT. Consequently, the D76N is potentially more prone to the cis → trans isomerization.
2.3 Molecular determinants of the differential isomer stability
Previous computational works32,35 underlined that the stability of cis in WT β2-m is strictly related to the hydrogen bond network around Pro32 and to the nature of the neighbor amino-acids. Such works identified the h-bond between the oxygen of Pro32 with the side chain of His8432,35 and the h-bond between the backbone NH of Arg3 with the backbone oxygen of His3132 as important determinants of the stability of cis in wild-type β2-m. In fact, such bonds are persistent in the cis isomer, while they are lost in the trans isomer.The geometry of the peptidyl–prolyl bond in both isomers of β2-m is shown as seen from above the N-term apical part in Fig. 3a, and it is super-imposable for both wild-type and D76N. The snapshots are taken from the most populated clusters of the wild-type free energy minima, calculated through the clustering algorithm by Daura et al.,60 implemented within the GROMACS software package (as GROMOS) with a cut-off of 2.5 Å. Table 2 reports the relevant differences in the Pro32 hydrogen bond network in both cis and trans.
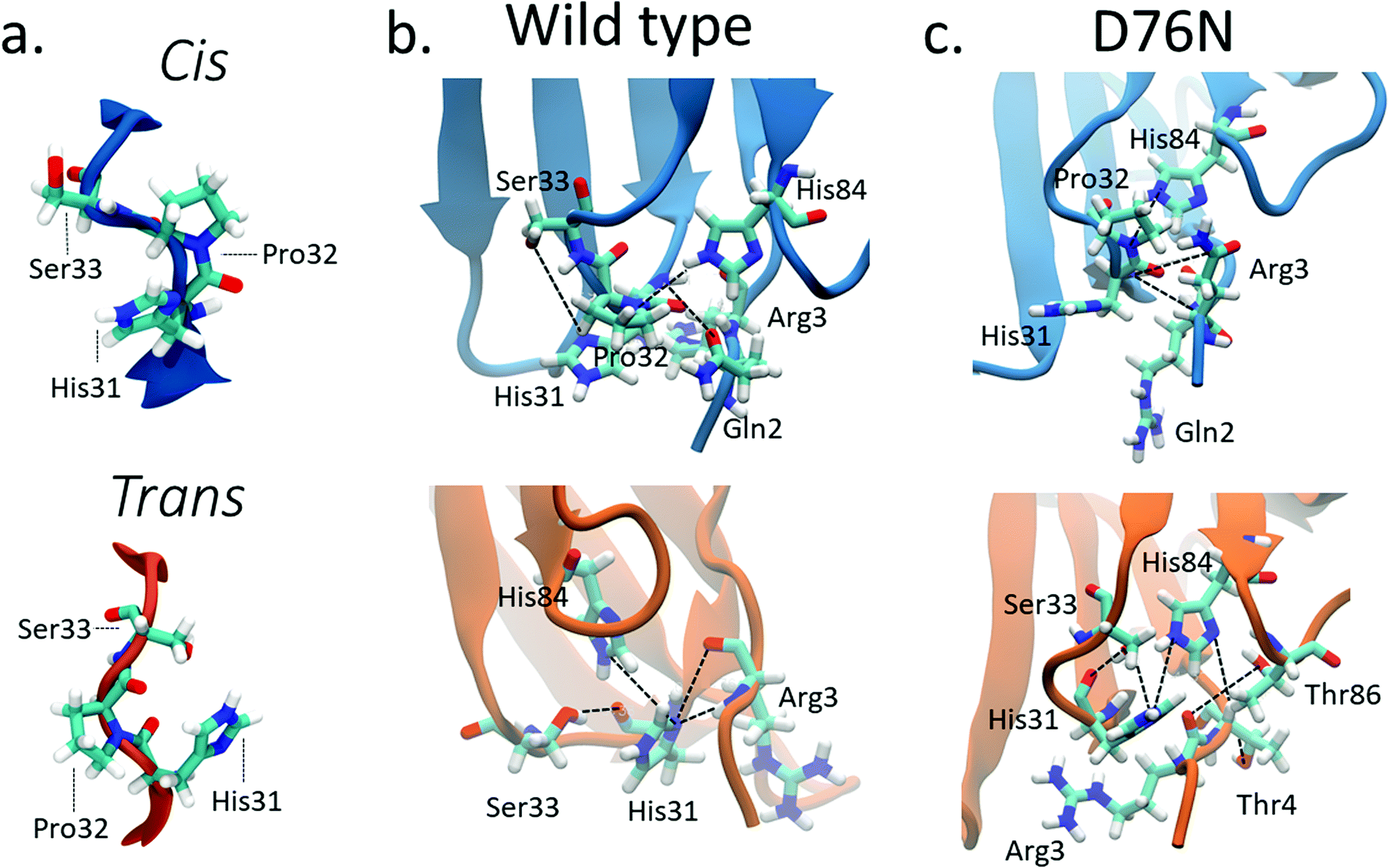 | ||
| Fig. 3 (a) Typical conformations assumed by Pro32 and its neighbors His31 and Ser33 in cis (blue) and trans (orange) isomers; hydrogen bonds stabilizing cis (blue) and trans (orange) isomers for wild-type β2-m (b) and D76N (c). Black lines connect the atoms involved in the hydrogen bonds reported in Table 2 although not all such bonds are properly formed in the figure, since some of them are mutually exclusive. | ||
| Donor | Acceptor | WT cis (%) | WT trans (%) | D76N cis (%) | D76N trans (%) |
|---|---|---|---|---|---|
| ARG3 main N | HIS31 main O | 56.09 | 0 | 45.40 | 0 |
| HIS 31 main N | ARG3 main O | 47.58 | 23.19 | 41.19 | 0 |
| HIS84 side NE2 | PR032 main O | 42.61 | 0 | 43.98 | 0 |
| HIS84 side NE2 | HIS31 main O | 0 | 28.58 | 10.46 | 37.87 |
| SER33 side OG | HIS31 main O | 0 | 21.22 | 0 | 27.87 |
| ARG3 main N | HIS31 side ND1 | 0 | 13.98 | 0 | 36.93 |
For WT, we observe the same h-bonds (see Table 2) previously reported,32,35 thus confirm previous findings. Moreover, we detect a h-bond between the backbone oxygen of Arg3 and the backbone NH of His31 that was not previously reported. Such interactions are lost (or anyway greatly reduced) in passing to the trans isomer, and are replaced by h-bonds that are less persistent than those in cis (14% to 29% in WT trans compared to the 43% to 56% in WT cis isomer, see Table 2). Such weakening of the h-bond network in trans compared to cis may well explain the inversion of isomer stability compared to proline dipeptide in solution.
The corresponding h-bond analysis for D76N shows that its cis isomer is also stabilized by the same h-bond network as WT, but such h-bonds are less persistent in D76N (41% to 45% for D76N compared to 43% to 56% for WT, see Table 2), pointing to a smaller stabilization of the cis D76N isomer compared to cis WT that agrees with the free energy data. Additionally, the h-bonds found in trans D76N are more persistent than those in trans WT, again supporting the calculated and the experimental free energy data. In both the cis and trans isomers for both WT and D76N, a series of hydrogen bonds within the Pro32 surroundings play a structural role, registering high occurrences for the entire simulation: Thr4 with Lys91 and Arg3 with Thr86 contribute to preserve the hydrophobic pocket characterizing β2-m, formed by the FG loop, BC loop and N-term. The histograms of the h-bond populations and the structure alignment are shown in Figs. S3–S5 of the ESI.† We also repeated the h-bond analysis on 500ns plain MD simulations starting from the cis WT and D76N experimental structures. Such analysis agrees with the results of metadynamics, although it suffers from a much poorer sampling. Indeed the MD simulation presents a strong dependence on the initial configuration: the population of the hydrogen bonds already present in the starting structure increases from 40–50% for the metadynamics to 65–90% for the MD. At the same time, a decrease in the population of the only monitored h-bond which forms spontaneously after >150 ns of simulations (Arg3-N to His31-O) is observed.
Along with providing a more extensive sampling, the metadynamics simulations also reveal the relevant role of hydrophobic interactions. The cis isomer is stabilized by the hydrophobic contact between the side chain of Pro32 and the side chain of Val85. This is clearly shown by the minimum distance distribution between the side chain of Pro32 and the side chain of Val85 in Fig. 4a and b. Such hydrophobic interaction is lost in the trans isomer, and thus contributes to make the native cis isomer more stable than the trans one. It is also noticeable that for trans D76N, the Pro32-Val85 interaction is less disrupted than for trans WT, contributing to the different cis–trans behavior of the two proteins. A hydrophobic contact stabilizing the trans conformers is also observed, between the side chain of Pro32 and that of Phe62 (see Fig. 4c and d). However the distance distributions show a more labile interaction than Pro32-Val85 in cis, and visual inspection of the trajectory shows that such interaction comes at the price of transiently pointing the oxygen of Pro32 toward the side chain of Phe62 with no possibility of forming other h-bond or even dipole–dipole interactions. In the PDB 2XKU structure obtained by Eichner et al.61 for the amylodogenic ΔN6 mutant, where Pro32 is in trans conformation, the structural displacement in the position of Pro32 going from cis to trans is along the same direction, although it is ampler.
 | ||
| Fig. 4 Violin plots describing the distances between the hydrophobic side chains of Pro32 and Val85 (panels a and b) and of Pro32 and Phe62 (panels c and d) for wild-type and D76N β2-m within the cis and trans basins, represented in blue and orange respectively. | ||
Remarkably, the interactions that are lost upon isomerization are also relevant for the stability of the protein against aggregation:32 losing the interaction between the backbone of His31 and Arg3 makes the N-terminus more loosely bound (for D76N the N-terminus is less bound already for the native cis conformation, see Table 2). The same happens for the FG unit whose interaction with the C strand is decreased by the loss of the His84-Pro32 h-bond, as well as by the Val85-Pro32 hydrophobic interaction (His84 and Val85 are at the end of the F strand). Val85 has been identified recently as an amyloidogenic hot spot, and the rationally designed mutant V85E was found to be both less amyloidogenic and also slightly less stable than WT.62 The decreased stability of V85E may be related to the loss of the hydrophobic interaction with Pro32 when Val is replaced with Glu. On the other hand, the hydrophobic interaction of Pro32 with Val85 (much stronger in the cis than in the trans form) may be a way to stabilize the Val85 aggregation hot-spot, a stabilization that is lost in the trans form. Remarkably, Val85 is fully solvent-exposed in the amyloidogenic ΔN6 structure 2XKU.61 Moreover, while P32G is more amyloidogenic than WT, P32V (where the hydrophobic interaction between residue 32 and Val85 is likely conserved by the mutation of Pro to Val) is not.21
In the evaluation of isomer stability, Stober and Abrams investigated the role of the protonation of His84 that takes place at low pH. It was found that upon such protonation, the trans isomer is strongly stabilized.35 To assess the role of the same protonation in the D76N variant, we performed a 1.5 μs metadynamics with protonated His84 (and leaving all the other residues unaltered). We remark that pKa of His84 is not expected to change substantially from WT to D76N.31 Based on such simulation, we confirm for D76N the same behaviour observed by Stober and Abrams for WT. We show the results for the free energy landscape in Fig. 5. Under normal protonation conditions of His84, the cis isomer of Pro32 is stabilized by the hydrogen bond to His84. Upon protonation of His84 (called His84* here), such hydrogen bond is disrupted in favour of a new His84*–Asp34 hydrogen bond, resulting in a destabilization of the cis-Pro32. As a consequence, trans D76N becomes substantially more stable than the cis form (ΔFt→c = 10.8 ± 6.7 kJ mol−1). The trans → cis counterclockwise barrier is rather unaltered with respect to the unprotonated case, measuring 78.2 ± 4.9 kJ mol−1. Reminiscent of the trans stabilization effect, the trans → cis isomerization is hindered while the backwards reaction is more favoured, with a c → t barrier of 67.4 ± 10.3 kJ mol−1.
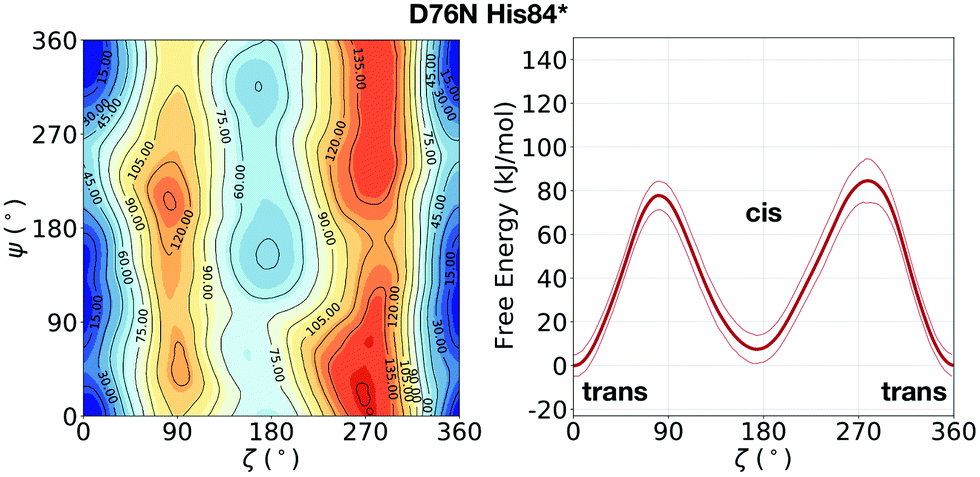 | ||
| Fig. 5 Left: FES of D76N with protonated His84 (His84*), mapped as a function of the torsional angles ζ and ψ. Right: The corresponding potential of mean force (PMF) as a function of ζ only. With respect to the previous result of D76N in Fig. 2, a significant stabilization of the trans conformer can be observed. | ||
It is not easy to understand the chain of molecular events that connects the mutation D76N to the rearrangement of h-bond network and hydrophobic contact around Pro32 that makes D76N more prone to the cis to trans transition. The mutation is in the loop connecting strand E to strand F, close to the beginning of strand F. The conformation of the loop is fluctuating, but it is somewhat different in WT and D76N: in D76N we observe that Asn76 is mostly oriented toward strands C–C′, where it is engaged in h-bond with Asn42 (in agreement with NMR findings),31 while in WT Asp76 is often found facing the C-terminus and creating a salt-bridge with Arg97. It is possible that this local perturbation extends through the F strand to reach His84 and Val85. Both these residues remain closer to Pro32 in the trans form (as shown by more persistent h-bonds and shorter Pro32-Val85 side chain distances for trans D76N compared to trans WT). The variation in the residue charge upon mutation (from negative to neutral) may also induce long range effects via electrostatic interactions through the protein. Although we could not pinpoint the exact mechanism connecting the mutation to the enhanced interactions around trans Pro32 in D76N compared to WT, the local changes around Pro32 discussed in this section are all coherent with the metadynamics free energy results reported in Section 2.2.
2.4 Comparative conformational analysis of the cis and trans isomers
Amyloidogenesis unfolds the proteins, meaning that the regular shape of the immunoglobulins domain is lost to form fibrils. In this section we analyze possible conformation changes associated to the isomerization that may point to a causal relation between isomerization and aggregation propensity.Relevant changes can be revealed by means of critical distances between adjacent structural elements. The distances between selected protein portions are monitored for each frame of the metadynamics by computing the average of the backbone distances between such portions and collecting them in histograms. The collected data are then split, depending on the ζ dihedral, into cis or trans. The distribution of the distances between each pair of sections of the protein are compared for both conformers with the aid of violin graphs.63 Each plot has been realized for both the wild-type and the D76N variant (Fig. 6). The areas of each conformer are normalized in each individual panel to show the relative distributions between cis and trans of the same variant.
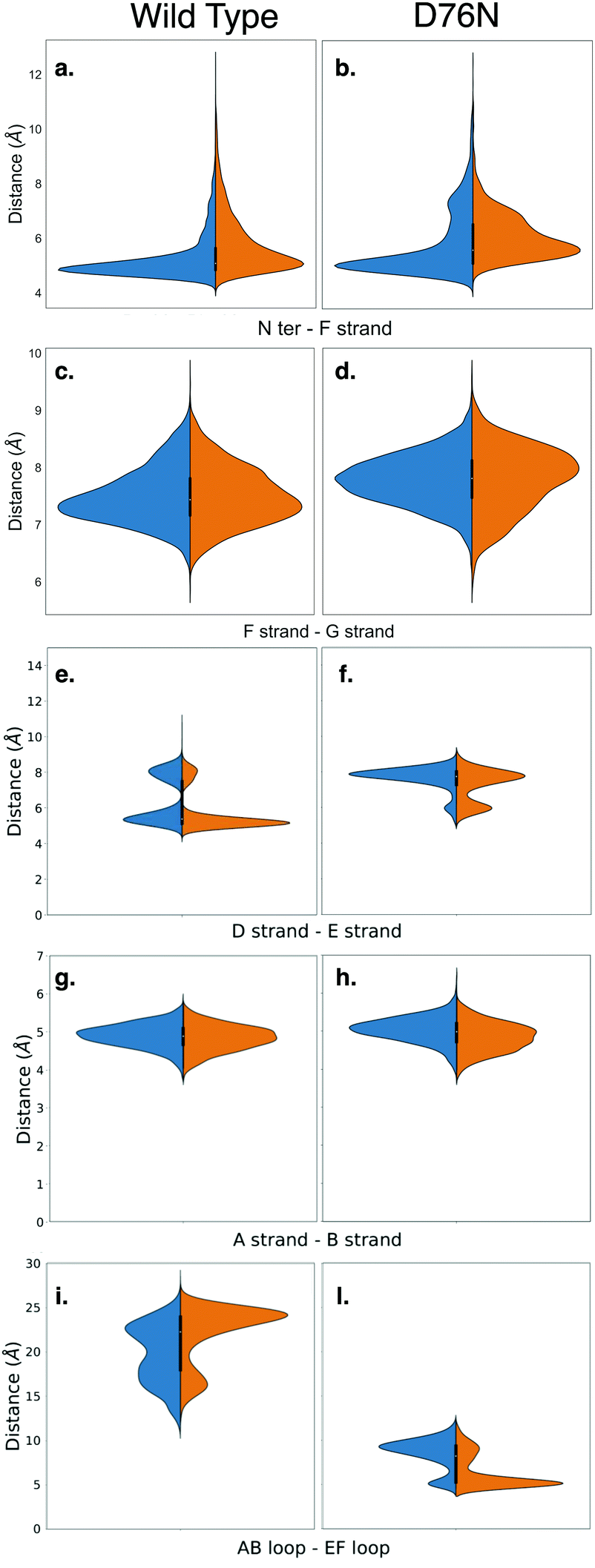 | ||
| Fig. 6 Violin plots describing the conformational changes of wild-type and D76N β2-m within the cis and trans basins, represented in blue and orange respectively. | ||
Based on the analysis of the previous section, we investigated the change in the distance between the N-terminus and the closest portion of the F strand (Arg3 Cα to His31 Cα) (Fig. 6a and b). By looking at the distribution of distances for WT (panel a), it is apparent that in passing from cis to trans there is a clear although moderate broadening of the distribution toward higher distances, i.e., the N-terminus is in fact less bound to the F-strand. This is in line with the findings on the h-bond network presented in the previous section, and marks the higher propensity of the N-terminus to open up. More evidence on the role of the opening of the N-terminus in the aggregation process has been reported for the ΔN6 variant,28,64 which corresponds to the removal of the first six residues. These studies highlight an increased propensity to amyloidogenical aggregation for such variant. The same N-terminus – F strand distance parameter for D76N (panel b) has an interesting trend. The native (cis) isomer has a main peak that coincides with that of cis WT, but it also has an additional peak showing that in the native D76N there is a sub-population of conformations where the N-terminus is already more detached from the F strand than in the WT. Upon isomerization to trans, there is a marked increase in the distance, and the distance gap between the two peaks is reduced.
We also investigate what happens at the F and the G strands nearby the C-terminus (distance between Glu77 Cα and Trp95 Cα). For the WT this is shown in Fig. 6-panel c, and the changes upon isomerization are clearly negligible. In passing to D76N (panel d), we observe first of all that the distribution is moved to higher distances compared to WT, showing that the local structure is affected by the mutation D76N in the direction of loosening the C-terminus interaction. Moreover, upon isomerization to trans, the peak of the distribution shifts to higher distances, although the distribution also becomes broader and shorter distances are more populated. Overall, this seems to indicate a higher mobility of the end of the G strand and the C-terminus. The ending G strand is considered65 a portion heavily affected during unfolding, as it may open in order to connect to another G strand of a similar protein.
Another structural element that is relevant for amyloidogenicity are the strands D and E. Their distance in wild-type β2-m has a bi-modal distribution (Fig. 6e), due to the existence of different conformations for the D strands, as known already.27 Upon isomerization the two peaks are both conserved. There is a sharpening and a slight height increase of the peak at smaller D–E distances, and a reduction of the other peak. The opening of the D–E strands has been recognized66 to be associated with a more amyloidogenic behaviour, so this change is going in the direction opposite to what expected. We have noted above that upon isomerization to trans, Pro32 is moving toward Phe62 which is within strand E. Probably this movement results in a stabilization of the D–E β-sheet, and additional conformational changes on longer time scales are required to take Pro32 in the internal position evidenced in structure 2XKU that also induces a destabilization of the D–E interaction.61 For D76N (panel f) the same bi-modal distribution is present, but now the sub-population at larger distances (around 8 Å) is higher than that at lower distances (around 6 Å). This is in line with the more amyloidogenic behaviour for the D76N, as Pro32 in trans seems to destabilize already in this conformation the D–E interaction.
We also analyze the A–B strand distance (panel g and h). It has a constant median of about 5 Å for both β2-m variants with Pro32 in both isomers, so it is not affected by either isomerization or mutation.
Finally, we characterize the distances between the AB and EF loops in the apical part of the protein (panels i and j). In the cis wild-type, the shape of the distribution of the distances between the AB loop and EF loop identifies an opening of the apical part with a median of 20 Å. The trans isomer is instead characterized by increased population characterized by a larger distance, of about 25 Å. In the amyloidogenic D76N, the cis isomer has more population characterized by a 10 Å distance between the AB and EF loops, which becomes even shorter upon Pro32 isomerization, resulting in a more closed AB and EF loops with respect to the wild-type.
All these results seem to suggest that Pro32 isomerization is only moderately affecting the structure of β2-m. Yet, some of the changes that we observe (chiefly the weakening of the h-bond network that keeps the N-terminus anchored to the protein core and the loss of hydrophobic interaction stabilizing Val85) suggest a higher amyloidogenic propensity of the trans isomer compared to the cis one. The higher amyloidogenic power of D76N is also (at least partially) explained by the higher amyloidogenic propensity of the trans isomer, since D76N is converting more easily to trans (less unfavorable isomerization free energy and lower kinetic barrier) in comparison to WT. Anyway, its higher amyloidogenic power may also depend on other intrinsic feature of the mutant, since we observe differences with WT for the cis form of D76N as well.
Each protein variant is also simulated with Pro32 in either the cis or trans isomer with molecular dynamics, in order to assess their stability over 500 ns. For the backbone of the protein, we calculate the RMSD averaged over time through the following relation:
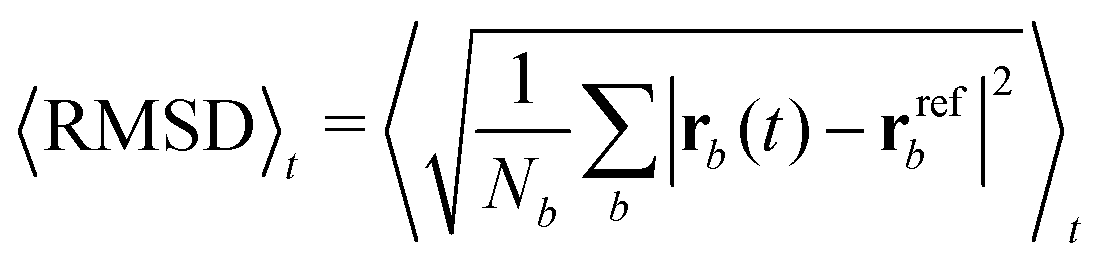 | (1) |
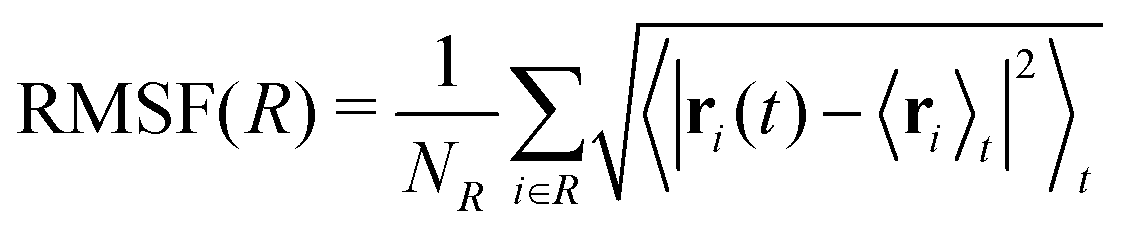 | (2) |
 | ||
| Fig. 7 Root mean square fluctuations (RMSF) for wild-type (left) and D76N (right) β2-m. Blue and orange lines represent cis and trans isomers respectively. Loops in the protein structure are highlighted with light grey vertical span. | ||
Few differences are shown in the RMSF for the wild-type β2-m, despite the configuration of Pro32 (either cis or trans): the main fluctuations are registered for the AB loop and the N- and C-termini. In the wild-type, the AB loop fluctuates more when the Pro32 is in its cis configuration. Conversely, the cis D76N shows an increase in the fluctuation of the AB loop after isomerization to trans occurs. The cause is once again to be sought in the hydrogen bond network, as the D76N tail when Pro32 is in the cis isomer is notably more involved in hydrogen bonds with respect to when it is in trans. Coherently with what reported in Table 2, upon isomerization, the D76N hydrogen bond network surrounding Pro32 is rearranged. In the cis configuration, Pro32 is bound to the terminal part of the F strand through His84. Such bond impairs the mobility of the whole BC loop. The rearrangement due to the isomerization disrupts this bond and forms one between residues within the same loop (Ser33-His31), making the whole loop loose. Finally, the mobility of the C-terminus in trans D76N is also enhanced, coherently with what was discussed above concerning the F-G distance distribution.
3 Conclusions
In summary, in this work we have characterized computationally at the molecular level the effects of the Pro32 isomerization in two variants of the amyloidogenic protein β2-microglobulin: the wild-type and the single point mutated D76N, simulated in water. This isomerization is known to occur on relatively long timescales, far beyond the computational limit of traditional MD. The method we chose to accelerate this rare event is well-tempered metadynamics, previously and successfully used to investigate the same phenomena in other biological systems.8By comparing with the behaviour of proline dipeptide in water, we have shown how the complex protein environment affects the free energy landscape as a function of two CVs involved in the isomerization of Pro32. In such environment, we estimated the free energy differences between the two isomers and the height of the barriers separating them. The relative stability of the conformers follows the same trend of the experimental results,31 according to which the cis conformer is more stable than the trans for wild-type. Also consistently with experiments, D76N decreases the stabilization of cis with respect to trans and the two isomers become almost degenerate. The height of the barriers for proline dipeptide obtained with the OPLS-AA force-field is overestimated with respect to the other force-fields used in literature. However, in comparison to the estimated barriers from DSC studies,6,31,58 our computational setup well reproduces the experimental values for all the systems investigated in the present work.
We confirm the correlation between the relative stability of each isomer to the Pro32–His84 hydrogen bond.32,35 This bond stabilizes the cis isomer in both variants, although to a different extent: more in the WT than in D76N, in agreement with computed and experimental free energy differences. A signature of the importance of His84 in the relative stability of the isomers also comes from the study of the protonated His84 (His84*). For WT, such a study was already performed,35 so here we simulated only the D76N mutant. We observed that the positive His84* is more prone to participate in a salt bridge with Asp34 rather than the hydrogen bond with the Pro32, and the stabilizing effect on the cis conformer is effectively lost as shown by the free energy results.
Moreover, we have highlighted the role of hydrophobic interactions involving the side chain of Pro32 in stabilizing the two isomers. In cis, the partner of such interaction is mostly Val85. Such residue has been identified as an aggregation hot-spot;62 the interaction with Pro32 may be a stabilization factor that is lost in the trans isomer, contributing to a larger amyloidogenicity of the trans form. Incidentally, a view that Val85 may be an aggregation hot-spot since it stabilizes the Pro32 cis conformation is unlikely, since the mutant V85E has a cis native form and less aggregation propensity than WT. Finally, we have analyzed conformational changes taking place in the protein upon isomerization to trans. Although we did not find major differences, we evidenced a loosening of the N-terminus interaction compatible with the h-bond analysis. The conformational analysis also points to a few indications of the larger amyloidogenicity of D76N already in the cis form (such as the more mobile N-terminus). The higher propensity of D76N to isomerize to the trans form may also contribute to its higher amyloidogenicity.
Overall, our results support the view that the isomerization of Pro32 to trans does favor aggregation. It is anyway difficult to create a consequential link from our results to the protein aggregation process. Yet, on a speculative ground, the mechanism that better fits our findings is not that such isomerization directly triggers conformational changes (misfolding) prodromic to aggregation. Rather, the isomerization is acting indirectly, in the sense that it creates a favorable scenario for the other processes necessary to aggregation (not accessible in the present investigation) to take place. D76N may jump into this scenario more easily than WT, which explains (or at least contributes to explain) its higher propensity to aggregate. What these further processes are is not easy to state. A combined look at our findings and literature reports suggest deeper investigations of the role of Val85 as a promising route to build another piece of the bridge connecting molecular events to formation of protein aggregates.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
MCM acknowleges the HPC Europa 3 Programme. We are grateful for computational support from the UK high performance computing service ARCHER, for which access was obtained via the UKCP consortium and funded by EPSRC grant EP/P022472/1. We also acknowledge the UK Materials and Molecular Modelling Hub for computational resources, which is partially funded by EPSRC (EP/P020194/1).References
- T. J. McCormack, C. Melis, J. Colon, E. A. Gay, A. Mike, R. Karoly, P. W. Lamb, C. Molteni and J. L. Yakel, J. Physiol., 2010, 588.22, 4415–4429 CrossRef PubMed.
- F. S. Cordes, J. N. Bright and M. S. P. Sansom, J. Mol. Biol., 2002, 323, 951–960 CrossRef CAS PubMed.
- A. A. Morgan and E. Rubenstein, PLoS One, 2013, 8, e53785 CrossRef CAS PubMed.
- S. Fischer, R. L. Dunbrack and M. Karplus, J. Am. Chem. Soc., 1994, 116, 11931–11937 CrossRef CAS.
- W. J. Wedemeyer, E. Welker and H. A. Scheraga, Biochemistry, 2002, 41, 14637–14644 CrossRef CAS PubMed.
- H. Cheng and F. Bovey, Biopolymers, 1977, 16, 1465–1472 CrossRef CAS PubMed.
- S. C. R. Lummis, D. L. Beene, L. W. Lee, H. A. Lester, R. W. Broadhurst and D. A. Dougherty, Nature, 2005, 438, 248–252 CrossRef CAS PubMed.
- A. Crnjar, F. Comitani, W. Hester and C. Molteni, J. Phys. Chem. Lett., 2019, 10, 694–700 CrossRef PubMed.
- L. Rognoni, T. Möst, G. Žoldák and M. Rief, Proc. Natl. Acad. Sci. U. S. A., 2014, 111, 5568–5573 CrossRef CAS PubMed.
- A. Kraus, Prion, 2016, 10, 57–62 CrossRef CAS PubMed.
- A. Ryo, T. Togo, T. Nakai, A. Hirai, M. Nishi, A. Yamaguchi, K. Suzuki, Y. Hirayasu, H. Kobayashi, K. Perrem, Y.-C. Liou and I. Aoki, J. Biol. Chem., 2006, 281, 4117–4125 CrossRef CAS PubMed.
- M. Stoppini and V. Bellotti, J. Biol. Chem., 2015, 290, 9951–9958 CrossRef CAS PubMed.
- S. L. Myers, S. Jones, T. R. Jahn, I. J. Morten, G. A. Tennent, E. W. Hewitt and S. E. Radford, Biochemistry, 2006, 45, 2311–2321 CrossRef CAS PubMed.
- S. Yamamoto, I. Yamaguchi, K. Hasegawa, S. Tsutsumi, Y. Goto, F. Gejyo and H. Naiki, J. Am. Soc. Nephrol., 2004, 15, 126–133 CrossRef CAS PubMed.
- M. F. Calabrese, C. M. Eakin, J. M. Wang and A. D. Miranker, Nat. Struct. Mol. Biol., 2008, 15, 965 CrossRef CAS PubMed.
- C. M. Eakin, A. J. Berman and A. D. Miranker, Nat. Struct. Mol. Biol., 2006, 13, 202 CrossRef CAS PubMed.
- V. J. McParland, N. M. Kad, A. P. Kalverda, A. Brown, P. Kirwin-Jones, M. G. Hunter, M. Sunde and S. E. Radford, Biochemistry, 2000, 39, 8735–8746 CrossRef CAS PubMed.
- T. Eichner and S. E. Radford, FEBS J., 2011, 278, 3868–3883 CrossRef CAS PubMed.
- F. Chiti, P. Mangione, A. Andreola, S. Giorgetti, M. Stefani, C. M. Dobson, V. Bellotti and N. Taddei, J. Mol. Biol., 2001, 307, 379–391 CrossRef CAS PubMed.
- A. Kameda, M. Hoshino, T. Higurashi, S. Takahashi, H. Naiki and Y. Goto, J. Mol. Biol., 2005, 348, 383–397 CrossRef CAS PubMed.
- M. Sakata, E. Chatani, A. Kameda, K. Sakurai, H. Naiki and Y. Goto, J. Mol. Biol., 2008, 382, 1242–1255 CrossRef CAS PubMed.
- T. R. Jahn, M. J. Parker, S. W. Homans and S. E. Radford, Nat. Struct. Mol. Biol., 2006, 13, 195 CrossRef CAS PubMed.
- E. Rennella, A. Corazza, S. Giorgetti, F. Fogolari, P. Viglino, R. Porcari, L. Verga, M. Stoppini, V. Bellotti and G. Esposito, J. Mol. Biol., 2010, 401, 286–297 CrossRef CAS PubMed.
- A. Corazza, E. Rennella, P. Schanda, M. C. Mimmi, T. Cutuil, S. Raimondi, S. Giorgetti, F. Fogolari, P. Viglino, L. Frydman, M. Gal, V. Bellotti, B. Brutscher and G. Esposito, J. Biol. Chem., 2010, 285, 5827–5835 CrossRef CAS PubMed.
- C. M. Eakin, A. J. Berman and A. D. Miranker, Nat. Struct. Mol. Biol., 2006, 13, 202 CrossRef CAS PubMed.
- M. F. Calabrese, C. M. Eakin, J. M. Wang and A. D. Miranker, Nat. Struct. Mol. Biol., 2008, 15, 965 CrossRef CAS PubMed.
- G. Esposito, A. Corazza and V. Bellotti, Protein Aggregation and Fibrillogenesis in Cerebral and Systemic Amyloid Disease, Springer, 2012, pp. 165–183 Search PubMed.
- G. Esposito, R. Michelutti, G. Verdone, P. Viglino, H. Hernández, C. V. Robinson, A. Amoresano, F. Dal Piaz, M. Monti, P. Pucci, P. Mangione, M. Stoppini, G. Merlini, G. Ferri and V. Bellotti, Protein Sci., 2000, 9, 831–845 CrossRef CAS PubMed.
- M. Monti, S. Principe, S. Giorgetti, P. Mangione, G. Merlini, A. Clark, V. Bellotti, A. Amoresano and P. Pucci, Protein Sci., 2002, 11, 2362–2369 CrossRef CAS PubMed.
- S. Valleix, J. D. Gillmore, F. Bridoux, P. P. Mangione, A. Dogan, B. Nedelec, M. Boimard, G. Touchard, J.-M. Goujon, C. Lacombe, P. Lozeron, D. Adams, C. Lacroix, T. Maisonobe, V. Plante-Bordeneuve, J. A. Vrana, J. D. Theis, S. Giorgetti, R. Porcari, S. Ricagno, M. Bolognesi, M. Stoppini, M. Delpech, M. B. Pepys, P. N. Hawkins and V. Bellotti, N. Engl. J. Med., 2012, 366, 2276–2283 CrossRef CAS PubMed.
- P. P. Mangione, G. Esposito, A. Relini, S. Raimondi, R. Porcari, S. Giorgetti, A. Corazza, F. Fogolari, A. Penco, Y. Goto, Y.-H. Lee, H. Yagi, C. Cecconi, M. M. Naqvi, J. D. Gillmore, P. N. Hawkins, F. Chiti, R. Rolandi, G. W. Taylor, M. B. Pepys, M. Stoppini and V. Bellotti, J. Biol. Chem., 2013, 288, 30917–30930 CrossRef CAS PubMed.
- F. Fogolari, A. Corazza, N. Varini, M. Rotter, D. Gumral, L. Codutti, E. Rennella, P. Viglino, V. Bellotti and G. Esposito, Proteins: Struct., Funct., Bioinf., 2011, 79, 986–1001 CrossRef CAS PubMed.
- A. D. MacKerell Jr, D. Bashford, M. Bellott, R. L. Dunbrack Jr, J. D. Evanseck, M. J. Field, S. Fischer, J. Gao, H. Guo and S. Ha, et al. , J. Phys. Chem. B, 1998, 102, 3586–3616 CrossRef PubMed.
- A. D. Mackerell Jr, M. Feig and C. L. Brooks III, J. Comput. Chem., 2004, 25, 1400–1415 CrossRef PubMed.
- S. T. Stober and C. F. Abrams, J. Phys. Chem. B, 2012, 116, 9371–9375 CrossRef CAS PubMed.
- A. Laio and F. L. Gervasio, Rep. Prog. Phys., 2008, 71, 126601 CrossRef.
- A. Laio and M. Parrinello, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 12562–12566 CrossRef CAS PubMed.
- A. Barducci, M. Bonomi and M. Parrinello, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2011, 1, 826–843 CAS.
- A. Barducci, G. Bussi and M. Parrinello, Phys. Rev. Lett., 2008, 100, 020603 CrossRef PubMed.
- C. Melis, G. Bussi, S. C. R. Lummis and C. Molteni, J. Phys. Chem. B, 2009, 113, 12148–12153 CrossRef CAS PubMed.
- V. Leone, G. Lattanzi, C. Molteni and P. Carloni, PLoS Comput. Biol., 2009, 5, e1000309 CrossRef PubMed.
- G. Verdone, A. Corazza, P. Viglino, F. Pettirossi, S. Giorgetti, P. Mangione, A. Andreola, M. Stoppini, V. Bellotti and G. Esposito, Protein Sci., 2002, 11, 487–499 CrossRef CAS PubMed.
- D. Van Der Spoel, E. Lindahl, B. Hess, G. Groenhof, A. E. Mark and H. J. C. Berendsen, J. Comput. Chem., 2005, 26, 1701–1718 CrossRef CAS PubMed.
- W. L. Jorgensen and J. Tirado-Rives, J. Am. Chem. Soc., 1988, 110, 1657–1723 CrossRef CAS PubMed.
- G. A. Tribello, M. Bonomi, D. Branduardi, C. Camilloni and G. Bussi, Comput. Phys. Commun., 2014, 185, 604–613 CrossRef CAS.
- M. Bonomi, G. Bussi, C. Camilloni, G. A. Tribello, P. Banás, A. Barducci, M. Bernetti, P. G. Bolhuis, S. Bottaro, D. Branduardi, R. Capelli, P. Carloni, M. Ceriotti, A. Cesari, H. Chen, W. Chen, F. Colizzi, S. De, M. De La Pierre, D. Donadio, V. Drobot, B. Ensing, A. L. Ferguson, M. Filizola, J. S. Fraser, H. Fu, P. Gasparotto, F. L. Gervasio, F. Giberti, A. Gil-Ley, T. Giorgino, G. T. Heller, G. M. Hocky, M. Iannuzzi, M. Invernizzi, K. E. Jelfs, A. Jussupow, E. Kirilin, A. Laio, V. Limongelli, K. Lindorff-Larsen, T. Löhr, F. Marinelli, L. Martin-Samos, M. Masetti, R. Meyer, A. Michaelides, C. Molteni, T. Morishita, M. Nava, C. Paissoni, E. Papaleo, M. Parrinello, J. Pfaendtner, P. Piaggi, G. Piccini, A. Pietropaolo, F. Pietrucci, S. Pipolo, D. Provasi, D. Quigley, P. Raiteri, S. Raniolo, J. Rydzewski, M. Salvalaglio, G. C. Sosso, V. Spiwok, J. Sponer, D. W. H. Swenson, P. Tiwary, O. Valsson, M. Vendruscolo, G. A. Voth, A. White and the Plumed Consortium, Nat. Methods, 2019, 16, 670–673 CrossRef PubMed.
- H. Berendsen, J. Grigera and T. Straatsma, J. Phys. Chem., 1987, 91, 6269–6271 CrossRef CAS.
- G. Brancolini, A. Corazza, M. Vuano, F. Fogolari, M. C. Mimmi, V. Bellotti, M. Stoppini, S. Corni and G. Esposito, ACS Nano, 2015, 9, 2600–2613 CrossRef CAS PubMed.
- C. Cantarutti, S. Raimondi, G. Brancolini, A. Corazza, S. Giorgetti, M. Ballico, S. Zanini, G. Palmisano, P. Bertoncin, L. Marchese, P. Patrizia Mangione, V. Bellotti, S. Corni, F. Fogolari and G. Esposito, Nanoscale, 2017, 9, 3941–3951 RSC.
- G. Brancolini, M. C. Maschio, C. Cantarutti, A. Corazza, F. Fogolari, V. Bellotti, S. Corni and G. Esposito, Nanoscale, 2018, 10, 4793–4806 RSC.
- M. C. Maschio, G. Brancolini and S. Corni, J. Self-Assem. Mol. Electron., 2018, 6, 35–60 CrossRef CAS.
- F. Comitani, K. Rossi, M. Ceriotti, M. E. Sanz and C. Molteni, J. Chem. Phys., 2017, 146, 145102 CrossRef PubMed.
- A. Crnjar, F. Comitani, C. Melis and C. Molteni, Interface Focus, 2019, 9, 20180067 CrossRef PubMed.
- G. Bussi, D. Donadio and M. Parrinello, J. Chem. Phys., 2007, 126, 014101 CrossRef.
- M. Parrinello and A. Rahman, J. Appl. Phys., 1981, 52, 7182–7190 CrossRef CAS.
- B. Hess, H. Bekker, H. J. C. Berendsen and J. G. E. M. Fraaije, J. Comput. Chem., 1997, 18, 1463–1472 CrossRef CAS.
- G. Bussi and A. Laio, Nat. Rev. Phys., 2020, 2, 200–212 CrossRef.
- E. Beausoleil and W. D. Lubell, J. Am. Chem. Soc., 1996, 118, 12902–12908 CrossRef CAS.
- C. M. Taylor, R. Hardré, P. J. Edwards and J. H. Park, Org. Lett., 2003, 5, 4413–4416 CrossRef CAS PubMed.
- X. Daura, K. Gademann, B. Jaun, D. Seebach, W. F. Van Gunsteren and A. E. Mark, Angew. Chem., Int. Ed., 1999, 38, 236–240 CrossRef CAS.
- T. Eichner, A. P. Kalverda, G. S. Thompson, S. W. Homans and S. E. Radford, Mol. Cell, 2011, 41, 161–172 CrossRef CAS PubMed.
- C. Camilloni, B. M. Sala, P. Sormanni, R. Porcari, A. Corazza, M. de Rosa, S. Zanini, A. Barbiroli, G. Esposito, M. Bolognesi, V. Bellotti, M. Vendruscolo and S. Ricagno, Sci. Rep., 2016, 6, 25559 CrossRef CAS PubMed.
- J. L. Hintze and R. D. Nelson, Am. Stat., 1998, 52, 181–184 Search PubMed.
- S. G. Estacio, H. Krobath, D. Vila-Vicosa, M. Machuqueiro, E. I. Shakhnovich and P. F. N. Faisca, PLoS Comput. Biol., 2014, 10, 1–17 CrossRef PubMed.
- K. Domanska, S. Vanderhaegen, V. Srinivasan, E. Pardon, F. Dupeux, J. A. Marquez, S. Giorgetti, M. Stoppini, L. Wyns, V. Bellotti and J. Steyaert, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 1314–1319 CrossRef CAS PubMed.
- A. Corazza, F. Pettirossi, P. Viglino, G. Verdone, J. Garcia, P. Dumy, S. Giorgetti, P. Mangione, S. Raimondi and M. Stoppini, et al. , J. Biol. Chem., 2004, 279, 9176–9189 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0cp04780e |
| This journal is © the Owner Societies 2021 |