
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
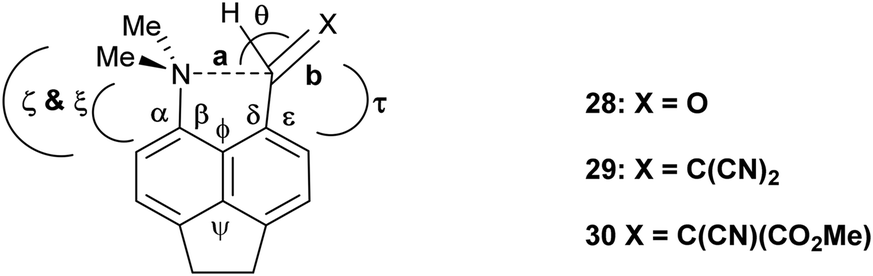
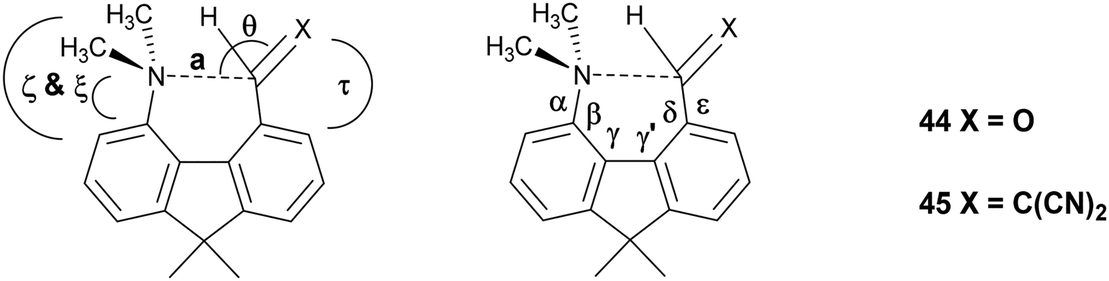
Interaction, bond formation or reaction between a dimethylamino group and an adjacent alkene or aldehyde group in aromatic systems controlled by remote molecular constraints†
Jonathan C.
Bristow
,
Stacey V. A.
Cliff
,
Songjie
Yang
and
John D.
Wallis
 *
*
School of Science and Technology, Nottingham Trent University, Clifton Lane, Nottingham NG11 8NS, UK. E-mail: john.wallis@ntu.ac.uk
First published on 27th May 2021
Abstract
Peri–peri interactions in naphthalene systems control the degree of bond formation between a peri-dimethylamino group and a polarised alkene or aldehyde group. Two peri-phenyl groups, which repel, induce closer N⋯C interactions or bond formation, while the ethylene link in the corresponding acenaphthene system has the opposite effect, and for the more electron-deficient alkenes lead to formation of a fused azepine ring initiated by the tert-amino effect. In related 1,8-fluorene derivatives N⋯C interactions occur for an aldehyde and a moderately polarised alkene, but fused azocines are formed when the alkene is more reactive.
Introduction
Intramolecular interactions between a nucleophile and an electrophile can be considered as representing different stages of the reaction between them, depending on their separation.1 This approach originated with structural studies on trans-annular N⋯C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O interactions in pyrrolizidine alkaloids.2 However, the peri-naphthalene skeleton has provided a more convenient system open to the study of a wide variety of such interactions starting with the X-ray structural determinations of Dunitz et al. on Me2N/CO, MeO/CO and HO/CO systems as in 1–3.3 Such interactions can change the chemical properties of the groups, e.g. the dimethylamino aldehyde 4 protonates on oxygen, not nitrogen, with HCl to give salt 5 with formation of a N–C bond.4,5 Studies have been extended to interactions between electron-rich centres and electrophilic polar multiple bonds such as alkynes, nitriles and alkenes in 6–8.6–10 The interactions between a dimethylamino group and a range of polarised alkenes has been studied the most intensively. For the most electrophilic alkenes a long bond (1.60–1.66 Å) can form between the two groups producing a zwitterionic doubly fused five-membered ring e.g. in 9 and 10. The constraint applied by the peri-naphthalene system has been used for studying other interactions such as unusual hydrogen bonding situations11 and through space magnetic coupling between specific elements.12 The 2,2′-disubstituted biphenyl system has also been employed, with the 1,5 interactions in naphthalenes replaced by 1,6 interactions with greater freedom of movement between the groups due to the possibility of rotation about the inter–ring bond. Thus, there is a long Me2N⋯CHO interaction in 11 (2.989(2) Å), but formation of a zwitterion with a six-membered ring in 12, with a Me2N+–C(CN)2− bond (1.586(3) and 1.604(3) Å).13
O interactions in pyrrolizidine alkaloids.2 However, the peri-naphthalene skeleton has provided a more convenient system open to the study of a wide variety of such interactions starting with the X-ray structural determinations of Dunitz et al. on Me2N/CO, MeO/CO and HO/CO systems as in 1–3.3 Such interactions can change the chemical properties of the groups, e.g. the dimethylamino aldehyde 4 protonates on oxygen, not nitrogen, with HCl to give salt 5 with formation of a N–C bond.4,5 Studies have been extended to interactions between electron-rich centres and electrophilic polar multiple bonds such as alkynes, nitriles and alkenes in 6–8.6–10 The interactions between a dimethylamino group and a range of polarised alkenes has been studied the most intensively. For the most electrophilic alkenes a long bond (1.60–1.66 Å) can form between the two groups producing a zwitterionic doubly fused five-membered ring e.g. in 9 and 10. The constraint applied by the peri-naphthalene system has been used for studying other interactions such as unusual hydrogen bonding situations11 and through space magnetic coupling between specific elements.12 The 2,2′-disubstituted biphenyl system has also been employed, with the 1,5 interactions in naphthalenes replaced by 1,6 interactions with greater freedom of movement between the groups due to the possibility of rotation about the inter–ring bond. Thus, there is a long Me2N⋯CHO interaction in 11 (2.989(2) Å), but formation of a zwitterion with a six-membered ring in 12, with a Me2N+–C(CN)2− bond (1.586(3) and 1.604(3) Å).13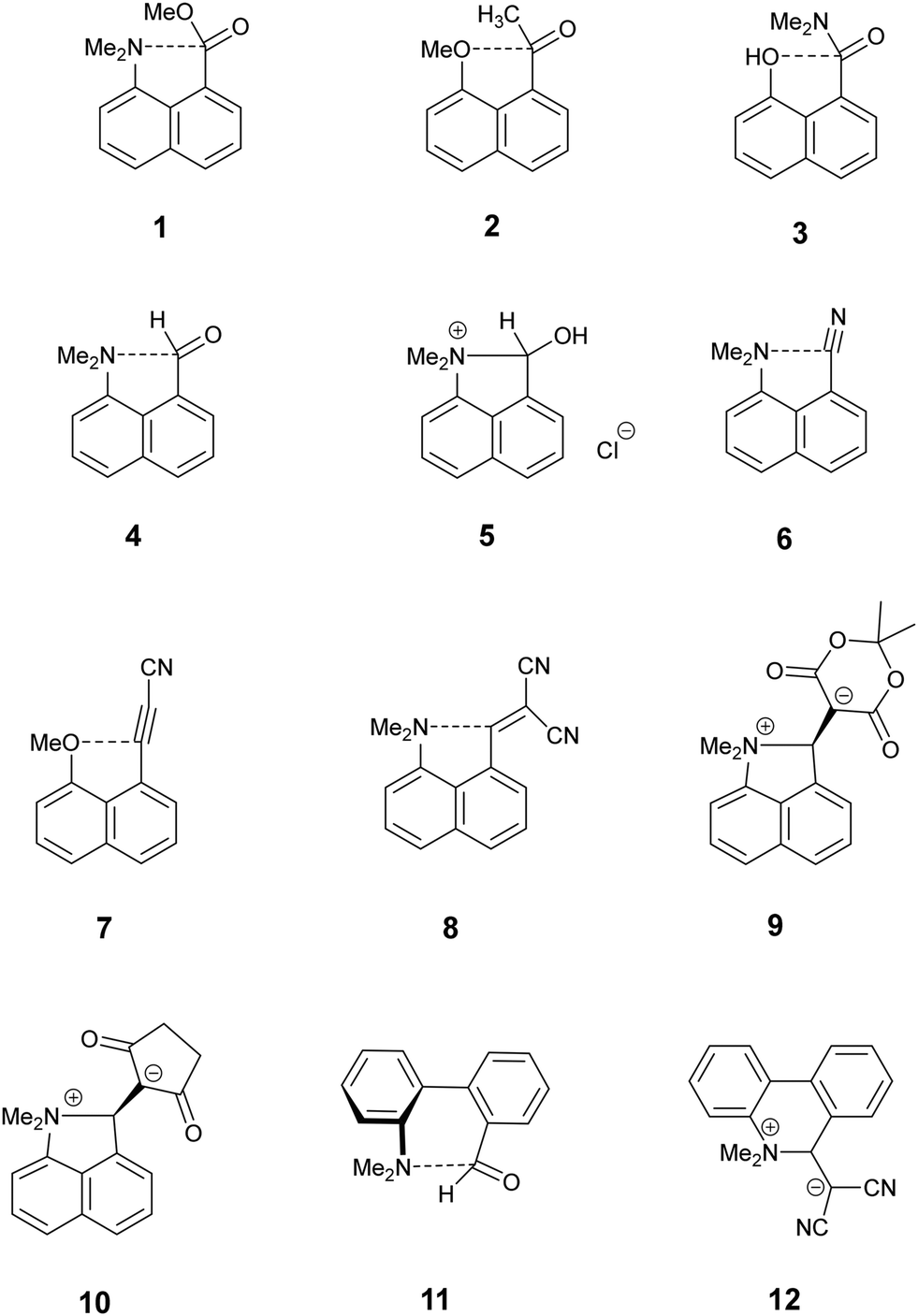
Recently we demonstrated that for peri-naphthalenes containing a Me2N- group adjacent to a –CHC(CN)2 group, the Me2N⋯CC separations can be controlled by substituents at the opposite pair of peri-positions, e.g. an ethylene bridge as in acenaphthene 13 opens up the separation between the two groups, while two peri-phenyl groups, which repel each other, reduces the separation in 14.14 Furthermore, we observed a temperature variable separation between the groups in the salt 15. Remarkably the Me2N⋯C separation at 200 K is 2.098(4) Å but reversibly contracts to 1.749(3) Å at 100 K. From all these data we were able to propose a preliminary reaction coordinate for the reaction between the groups. Here we now report the structures of two families of peri-naphthalenes with a dimethylamino group next to different electrophilic groups with either an ethylene bridge or two phenyl groups at the opposite peri-positions. Furthermore, we report the structures of a small family of 9,9-dimethylfluorenes with dimethylamino and electrophilic groups adjacent, which are designed as modified biphenyl systems in which the phenyl rings are constrained to be close to coplanar but pulled away from each other by the single carbon link between the two rings. A peri-disubstituted acenaphthene system has been used to investigate nucleophilic attack at silicon15 and to prepare frustrated lone pair systems.16
Discussion
Peri-Diphenyls derivatives
1,8-Diphenylnaphthalene17 was converted in three steps to its 4-dimethylamino derivative 16, which was peri-lithiated using n-butyl lithium, and converted to the aldehyde 17 with DMF in 34% yield. The aldehyde underwent Knoevenagel condensations with malonitrile, methyl cyanoacetate and cyclohexan-1,3-dione to give three derivatives 18, 20 and 21 with two nitriles, a nitrile and an ester or a cyclic diketone, respectively, which activate the alkene to nucleophilic attack from the adjacent dimethylamino group (Scheme 1). Solution NMR studies suggests that only 21 has a bond between the peri-groups, since there is no low field 13C NMR resonance at ca. 160 ppm for the peri-alkene carbon but a signal instead at 89.8 ppm. The crystal structures of the dimethylamino derivative 16, the peri-aldehyde 17 and the Knoevenagel products were determined by X-ray crystallography to study the interaction between the functional groups. We have already reported the structure of 18.14 | ||
| Scheme 1 (a) n-BuLi/hexane/80 °C/72 h; DMF/THF/−78 °C; (b)–(d) malonitrile, methyl cyanoacetate or cyclohexane-1,3-dione/ethylenediammonium diacetate cat./methanol/reflux. | ||
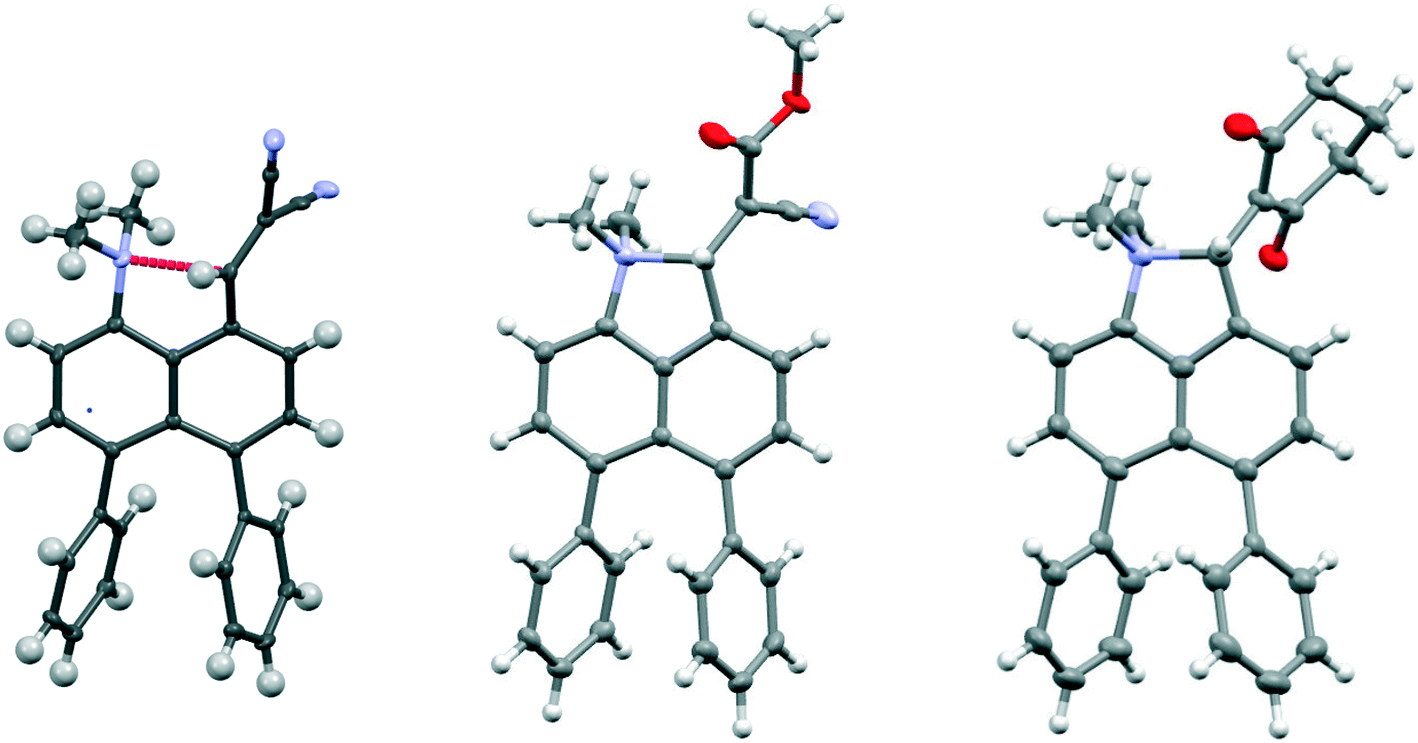
Comparison of the structures of the dimethylamino derivative 16 and the peri-dimethylamino-aldehyde 17 show how the nitrogen lone pair which is partially conjugated with the naphthalene ring in 16 has oriented towards the aldehyde carbon in 17 (Fig. 1). Thus, the torsion angles of the N–CH3 bonds with the aromatic ring in aldehyde 17 are much less asymmetric: 51.6(3)/−78.0(3) cf. 23.9(2)/−105.1(2)° in 16, as the lone pair is rotated further away from alignment with the p orbitals of the aromatic ring (Table 1). The two phenyl rings in aldehyde 17 are tilted at 56.7 and 59.2° in the same sense from the best naphthalene plane, and lie at 20.7° to each other (ESI:† Table S2). The phenyl groups are splayed apart in the naphthalene plane, with displacements of 4–4.5° from their symmetrical positions at their peri-attachment positions, and a contact of 3.012 Å between their ipso carbon atoms. The nearby exo angle ψ between the fused rings in the naphthalene core is expanded to 126.1(2)°. The latter leads to the corresponding exo angle ϕ at the naphthalene between the Me2N and CHO groups being contracted to 117.5(2)° to produce a closer Me2N⋯CHO separation of 2.309(3) Å. This is 0.18 Å shorter than in the case of 44 without the two phenyl groups (2.489(6) Å). In other respects the molecular geometries of 17 and 4 are very similar (Table 1), for example the Me2N⋯CO angles are 112.56(17) and 113.5(3)° respectively. It is of note that the difference between the exo angles ϕ and ψ in the peri-amino-aldehyde 17 is larger than in the amine 16 which lacks an electrophilic peri-neighbour (8.6 v 5.4°) suggesting that the attractive Me2N/CHO interaction contributes to this asymmetry in the exo angles.
 | ||
| Fig. 1 Face-on and side-on views of the molecular structure of 1-dimethyamino-4,5-diphenylnaphthalene 16 (top), and of the corresponding peri-dimethyamino-aldehyde 17 (middle) showing the change in orientation of the dimethylamino group; side-on view of peri-aldehyde 17 (bottom) showing the splaying apart of the phenyl groups. | ||
|
|
||||||
|---|---|---|---|---|---|---|
| a/Å | b/Å | θ/° | ζ & ξ/°a | τ/°a | ΔN, C/Åb | |
| 16 | — | — | — | 23.9(2)/−105.1(2) | — | 0.0646(19), — |
| 17 | 2.309(3) | 1.216(3) | 112.56(17) | 51.6(3)/−78.0(3) | 58.9(3) | 0.0610(19), −0.204(3) |
| 18 | 2.3603(19) | 1.356(2) | 114.21(11) | 36.9(2)/−94.95(18) | 51.0(2) | 0.2526(19), −0.4976(19) |
| 4 4 | 2.489(6) | 1.213(6) | 113.5(3) | 44.5(5)/−85.2(5) | 57.3(5) | 0.220(3), −0.234(4) |
| 8 8 | 2.413(2) | 1.354(2) | 112.51(12) | 49.7(2)/−81.5(2) | 56.5(2) | 0.166(2), −0.270(2) |

| α/° | β/° | δ/° | ε/° | ϕ/ψ/° | ||
|---|---|---|---|---|---|---|
| a ζ, ξ and τ: torsion angles. b ΔN, C: deviations of peri substituent atoms from the naphthalene's best plane. c Parent = 1,8-diphenylnaphthalene.18 | ||||||
| Parentc | — | — | — | — | 119.9(2)/126.2(2) | |
| 16 | 123.19(14) | 117.65(14) | — | — | 119.42(15)/124.78(14) | |
| 17 | 124.8(2) | 114.8(2) | 121.7(2) | 118.1(2) | 117.5(2)/126.1(2) | |
| 18 | 124.56(13) | 115.34(12) | 119.56(12) | 119.83(12) | 117.74(12)/126.24(13) | |
| 4 4 | 124.3(4) | 116.0(3) | 122.2(4) | 118.5(4) | 120.6(3)/121.5(4) | |
| 8 8 | 124.30(16) | 115.92(14) | 120.15(14) | 120.27(14) | 120.42(13)/122.65(15) | |
We have already reported that a similar effect is shown in a crystal of the toluene solvate of the dinitrile 18, where the installation of the two phenyl groups led to the reduction in the Me2N⋯CHC(CN)2 by 0.054 Å to 2.3603(19) Å14 compared to the corresponding molecule 88 without the phenyl substituents (Table 1). The phenyl groups take similar orientations to those in 16 and 17, and the exo angles, ϕ and ψ, change to 117.74(12) and 126.24(13)° (Fig. 2). The larger separation between the two functional groups in dinitrile 18, compared to the aldehyde 17 (2.359 cf. 2.309 Å) is mainly attributable to the larger displacements of the functional groups out of the naphthalene plane in opposite directions. It is important to note that the exact molecular structure is dependent on both the attraction between the groups and optimisation of crystal packing, thus for the peri-diphenyl series the Me2N⋯C is shorter for the aldehyde than for the dinitrile (2.309 v 2.359 Å), but it is the other way round for the series without the phenyl groups (2.489 v 2.413 Å), though the differences are quite small.
 | ||
| Fig. 2 Molecular structures of the peri-diphenyl naphthalene derivatives 18 (left), 19 (middle) and 21 (right). | ||
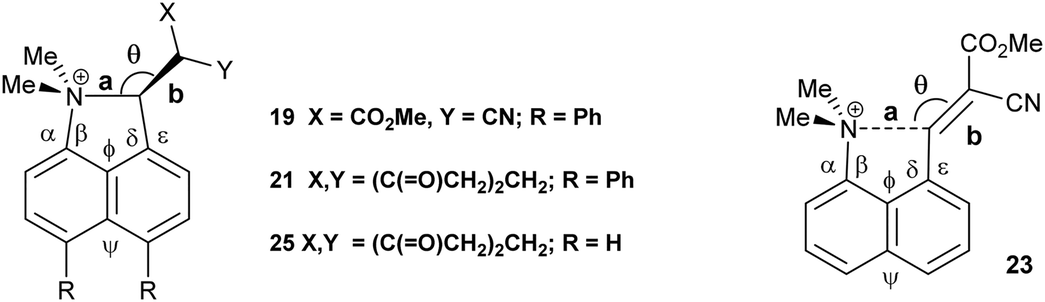
Replacement of one of the nitriles in 18 with a methyl ester group has surprising consequences. Although, this compound has a similar open structure 20 in CDCl3 solution according to NMR, in the crystal structure it adopts a closed zwitterionic structure 19 where the dimethylamino group has added to the alkene (Fig. 2), promoted by the presence of the diphenyl groups. In contrast, the analogue without the two phenyl groups, cyanoester 23, adopts an open structure in the crystalline state with a Me2N⋯CHC(CN)CO2Me separation of 2.595(2) Å, an even larger value than in the corresponding dinitrile 8 (Table 2).8 The difference in exo angles for the closed structure is, of course, considerably larger than for the aldehyde 17 and dinitrile 18: 17.6° v 8.6–8.9°. The formation of the five-membered ring reduces angle ϕ more and opens angle ψ further, illustrating the interdependence of these two angles. The disposition of the phenyl groups is similar to other diphenyl derivatives (Table S2†).
|
|
|||||
|---|---|---|---|---|---|
| a/Å | b/Å | θ/° | N–CH3/Å | φ/ψ/° | |
| 19 | 1.6719(14) | 1.4543(14) | 115.94(9) | 1.4899(16)/1.4932(15) | 112.28(9)/129.88(9) |
| 21 | 1.620(4) | 1.484(4) | 116.0(3) | 1.503(4)/1.496(4) | 111.6(3)/130.0(3) |
| 23 14 | 2.595(2) | 1.3485(18) | 116.23(10) | 1.4586(18)/1.4639(17) | 121.39(13)/122.56(12) |
| 25 10 | 1.6310(19) | 1.4863(19) | 113.99(10) | 1.4995(17)/1.5020(18) | 113.16(12)/128.31(14) |
| α/° | β/° | δ/° | ε/° | ||
|---|---|---|---|---|---|
| 19 | 127.80(9) | 109.84(9) | 110.75(9) | 129.78(9) | |
| 21 | 126.7(3) | 110.7(3) | 110.3(3) | 130.5(3) | |
| 23 | 123.15(13) | 116.82(11) | 122.53(12) | 117.89(12) | |
| 25 | 128.64(12) | 109.27(13) | 109.46(12) | 131.62(14) | |
The Me2N–C bond formed between peri-substituents in 19 is 1.6719(14) Å long and the former alkene bond is now 1.4543(14) Å. In the two independent molecules of the biphenyl derivative 1213 where a dimethylamino group has added to an alkenedinitrile to form a less strained six-membered ring, the Me2N–C bonds (1.586(3) and 1.604(3) Å) are more than 0.07 Å shorter than in 19, and the broken alkene bonds (1.493(3) and 1.487(3) Å) are 0.04 Å longer than in 19. Thus, in the naphthalene 19 the Me2N–C bond can be considered as not fully formed, and the alkene bond as not fully broken. An earlier stage in the Michael reaction between the two groups is illustrated in the chloride salt of naphthalene 24 which has one peri-dimethylammonium group in place of the phenyl groups. In this case the Me2N–C bond is even longer (1.754(6) Å) than in 19, and the former alkene slightly shorter (1.442(5) Å).14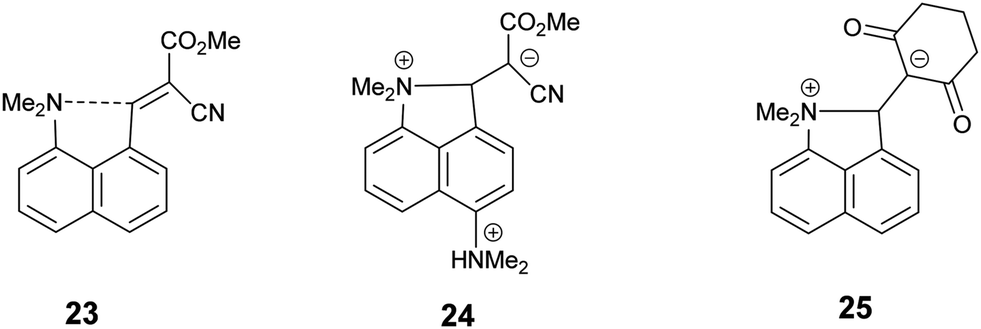
The accumulation of positive charge on the dimethylamino group in 19 leads to longer N–CH3 bonds (1.4899(16) and 1.4932(15) Å cf. 1.4586(18) and 1.4639(17) in 23 without phenyl groups, and the development of negative charge on the carbon atom between the nitrile and ester groups causes the bonds from the carbanionic centre to these groups to be shortened (C–CN, C–C(O): 1.4100(15) and 1.4289(14) Å cf. 1.4420(18) and 1.4851(19) Å in 23). Furthermore, the lengths of both bonds from the naphthalene ring to the peri-groups are increased on the formation of the bond between them: (C(nap)–N, C(nap)–C: 1.4720(12), 1.5058(14) Å cf. 1.4259(17) and 1.4772(19) Å in 23).
Study of a crystal of the chloroform solvate of 21, the Knoevenagel product formed from the diphenyl aldehyde 17 with cyclohexan-1,3-dione, showed that it also adopts a closed structure, as does its analogue without peri-phenyl groups 25.10 The Me2N–C bond (1.620(4) Å) is 0.05 Å shorter than in the corresponding cyanoester 19 and the bond to the carbanion centre (1.484(4) Å) is 0.03 Å longer due to the stronger carbanion stabilising ability of two ketone groups compared to an ester and a nitrile group. The presence of the two phenyls has made little difference to the Me2N–C bond compared to that the analogue 25 without phenyl groups. The expected small increase and decrease in the exo angles ψ and ϕ respectively, are compensated by changes in the angles at the peri-substituents (α, β, δ and ε). It is of note that for the diphenylnaphthalene derivatives whose crystals do not include solvent, 16, 17, and 19, there is common packing motif, in which two molecules lie such that the two phenyl groups of one lie over the naphthalene of the other and vice-versa (Fig. S1, ESI†).
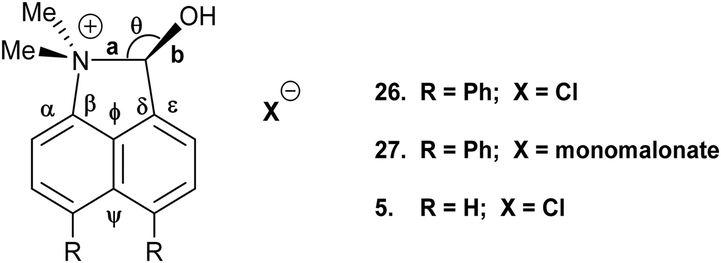
Addition of acid to the aldehyde 17 leads to protonation of the carbonyl group and formation of a N–C bond between the peri-groups. Thus, addition of HCl gave the chloride salt 26 as a DCM solvate, while an attempted Knoevenagel reaction with Meldrum's acid gave the analogous salt with a monomalonate anion 27 (Scheme 2). The crystal structures of both salts were determined (Fig. 3, Table 3). The phenyl groups have enhanced the difference in the exo angles ψ and φ between the peri-positions from 14.4° in naphthalene salt 55 which has no peri-phenyl groups, to 18.2–18.4° in cations 26 and 27. This leads to shorter Me2N–C(OH) bonds, 1.617(5) and 1.621(2) Å, compared to 5 (1.638(2) Å) without the phenyl groups, and correspondingly the C–OH bonds are slightly longer: 1.360(4) and 1.363(2) v 1.353(2) Å. The anion in each salts form a hydrogen bond to the cation's hydroxyl group (Fig. 3). All these structures with two peri-phenyl groups show evidence of the effect of the repulsion between the two phenyl groups on shortening the interaction or bond between the two opposite peri-groups, and in the case of 19 forcing the formation of the bond in the solid state.
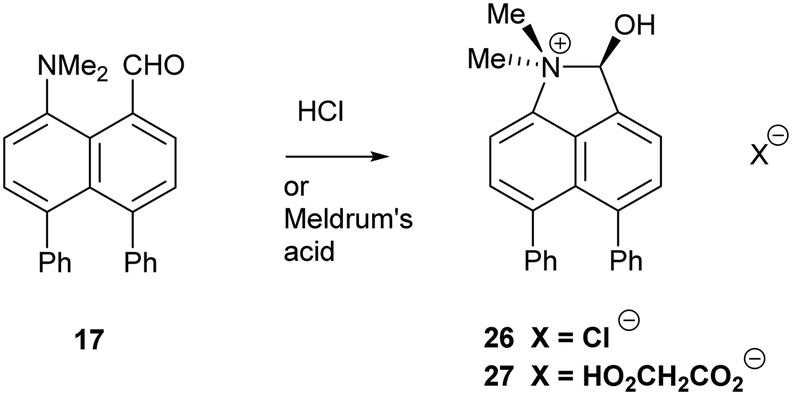 | ||
| Scheme 2 Preparation of salts of 17. | ||
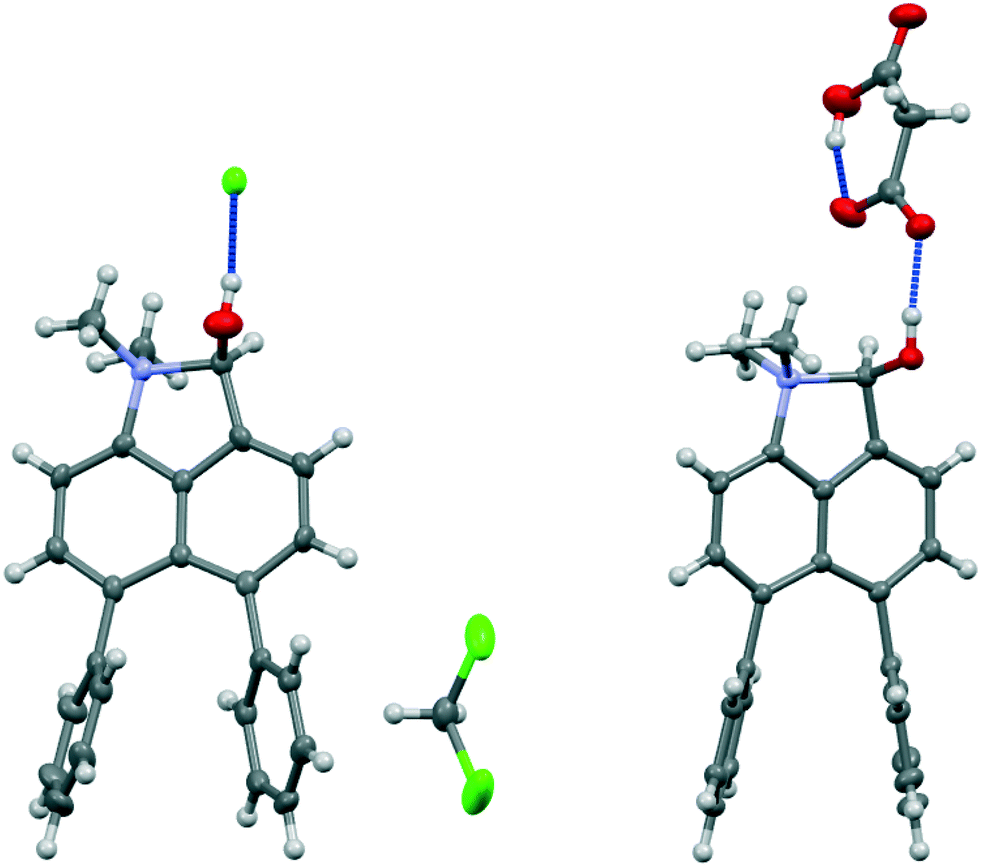 | ||
| Fig. 3 Salts formed from aldehyde 17 by O-protonation: a chloride salt 26 as a DCM solvate (left) and a monomalonate salt 27 (right). | ||
|
|
|||||
|---|---|---|---|---|---|
| a/Å | b/Å | N–CH3/Å | θ/° | φ/ψ/° | |
| 26 | 1.617(5) | 1.360(4) | 1.491(5)/1.499(5) | 111.3(3) | 112.1(3)/130.3(4) |
| 27 | 1.621(2) | 1.363(2) | 1.487(3)/1.499(2) | 110.92(15) | 112.03(17)/130.42(17) |
| 5 5 | 1.638(2) | 1.353(2) | 1.497(2)/1.498(2) | 111.64(12) | 113.92(15)/128.32(15) |
| α/° | β/° | δ/° | ε/° | ϕ/ψ/° | |
|---|---|---|---|---|---|
| a ζ, ξ and τ: torsion angles. | |||||
| 28 | 121.74(13) | 119.27(13) | 124.14(13) | 116.08(13) | 129.50(14)/111.38(13) |
| 29 | 121.81(18) | 119.57(17) | 121.32(18) | 119.88(18) | 128.54(17)/111.25(18) |
| 122.77(18) | 118.23(16) | 121.87(12) | 119.47(17) | 127.30(18)/111.73(17) | |
| 30 | 121.76(13) | 119.53(12) | 121.36(12) | 119.72(12) | 128.62(13)/111.47(13) |
Attempted reaction of aldehyde 17 with nitromethane in the presence of ethylenediammonium diacetate led to a small amount of the crystalline bis-imine 22, formed by reaction between two equivalents of the aldehyde 17 and one of the catalyst, whose structure was determined by X-ray crystallography (Fig. 4). The structure is particularly informative with respect to the influence of the peri-phenyl groups. The imine and Me2N- groups are not well oriented for mutual interaction, both preferring to conjugate with the naphthalene ring. The two imine bonds make torsions of 36.4(3) and 42.0(3)° to their nearest aromatic C,C(H) bonds, and for both of the pyramidal –NMe2 groups one N-methyl bond make a torsion angle of just 23.1(3) and 26.0(3)° to the nearest aromatic C,C(H) bond.
 | ||
| Fig. 4 Face-on view of bis-imine 22 (top) and a view through one naphthalene ring's plane showing the strong displacement of all pairs of peri-substituents out of their naphthalene planes (bottom). | ||
The Me2N⋯C separations are quite long (2.667(3) and 2.711(3) Å) and their angles of interaction (Me2)N⋯CN are not favourable (125.84(15) and 129.12(15)°). In contrast to the other structures discussed above, the phenyl groups have a much lower distorting effect on the two exo angles at either side of each naphthalenes. Thus, instead of a difference of 8.5/8.6° between exo angles ψ and φ seen in 17 and 18, this is reduced to 1.8 and 2.6° in the two naphthalenes of the bis-imine. However, the naphthalene rings are strongly twisted, with angles of 9.9 and 11.5° between their benzene rings' best planes, so that all four sets of peri-substituent atoms are strongly displaced out of their best naphthalene planes, to opposite sides, by 0.271(2)–0.605(2) Å. The relative dispositions of the phenyl groups relative to the naphthalene plane and at the peri-positions, however, remain similar to those in the other diphenyl derivatives (Table S1†). In the case of bis-imine 22 the phenyl groups do not exert their normal effect because the Me2N/CNR interactions are not attractive enough, or possibly repulsive, at ca. 2.5 Å.19 In this case, faced with two unfavourable peri interactions, the naphthalene rings distort strongly out of the aromatic plane, rather than within it, and so move the peri-substituents apart. Thus, there are limits to the compressive influence of the two peri-phenyl groups on an opposite set of peri-substituents.
Acenaphthene derivatives
To test the effect of tightening the opposite exo angle ψ on the separation of the interacting functional groups a series of acenaphthene derivatives which have an ethylene bridge between the second set of peri-positions was synthesized (Scheme 3). Aldehyde 28 was prepared from the known peri-dimethylamino-bromoacenaphthene16 by halegon-lithium exchange and treatment with DMF in 55% yield. Knoevenagel condensations of aldehyde 28 with malonitrile and methyl cyanoacetate, catalysed by ethylenediamine diacetate in refluxing methanol yielded 2914 and 30 in 88–91% yields. However, reaction with cyclic dicarbonyl compounds: Meldrum's acid, cyclohexane-1,3-dione and cyclopentane-1,3-dione in DMSO at room temperature with no catalyst, gave directly the fused azepines 34–36, formed by reaction between the functional groups of the initially formed Knoevenagel products 31–33. The structures of 28–30 and 34–36 were determined by X-ray crystallography. For 28–30 the small exo angle ψ within the five membered ring of the acenaphthene (111.25(18)–111.73(17)°) leads to a larger exo angle ϕ between the two functional groups (127.03(18)–129.50(14)°), which increases the distance between them, the opposite to what was observed for the diphenyl derivatives. In the same way, it is the added space between the functional groups in the expected Knoevenagel products 31–33 which leads to a ready reaction between them which is initiated by hydride transfer from the N–CH3 group to the polarised alkene according to the tertiary amino effect.20,21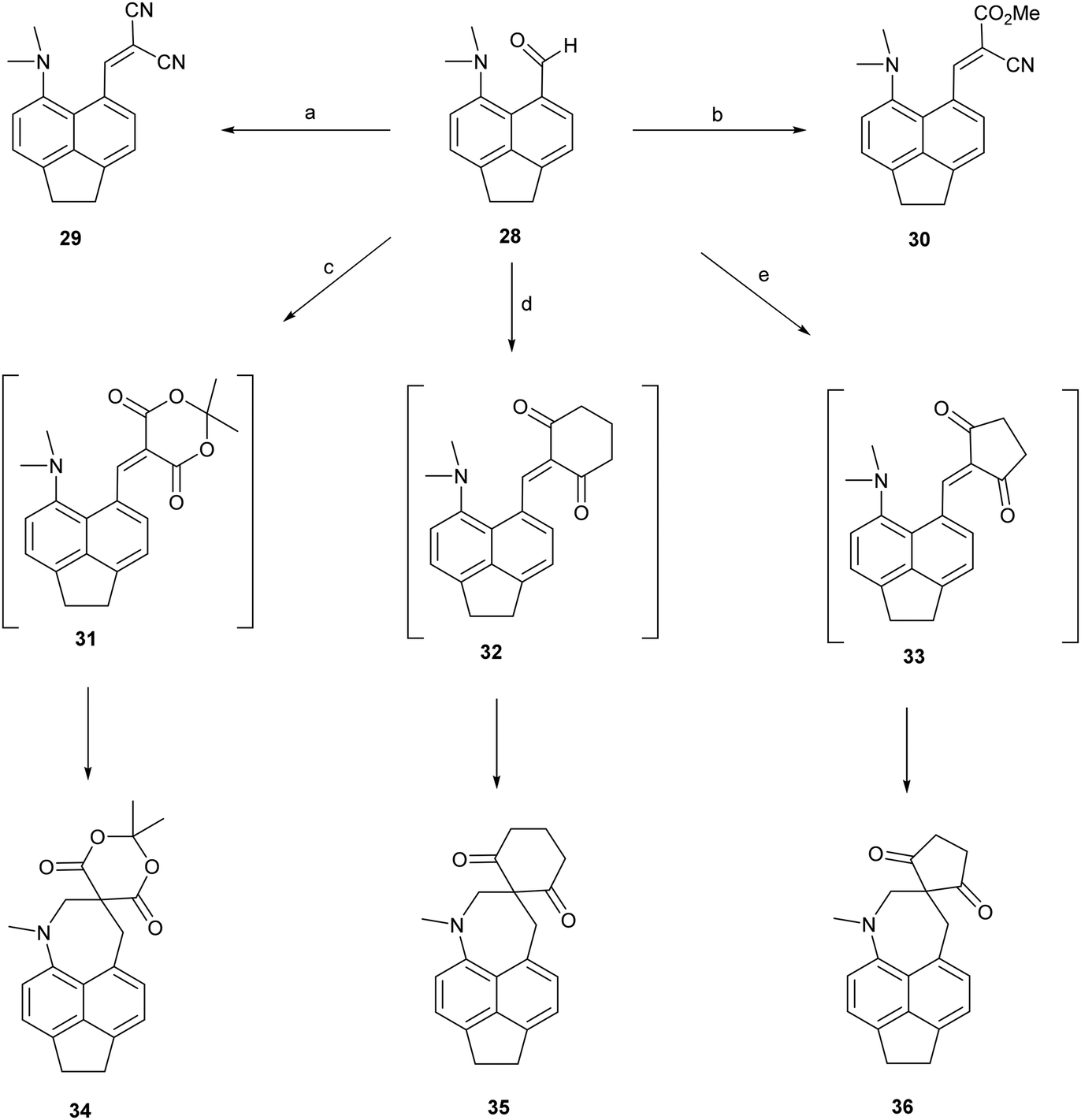 | ||
| Scheme 3 (a) and (b): Malonitrile or methyl cyanoacetate/ethylenediammonium diacetate cat./methanol/reflux; (c)–(e) Meldrum's acid, cyclohexane-1,3-dione or cyclopentane-1,3-dione, DMSO, RT. | ||
The structure of the aldehyde 28 shows a very significant difference to that of the corresponding naphthalene derivative 4 without the ethylene bridge (Fig. 5, Table 4). The two groups are splayed apart to a Me2N⋯C separation of 2.953(2) Å, cf. 2.489(6) Å in 4, due mainly to a widening of the ϕ exo angle to 129.50(14)°, and the aldehyde group has rotated so that it now it lies at just 16.4° to the nearest C,C(H) bond of the aromatic system. Thus, it is not involved in a n–π* interaction of type: Me2N⋯CO as it is in 4. The nitrogen atom lies at 2.37 Å from the aldehyde hydrogen atom, and its theoretical lone pair axis lies at 27° to the N⋯H vector. Both the aldehyde and dimethylamino groups are oriented to optimise their conjugation with the acenaphthene ring, though the bond lengths between these groups and the ring are not significantly shortened compared to 4: Me2N–C: 1.4221(19) v 1.420(6) Å, and (O)C–C: 1.480(2) v 1.490(6) Å for 28 and 4 respectively. The dimethylamino group is displaced slightly towards the aldehyde, and the aldehyde is displaced more strongly away, but it is the larger exo angle which is the main cause of their increased separation.
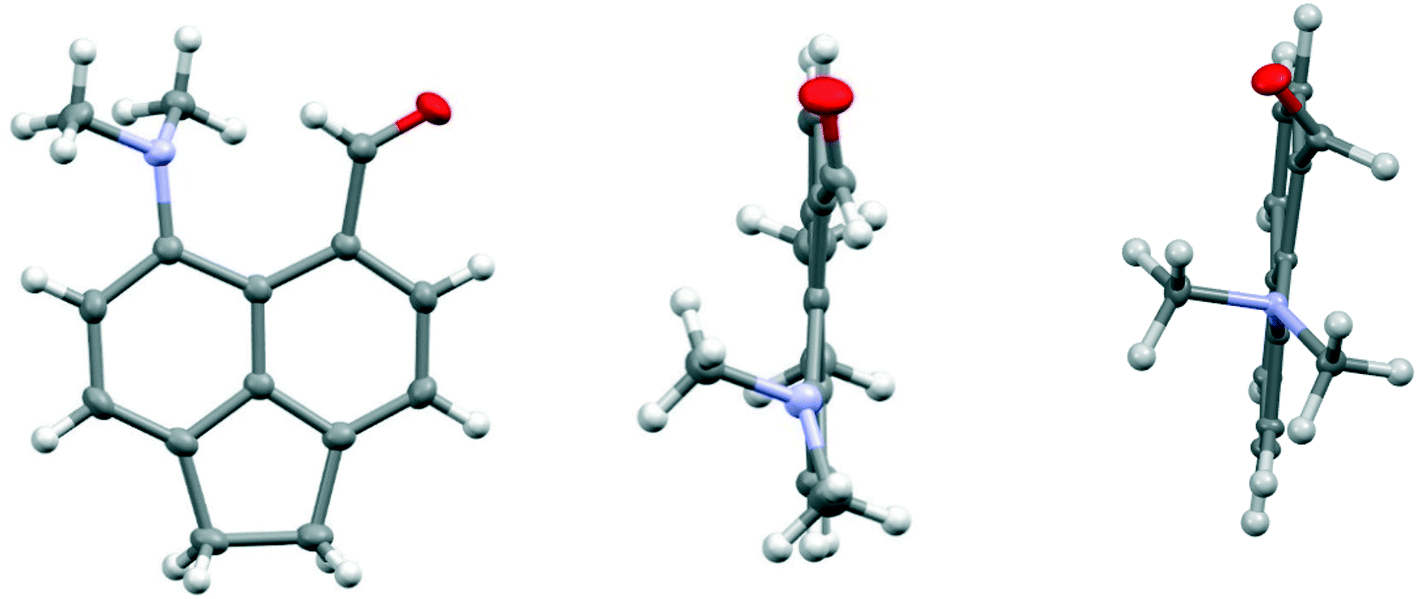 | ||
| Fig. 5 Two views of the acenaphthene aldehyde 28 (left and middle), showing how the aldehyde group lies close to the aromatic plane and the pyramidal dimethylamino group is oriented to conjugate with the aromatic ring, in contrast to the corresponding naphthalene without an ethylene bridge 4 (right).4 | ||
The structures of the Knoevenagel products 29 and 30 have a similar pattern of in-plane displacements as in the aldehyde 28 due to the widening of the ϕ exo angle to 127.03(18)–128.62(13)/Å, but the alkenes lie at greater angles (35.9(3)–51.9(3)°) to their acenaphthene rings (Fig. 6, Table 4). The Me2N⋯C separations lie in the range 2.755(3)–2.846(3) Å with the shortest for one of two crystallographically independent molecule of dinitrile 29 which has the largest rotation of the alkene group away from the acenaphthene. In this case the Me2N⋯CC angle is reduced to 122.68(14)° (cf. 129.28(11) and 133.00(15)° in the other cases), and this can be considered as a rather long n–π* interaction.
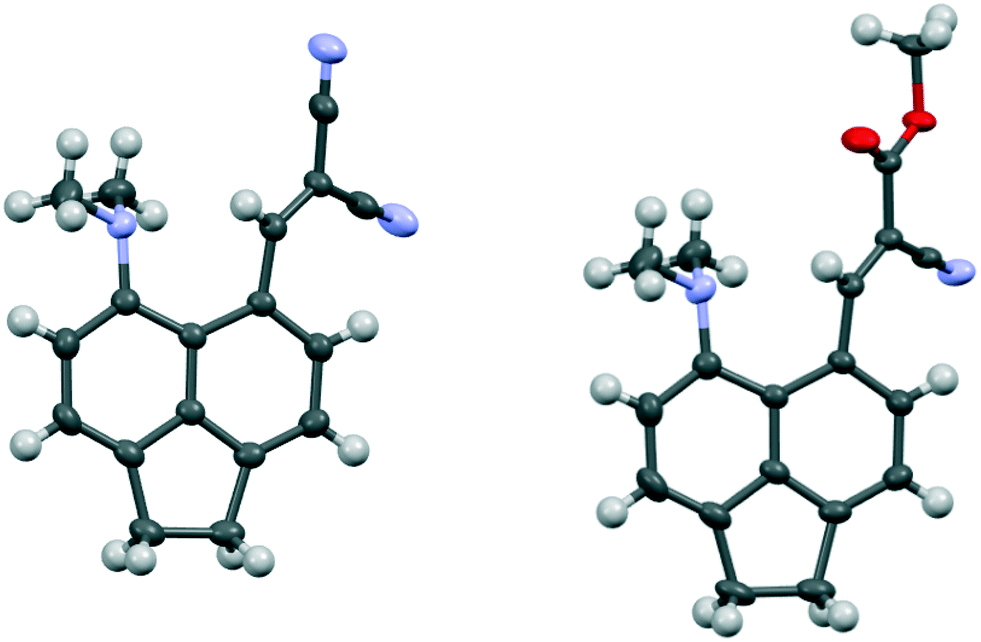 | ||
| Fig. 6 Molecular structures of one of the two molecules of dinitrile 29, with the larger rotation of the dinitrile side chain, (left) and of cyanoester 30 (right). | ||
It is known that for a naphthalene with a peri-dimethylamino group located next to an electron deficient alkene, on heating in DMSO at 60° C the groups react to form a fused azepine, for example from the dinitrile 8 or the N,N-dimethylbarbiturate derivative 38 to the fused azepines 37 and 39 (Scheme 4).22,23 Furthermore, recent related work has reported how 2-naphthol reacts with the peri-pyrrolidinyl aldehyde 40 to give 41.24 These reactions are triggered by the tertiary amino effect20,21 whereby a hydride from the N–CH2 or N–CH3 group adds to the polarised alkene, and then the iminium cation and the carbanion formed add to each other (Scheme 5). In the case of the attempted preparation of the acenaphthene Knoevenagel products 31–33, the reaction goes directly to the azepine by stirring aldehyde with the dicarbonyl compound in DMSO at room temperature in 40 to 80% yields. The widening of the exo angle allows the groups to get into positions to react more easily. The structures of the resulting three fused azepines 34–36 with various spiro cyclic dicarbonyl systems are shown in Fig. 7, with selected geometric data in Tables S4 and S5 (ESI†).
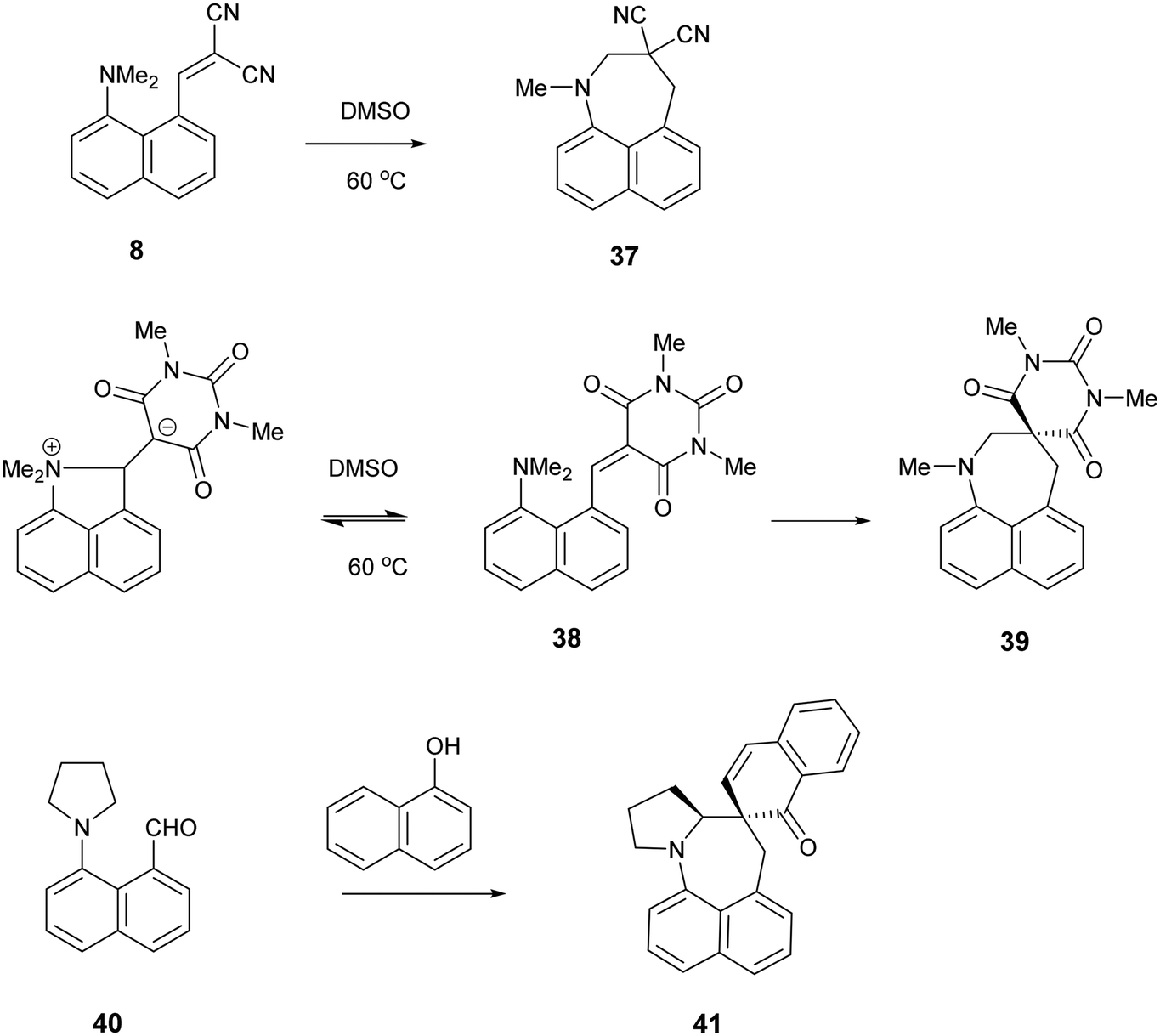 | ||
| Scheme 4 Examples of the conversion of peri-aminonaphthalene systems 8, 38 and 4022–24 to spiro derivatives initiated by the tertiary amino effect. | ||
 | ||
| Scheme 5 Mechanism of formation of spiro system 34 from Knoevenagel product 31 initiated by hydride transfer from the N-methyl group to the activated alkene. | ||
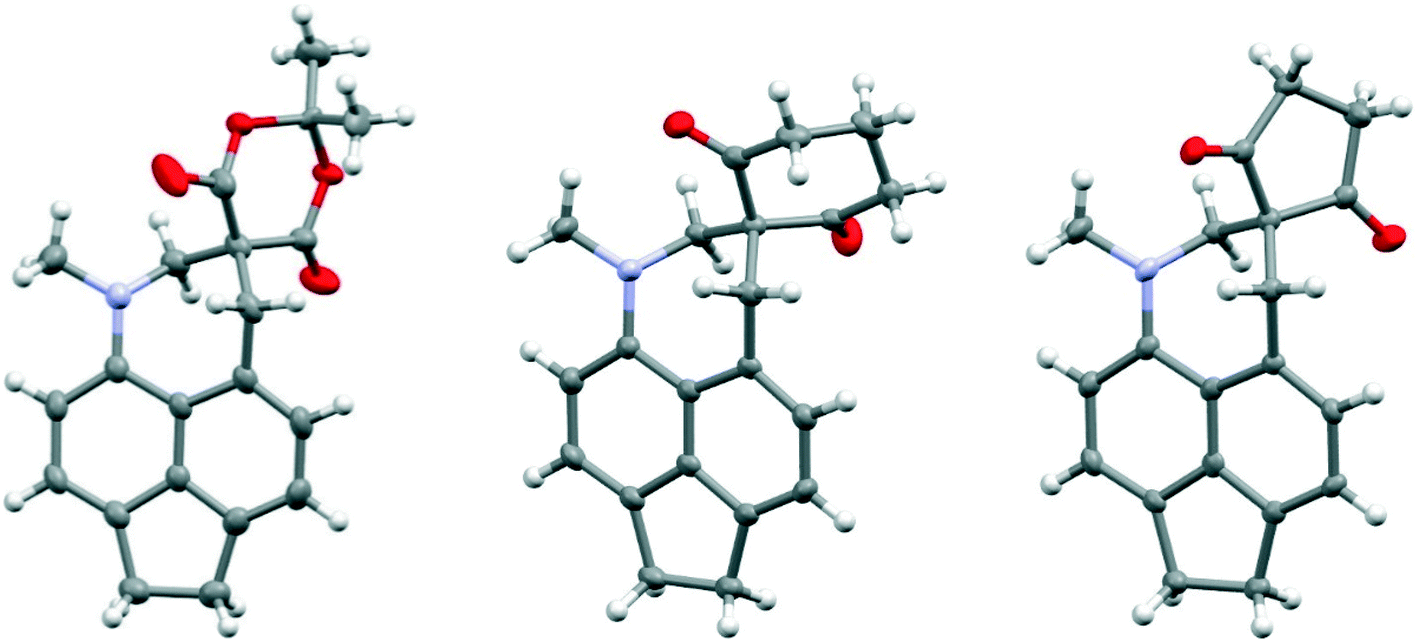 | ||
| Fig. 7 Molecular structures of the spiro acenaphtho-azepines 34, 35 and 36 (left to right). | ||
The structures are similar to related naphthalene based systems10 except that the widening of the ϕ angle is retained. The N–C(H2)–C(spiro) ring atoms are displaced to the same side of the acenaphthene ring, with the strongest displacements for the methylene and spiro carbons (1.163–1.354 and 0.551–0.919 Å respectively). The remaining methylene carbon is displaced to a smaller degree in the opposite sense (Table S5†). Within the azepine ring, the nitrogen atom adopts partial pyramidal bonding geometry (sum of angles: 343.6–349.6°), one bond to the spiro centre is strained (1.551–1.562 Å), and the angles at nitrogen and the methylene carbons show notable angle strain (113–117°). These are the first reported spiro derivatives of this fused azepine system.
Fluorene derivatives
To provide a skeleton to contrast with the biphenyl system but with the phenyl rings constrained to be near coplanar and with the separation between ortho substituents increased, the 9,9-dimethyl-fluorene skeleton 42 was selected. The parent dimethyl-hydrocarbon was converted to the ortho-diiodo compound 43 by bis-lithiation and treatment with iodine in 25% yield. The structure of the diiodo compound was confirmed by X-ray crystallography, and it shows considerably distortion of the fluorene system to accommodate an intramolecular I⋯I contact distance of 3.6392(4) Å (further details in the ESI†). Mono-Lithiation of diiodo compound 43 and treatment with DMF gave the dimethylamino aldehyde 44 in one step in 42% yield. The reaction proceeds by addition of DMF to the mono-lithiated aromatic, followed by expulsion of dimethylamide which substitutes the adjacent iodide. Barbasiewicz et al. has observed a similar reaction with peri-diodonaphthalene.25The aldehyde 44 gave the expected alkenes 45 and 46 by Knoevenagel reaction with malonitrile or nitromethane under reflux in methanol with ethylenediammonium diacetate as catalyst (Scheme 6). In contrast, just stirring aldehyde 44 with benzoyl-nitromethane, Meldrum's acid or cyclopentane-1,3-dione in DMSO at 20 °C gave fused azocine products 47–49, analogous to the behaviour of the more reactive acenaphthene derivatives (Scheme 7). Interestingly, on recrystallisation, some of the gem-benzoyl-nitro derivative 49 lost the benzoyl group, presumably due to the effect of water in the solvent, to give the fused nitro-azocine 50 (Scheme 7). Similar types of azocines have been reported from disubstituted biphenyls,21 however, we are not aware of any such derivatives of the fluoreno-azocine ring system in 47–50. The crystal structures of 44 and 45 were determined to examine the interaction between functional groups, and of 48 and 50 to confirm their molecular structures.
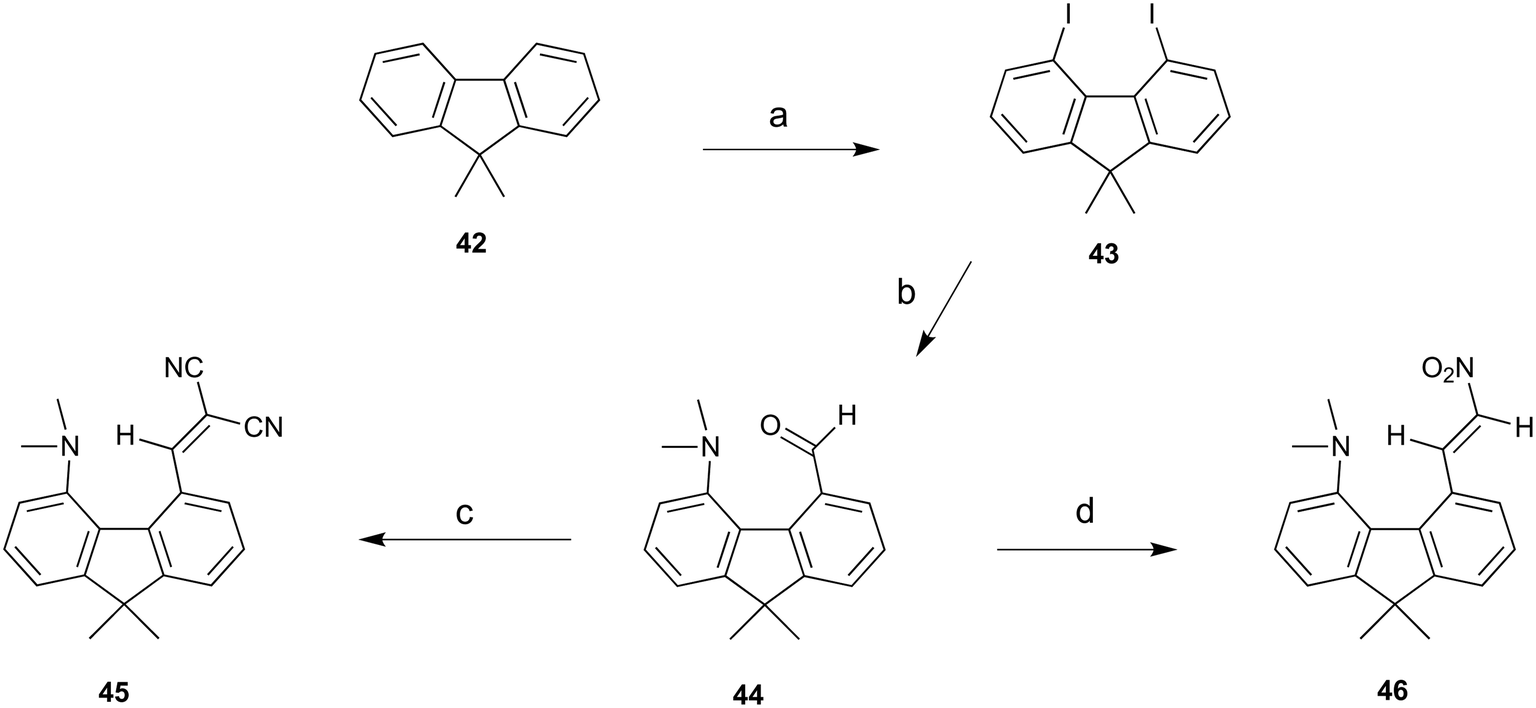 | ||
| Scheme 6 a) n-BuLi/TMEDA/60 °C/5 h, then I2/THF −78 °C; b) n-BuLi/ether/−78 °C then DMF, warm to 20 °C; (c) and (d) malonitrile or nitromethane/ethylenediammonium diacetate cat./methanol/reflux. | ||
 | ||
| Scheme 7 (a)–(c): Cyclopentane-1,3-dione, PhCOCH2NO2, or Meldrum's acid/DMSO/20 °C; (d): heat in DCM/hexane in air. | ||
The aldehyde and ethenedinitrile derivatives 44 and 45 adopt similar molecular conformations, with Me2N⋯C separations of 2.691(3) and 2.8304(17) Å (Fig. 8, Table 5). In contrast, in the biphenyl series, the corresponding separations are quite different: 2.989(2) and 1.586(3)/1.604(3) Å respectively. In 44 the lack of rotational freedom in the fluorene has brought the Me2N- and –CHO functional groups closer together, and the Me2N⋯CO angle is 111.04(16)°, as would be expected for a n–π* interaction. The axis of the nitrogen lone pair lies at 9.5° to the Me2N⋯C(O) vector. The greater separation of Me2N- and –CHC(CN)2 groups in the constrained fluorene system 45, prevents the bond formation seen in the biphenyl series. The Me2N⋯CC angle is favourable for n–π* interaction (110.80(10)°), though the Me2N⋯C distance is particularly long 2.8304(17) Å and the angle between the axis of the N lone pair and the Me2N⋯C vector is 8.5°. In the fluorene plane the Me2N- group is displaced towards the alkene which is displaced away, but the favourable alignment of groups in 44 and 45 are achieved by different combinations of (a) displacements of the groups to the opposite sides of the fluorene plane and (b) widening of the exo angles at the intervening ring fusions (Table 5).
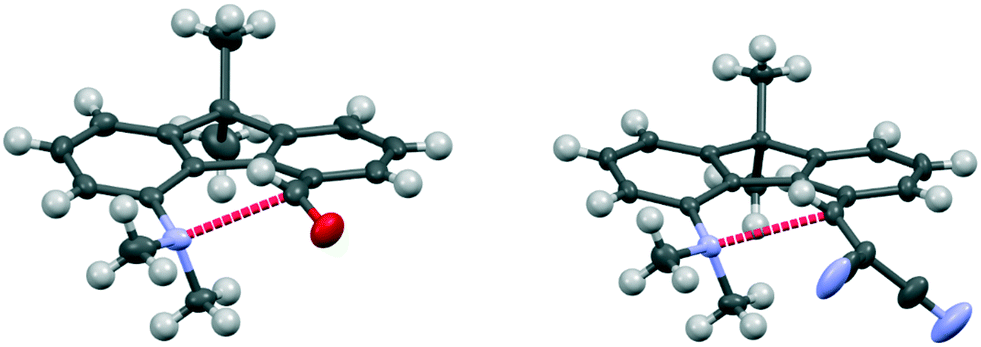 | ||
| Fig. 8 Molecular structures of fluorenes 44 (left) and 45 (right), with the long n–π* interaction shown in red. | ||
X-ray crystallography also confirmed the structures of two of the fused azocines, 48 and 50 which are shown in Fig. 9 with selected molecular geometry in Tables S6 and S7 (ESI†). The spiro junction in the azocine ring of 48 leads to more sp3C–sp3C bond strain with C–C bonds of 1.561(2) and 1.565(2) Å to the spiro atom, than in the nitro derivative 50 (1.506(4) and 1.525(4) Å). In the azocine ring the nitrogen atom adopts a moderately pyramidal bonding geometry (sum of angles: 48: 350.60(13)°; 50: 348.8(2)°), and there is significant angular strain along the N–CH2–C–CH2 fragment (112.0–118.26°) with exception of the spiro carbon of 48. The N–C(H2)–C ring atoms are displaced to the same side of the fluorene ring, with the strongest displacements for the two carbons (1.430–1.458 and 0.847–0.953 Å). The remaining methylene carbon is displaced to a smaller degree in the opposite sense (Table S7†). These out of plane displacements are larger than in the related acenaphthene derivatives.
 | ||
| Fig. 9 Molecular structures of fused azocines 48 (left) and 50 (right). | ||
Conclusions
These investigations show that the interaction between an electrophile and a nucleophile in the peri-positions of a naphthalene can be modified by the choice of substituents at the opposite peri-positions. Two phenyl groups repel each other and force the other substituents closer, and in one case, the cyanoester derivative 19, led to them forming a bond, which in the absence of the phenyls they did not. In contrast, in acenaphthene systems, the ethylene group, acts as a short constraint between the peri-positions, widening the separation between the interacting groups, with various consequences. The aldehyde group in compound 28 now has sufficient space to rotate into the plane of the aromatic system so the n–π* interaction is lost, while for the ethenedinitrile in 29 the Me2N⋯C interaction in lengthened, and for more reactive electrophiles cyclisation between the N-methyl group and the alkene occurs forming a fused azepine ring as in 34–36, initiated by intramolecular hydride transfer, which the increased space between groups permits. For the 1,6 interactions in the fluorene derivatives studied, two long n–π* interactions were observed for the less reactive electrophiles in 45 and 46, but for the more reactive the corresponding cyclisation to form fused azocine rings in 48 and 50 took place. The peri-diphenylnaphthalene system is the most promising for exploring further nucleophile–electrophile interactions and, in particular, at accessing those separations nearer to the transition state for the direct bond formation. Additions of small groups to the phenyl rings to increase the repulsion between them may lead to even closer peri-interactions at the opposite positions, and complement the very few N⋯C interactions known in the 1.7–2.3 Å range.14 In this work we did not find, but also did not deliberately look for, polymorphs of the compounds whose crystal structures were determined. It is possible that different polymorphs or solvates may show somewhat different interactions between the groups, as a result of the effects of the different packing arrangements, just as in the crystal structure of the acenaphthene 29 the two independent molecules have differences in their conformations. In the diphenylnaphthalene series, in particular, this may be worth pursuing.Experimental
Full details of the synthesis and characterisation of new substances and the determination of crystal structures by X-ray diffraction and their crystal data are provided in the ESI.† Crystallographic data are deposited at the Cambridge Crystallographic Data Centre with code numbers CCDC: 2069090–2069106 and 2069108.Conflicts of interest
There are no conflicts of interest to report.Acknowledgements
We thank Nottingham Trent University for a studentship (JCB), and for financial support. We thank Dr. Richard Grainger, Birmingham University for interesting discussions.References
- Structure Correlation, ed. H.-B. Bürgi and J. D. Dunitz, VCH, Weinheim, 1994, vol. 1 Search PubMed.
- H.-B. Bürgi, J. D. Dunitz and E. Schefter, J. Am. Chem. Soc., 1973, 95, 5065–5067 CrossRef.
- W. B. Schweizer, G. Procter, M. Kaftory and J. D. Dunitz, Helv. Chim. Acta, 1978, 61, 2783–2808 CrossRef CAS.
- D. R. W. Hodgson, A. J. Kirby and N. Feeder, J. Chem. Soc., Perkin Trans. 1, 1999, 949–954 RSC.
- A. Wannebroucq, A. P. Jarmyn, M. B. Pitak, S. J. Coles and J. D. Wallis, Pure Appl. Chem., 2016, 88, 317–331 CAS.
- P. C. Bell, W. Skranc, X. Formosa, J. O'Leary and J. D. Wallis, J. Chem. Soc., Perkin Trans. 2, 2002, 878–886 RSC.
- M. Parvez and I. I. Schuster, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 1990, 46, 947–948 CrossRef.
- P. C. Bell and J. D. Wallis, Chem. Commun., 1999, 257–258 RSC.
- J. O'Leary, W. Skranc, X. Formosa and J. D. Wallis, Org. Biomol. Chem., 2005, 3, 3273–3283 RSC.
- A. Lari, M. B. Pitak, S. J. Coles, G. J. Rees, S. P. Day, M. E. Smith, J. V. Hanna and J. D. Wallis, Org. Biomol. Chem., 2012, 10, 7763–7779 RSC.
- A. F. Pozharskii, O. V. Dyablo, O. G. Pogosova, V. A. Ozeryanskii, A. Filarowski, K. M. Vasilikhina and N. A. Dzhangiryan, J. Org. Chem., 2020, 85, 12468–12481 CrossRef CAS PubMed; C. Cox, H. Wack and T. Lectka, Angew. Chem., Int. Ed., 1999, 38, 798–800 CrossRef.
- M. W. Stanford, F. R. Knight, K. S. Athukorala Arachchige, P. Sanz Camacho, S. E. Ashbrook, M. Bühl, A. M. Z. Slawin and J. D. Woollins, Dalton Trans., 2014, 43, 6548–6560 RSC; F. R. Knight, R. A. M. Randall, K. S. Athukorala Arachchige, L. Wakefield, J. M. Griffin, S. E. Ashbrook, M. Bühl, A. M. Z. Slawin and J. D. Woollins, Inorg. Chem., 2012, 51, 11087–11097 CrossRef CAS PubMed.
- J. O'Leary and J. D. Wallis, Org. Biomol. Chem., 2009, 7, 225–228 RSC.
- J. C. Bristow, I. Naftalin, S. V. A. Cliff, S. Yang, M. Carravetta, R. Stern, I. Heinmaa and J. D. Wallis, CrystEngComm, 2020, 22, 6783–6795 RSC.
- E. Hupf, N. Olaru, C. I. Rat, M. Fugel, C. B. Hübschle, E. Lork, S. Grabowsky, S. Mebs and J. Beckmann, Chem. – Eur. J., 2017, 23, 10568–10579 CrossRef CAS.
- D. Pla, O. Sadek, S. Cadet, B. Mestre-Voegtlé and E. Gras, Dalton Trans., 2015, 44, 18340–18346 RSC.
- C. F. R. A. C. Lima, J. E. Rodriguez-Borges and L. M. N. B. F. Santos, Tetrahedron, 2011, 67, 689–697 CrossRef CAS.
- K. Komatsu, K. Taekuchi, M. Shiro, S. Cohen and M. Rabinovitz, J. Phys. Org. Chem., 1993, 6, 435–444 CrossRef.
- N. Asaad, J. E. Davies, D. R. W. Hodgson, A. J. Kirby, L. van Vliet and L. Ottavi, J. Phys. Org. Chem., 2005, 18, 101–109 CrossRef CAS.
- L. Wang and J. Xiao, Top. Curr. Chem., 2016, 17 CrossRef; O. Meth-Cohen and H. Suschitsky, Adv. Heterocycl. Chem., 1972, 14, 211–278 CrossRef.
- Á. Polonka-Bálint, C. Saraceno, K. Ludányi, A. C. Bényei and P. Mátyus, Synlett, 2008, 2846–2850 Search PubMed.
- Á. A. Földi, K. Ludányi, A. C. Bényei and P. Mátyus, Synlett, 2010, 2109–2113 Search PubMed.
- A. F. Pozharskii, M. A. Povalyakhina, A. V. Degtyarev, O. V. Ryabtsova, V. A. Ozeryanskii, O. V. Dyablo, A. V. Tkachuk, O. N. Kazheva, A. N. Chekhlov and O. A. Dyachenko, Org. Biomol. Chem., 2011, 9, 1887–1900 RSC.
- S. S. Li, X. Lv, D. Ren, C.-L. Shao, Q. Liu and J. Xiao, Chem. Sci., 2018, 9, 8253–8259 RSC.
- K. Grudzień, K. Żukowska, M. Malińska, K. Woźniak and M. Barbasiewicz, Chem. – Eur. J., 2014, 20, 2819–2828 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2069090–2069106 and 2069108. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d1ce00377a |
| This journal is © The Royal Society of Chemistry 2021 |