
Graphene-induced growth of Co3O4 nanoplates with modulable oxygen vacancies for improved OER properties†
Xinheng
Li  *a
*a
*a
Abstract
Transition metal oxide/hydroxide is intensively studied for the oxygen evolution reaction (OER). Herein, the graphene-induced growth of Co3O4 nanoplates with modulable oxygen vacancies via a hydrothermal treatment is reported. With the increase of reaction time before the formation of Co(OH)2, the oxygen vacancies and conductivity of Co3O4 nanoplates continued to increase resulting in dramatically enhanced OER performances. An ultralow overpotential of 354 mV at a current density of 100 mA cm−2 and a Tafel slope as low as 63.24 mV dec−1 in 1 M KOH solution were obtained, superior to those of most reported oxides and RuO2.
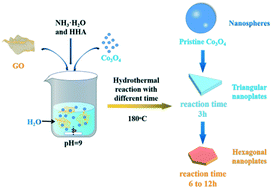
- This article is part of the themed collection: Crystal growth of nanomaterials
 Please wait while we load your content...
Please wait while we load your content...

