
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
From pipemidic acid molecular salts to metal complexes and BioMOFs using mechanochemistry†
Martin
Zábranský
ab,
Paula C.
Alves
ac,
Catarina
Bravo
 ac,
M. Teresa
Duarte
*ad and
Vânia
André
*ac
ac,
M. Teresa
Duarte
*ad and
Vânia
André
*ac
aCentro de Química Estrutural, Instituto Superior Técnico, Universidade de Lisboa, Av. Rovisco Pais, 1049-001 Lisboa, Portugal. E-mail: vaniandre@tecnico.ulisboa.pt; teresa.duarte@tecnico.ulisboa.pt
bDepartment of Inorganic Chemistry, Faculty of Science, Charles University, Hlavova 2030, 128 40 Prague, Czech Republic
cAssociação do Instituto Superior Técnico para a Investigação e Desenvolvimento (IST-ID), Av. Rovisco Pais, 1049-003 Lisboa, Portugal
dDepartamento de Engenharia Química, Instituto Superior Técnico, Universidade de Lisboa, Av. Rovisco Pais, 1049-001 Lisboa, Portugal
First published on 23rd December 2020
Abstract
Mechanochemistry has been assuming a major role in chemistry over the last years. Its potential in a wide variety of reactions is undeniable nowadays, contributing to sustainable chemistry and, at the same time, leading to lower reaction time, higher selectivity and novel reactivity. Herein we disclose new solid forms of pipemidic acid, an antibiotic against which bacteria have been developing resistance mechanisms, obtained by mechanochemistry. These forms are molecular salts with glycolic, oxalic and (R)- and (S)-camphorsulfonic acids, as well as a Cu(II) complex exhibiting a 1D hydrogen bonded network. To prove the importance of the technique, we present here also the rapid and high yielding mechanochemical synthesis of a previously reported Ag metal–organic framework with pipemidic acid.
Introduction
Mechanochemistry is a green and eco-friendly synthetic technique, whose application has tremendously grown in the last two decades, and become recently one of the ten IUPAC technologies foreseen to change the world.1 The technique of millenary use, regained importance in the area of crystal engineering and polymorphism in the 1980's and is nowadays reemerging as an easy, clean and straightforward synthetic method for organic, metal–organic, coordination and supramolecular syntheses.2–5 It evolved from being simply a solvent free alternative to a main synthetic technique leading to reduced reaction times, higher yields, product selectivity and completely unexpected reaction products, impossible to attain in solution.2,5,6A mechanochemical reaction, defined as “a chemical reaction that is induced by the direct absorption of mechanical energy” when grinding together two or more compounds,2,7 is also affected by the addition of solvents, ions, ionic liquids and other additives used to augment, direct or enable reactivity.2 The most commonly used mechanochemical techniques are: neat grinding (NG), in which no solvent is added;2,7,8 liquid-assisted grinding (LAG), in which a catalytic amount of solvent is added;2,7,9 ion and liquid-assisted grinding (ILAG), in which catalytic amounts of solvent and an ionic salt are added to the reaction;2,4,7,10 and lastly and most recently, polymer-assisted grinding (POLAG), which makes use of polymers in order to stimulate the reaction.2,7,9,11
As previously said, mechanochemistry gained importance in the field of Crystal Engineering, applied with great emphasis in pharmaceutical sciences for the development of novel crystalline forms towards improved properties of active pharmaceutical ingredients (API) already in the market.12,13 The development of novel polymorphs, salts, and cocrystals has been envisaged as an excellent alternative, proving to be able to tackle stability, solubility and flowability problems among others.14–16 At the same time, reports that coordination to metals could also improve these properties, and furthermore enhance their performance/activity have been published.17–19 Also the mechanochemical preparation of metallodrugs enclosing Cu (ref. 20) and Ag (ref. 21) have been recently reported. The interest in metal–organic frameworks (MOFs) towards pharmacological applications has also been increasing, especially for controlled drug delivery and release.22 We have recently proven that antibiotic coordination frameworks resulting from the direct coordination of commercially available antibiotics to safe metals can increase their antibacterial effect.3
Herein we present results using pipemidic acid (PA) (Fig. 1), a first-generation quinolone antibiotic used to treat urinary tract infections owing to its antibacterial activity against both Gram-negative23 and Gram-positive bacteria.24,25 Quinolone antibiotics are broad-spectrum synthetic antibacterial compounds presenting suitable oral absorption and bioavailability,26,27 results on their antitumor, anticancer and antiviral activities have also been reported.28–30 PA is obtained through the incorporation of a piperazinyl side chain in the main quinolone skeleton,31–33 thus increasing the lipophilicity of the compound, and consequently improving its ability to go through the bacterial cell wall.33,34
 | ||
| Fig. 1 Pipemidic acid. | ||
However, due to the rapid adaptation and resistance development that microorganisms have been showing, antibiotics might become ineffective to treat some bacterial infections.31,35 Coordination of quinolones to biocompatible metals emerged as an alternative approach to improve their biological and/or pharmaceutical activities while tuning their physicochemical properties.36,37 Recently, our group published results on bio-inspired metal–organic frameworks (BioMOFs) of nalidixic acid, and has successfully demonstrated their increased solubility and bioactivity compared to the free antibiotic.3,38 Using PA, we have also published results on hydrogen bonded frameworks based on Mn, Ca and Zn metal centers, presenting suitable shelf and thermal stability, similar cytotoxicity and increased antibacterial activity compared to the free API.39
Based on our results and a thorough search in the Cambridge Structural Database (CSD),40,41 we were able to identify two PA polymorphic forms,42 two polymorphs of the trihydrate derivative43,44 and two salts, pipemidic acid hydrochloride45 and pipemidic acid:1,2,4,5-benzenetetracarboxylic acid.46 Also several metal complexes and metal–organic frameworks,47,48 enclosing cadmium,49 nickel,50 silver,51,52 zinc,53–56 manganese,54,57 cobalt54 and copper58–61 were reported.
Here we present and discuss our results on five novel molecular salts, one metal complex and an Ag-MOF containing pipemidic acid. All these compounds were obtained by grinding using a mortar and pestle, or by mechanochemistry in a ball-mill.
Results and discussion
Mechanochemistry is a sustainable approach that perfectly suits the demand for the development of alternative forms of drugs. Thus, it was chosen herein for the investigation of novel crystalline forms of pipemidic acid (salts, complexes and MOFs) that may lead to improved physicochemical properties and performance of than those of the free antibiotic.A thorough datamining revealed that the development of new multicomponent crystal forms of pipemidic acid is still underexplored, as detailed hitherto. Thus, crystal engineering principles were applied for the design of new salts and the coformers chosen were those enclosing carboxylic and sulfonic acid moieties, known to be prone to interact with the functional groups present in pipemidic acid via strong hydrogen bond synthons. Amongst several compounds tested without successful results, such as gluconic, ethanesulfonic and citric acids, we present here novel salts of pipemidic acid with glycolic, oxalic and (R)- and (S)-camphorsulfonic acids. Even though the interaction of protonated amines with sulfonates/sulfonic acid (N+–H⋯OSO3−) is not one of the most abundant (286 hits at CSD), especially compared with the interaction with carboxylate moieties (N+–H⋯OCOO−, 1687 hits at CSD), it was decided to include camphorsulfonic acid in this study, which is used in some pharmaceutical formulations, yielding successful results.
Besides this multicomponent approach, we also pursued the development of novel complexes with Cu, looking for synergistic effects with the metal. Also, Ag was used in our studies and an Ag-pipemidic acid MOF previously prepared by time-consuming solution methods51,52 was reproduced with higher yields and short reaction time by mechanochemistry.
All these forms are represented in Scheme 1 and will be carefully discussed.
 | ||
| Scheme 1 List of structures. | ||
Molecular salt structures
In all the five salt structures, the cation is the pipemidic acid protonated at its secondary amine site. The adopted conformation is similar to the one observed in the structure of the octahydrate of its 2,5-dicarboxybenzene-1,4-dicarboxylate salt.46 The carboxylic group lies in the plane of the aromatic pyridopyrimidine system and forms an intramolecular hydrogen bond of O–H⋯OC![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O type with the oxygen of the ketogroup (graph set62 descriptor S(6)), also observed in the 2,5-dicarboxybenzene-1,4-dicarboxylate salt. The corresponding O⋯O bond distances in the prepared salts range from 2.499(1) Å to 2.559(2) Å, for 1·2H2O and for one of the symmetrically independent cations in 3·5H2O, respectively. The apex angles at the hydrogen atom fall between 161(2)° for both 1·2H2O and 2 and 166(6)° for 4S (see Tables S1–S5†).
O type with the oxygen of the ketogroup (graph set62 descriptor S(6)), also observed in the 2,5-dicarboxybenzene-1,4-dicarboxylate salt. The corresponding O⋯O bond distances in the prepared salts range from 2.499(1) Å to 2.559(2) Å, for 1·2H2O and for one of the symmetrically independent cations in 3·5H2O, respectively. The apex angles at the hydrogen atom fall between 161(2)° for both 1·2H2O and 2 and 166(6)° for 4S (see Tables S1–S5†).
The piperazine ring of the protonated pipemidic moiety assumes a chair conformation in all the structures with the Cremer–Pople θ angle63 values approaching either 0° or 180° and the total puckering amplitudes, Q,63 ranging from 0.549(2) Å for one of the symmetrically independent cations in 3·5H2O to 0.568(2) Å for 2. The ethyl group of the cation adopts a staggered conformation and points in the same direction as the piperazine ring with respect to the pyridopyrimidine plane in all the reported structures.
The protonated pipemidic secondary amine site in the piperazine ring participates as the donor in charge-assisted hydrogen bonds of the type N+–H⋯O−COO/SO3− with the carboxylate or sulfonate groups of the counterion in the structure of each salt. These bonds play a significant role in the observed supramolecular assemblies, predominantly one-dimensional in the anhydrous compounds. The hydrogen bonding stemming from the presence of solvating water molecules in the structures of the hydrates further extend the arrangement into a layered structure.
The asymmetric unit of 1·2H2O consists of one protonated pipemidic acid, one glycolate anion and two water molecules (Fig. S1†). The hydrogen bonded network in the supramolecular structure of 1·2H2O (Fig. 2) is built by seven intermolecular interactions (ii–viii, Table 1). The glycolate anion, ammonium group of the protonated pipemidic acid and the two solvating water molecules form a chain of hydrogen-bonded rings that propagate along b (Fig. 2a). The patterns observed in this network include binary motifs, R44(12) (iii, iv) and R44(14) (vi, vii), and higher order motifs, R34(11) and R44(13), both consisting of four hydrogen bonds (iii, iv, vi, vii), and R66(18) (i, vi, v) that incorporate four water molecules and two anions. The neighboring chains interact via hydrogen bond viii that gives rise to an infinite chain, C33(19), together with bonds ii and iii. This chain propagates in the [−1 1 1] direction (Fig. 2b). Furthermore, π–π stacking interactions can be observed between pyridopyrimidine systems (ring C1–C2–C3–N3–C7–N4–C4–N5–C5–C6, distance between centroids 3.4852(5) Å, distance between planes 3.3308(3) Å) of centrosymmetrically related cations of adjacent chains (Fig. 2b).
 | ||
| Fig. 2 Hydrogen bonded network in the structure of 1·2H2O viewed (a) along a, and (b) along b. The π–π stacking is highlighted by green dotted lines. | ||
| Bond | D–H⋯A | Equivalent position code | Distance H⋯A, [Å] | Distance D⋯A, [Å] | Angle DHA, [°] |
|---|---|---|---|---|---|
| i | O1–H1⋯O3 | x, y, z | 1.60(2) | 2.499(1) | 161(2) |
| ii | O1W–H1A⋯O22 | x, y, z | 1.76(2) | 2.705(1) | 178(2) |
| iii | N1–H4⋯O21 | 1 − x, 1 − y, 1 − z | 1.78(2) | 2.699(1) | 159(2) |
| iv | N1–H2⋯O22 | x − 1, y, z | 1.83(2) | 2.789(2) | 167(1) |
| v | O2W–H2B⋯O1W | x, 1 + y, z | 1.89(2) | 2.789(2) | 169(2) |
| vi | O23–H23⋯O2W | −x + 2, −y + 1, −z + 1 | 1.89(2) | 2.720(2) | 172(2) |
| vii | O2W–H2A⋯O21 | x, y, z | 2.02(2) | 2.777(2) | 158(2) |
| viii | O1W–H1B⋯O1 | −x + 2, −y, −z | 2.11(2) | 2.922(1) | 169(2) |
The asymmetric unit of 2 consists of one protonated pipemidic acid and one hydrogenoxalate anion (Fig. S2†). Four (i–iv) intermolecular hydrogen bonds were identified in the hydrogen-bonded network of 2 (Table 2). Apart from participating in the charge-assisted hydrogen bond iv, the hydrogen atom H4 is in this structure approached additionally by oxygen atom O22 of a nearby hydrogenoxalate anion (symmetry code x, −y + 3/2, z − 1/2) with a relatively long associated H⋯O distance (2.36(2) Å) and acute angle N1–H4⋯O22 (118(2)°). Oxygen atom O2 of nearby cation (symmetry code x − 1, −y + 3/2, z − 1/2) also lies in the vicinity of this hydrogen atom with even longer associated H⋯O distance (2.56(2) Å) and a more acute angle N1–H4⋯O2 (110(2)°). The bonds iii and iv form a zig-zag pattern C22(4) that propagates along b and that is built up by the two-fold screw axis (Fig. 3a). The chains are interconnected by hydrogen bond ii between pairs of centrosymmetrically related hydrogenoxalate anions which form pattern R22(10) (Fig. 3b). This interaction also brings hydrogen atom H21 into the vicinity of oxygen atom O24. The parameters of the corresponding potential intramolecular hydrogen bond are H⋯O24 2.14(3) Å, O21⋯O24 2.645(2) Å, O21–H21–O24 115(2)°. Pyrimidine rings (N4–C4–N5–C5–C6–C7) of the cations of neighboring hydrogen-bonded chains that are related by inversion participate in π–π stacking (distance between centroids 3.3773(9) Å, distance between planes 3.2039(6) Å, Fig. 3c).
| Bond | D–H⋯A | Equivalent position code | Distance H⋯A, [Å] | Distance D⋯A, [Å] | Angle DHA, [°] |
|---|---|---|---|---|---|
| i | O1–H1⋯O3 | x, y, z | 1.56(3) | 2.524(2) | 161(2) |
| ii | O21–H21⋯O24 | −x, −y + 1, 1 − z | 1.84(2) | 2.634(2) | 145(2) |
| iii | N1–H2⋯O23 | −x, −y + 1, 1 − z | 2.05(2) | 2.923(2) | 164(2) |
| iv | N1–H4⋯O23 | x, −y + 3/2, z − 1/2 | 2.16(2) | 2.986(2) | 163(2) |
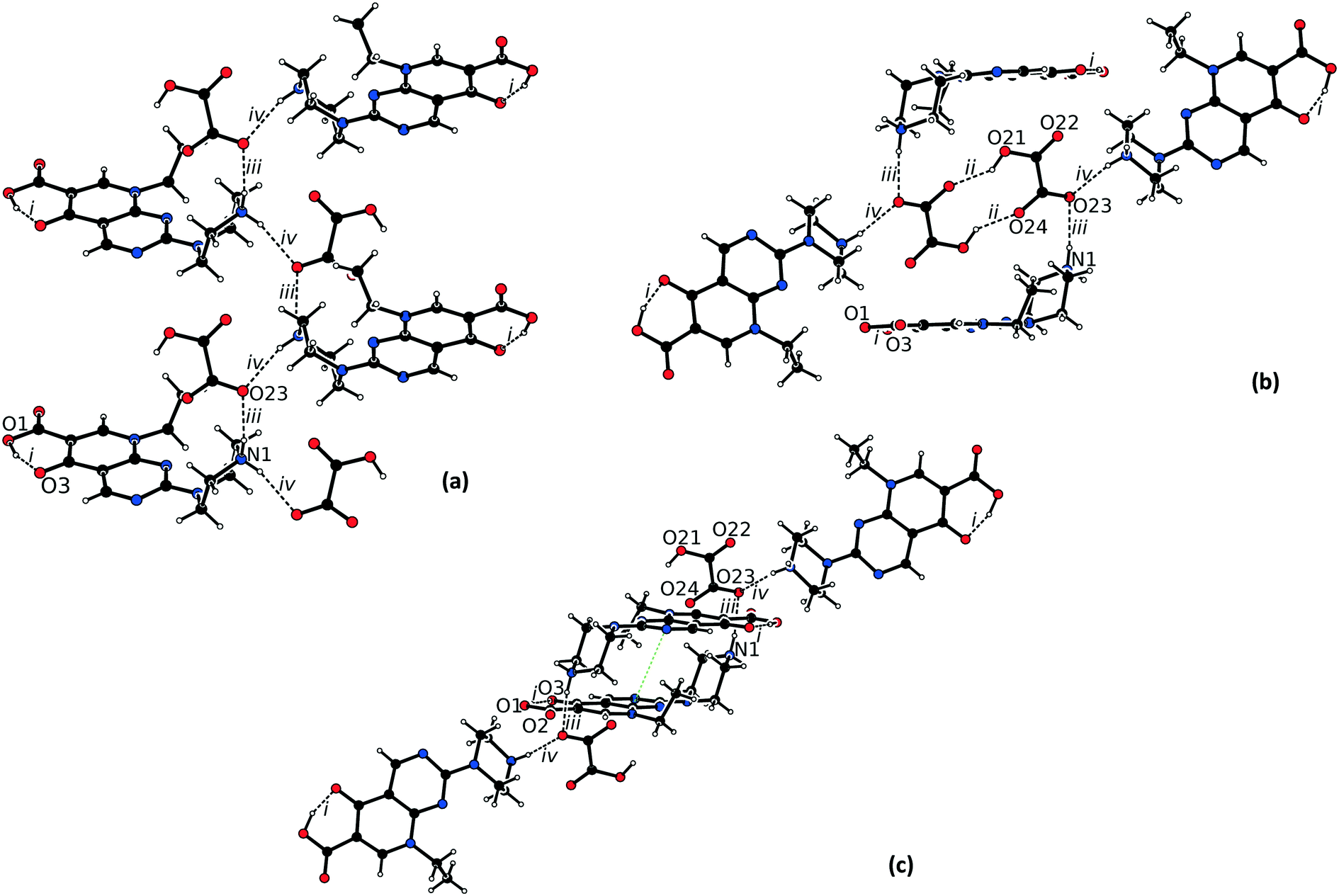 | ||
| Fig. 3 (a) Hydrogen bonded network in the structure of 2 viewed along a; (b) view of the centrosymmetric dimers formed by the hydrogen bond O21–H21⋯O24 between the hydrogenoxalate anions in the structure of 2; (c) view of the π–π stacking (green dotted line) in the structure of 2. | ||
The asymmetric unit of 3·5H2O consists of two protonated pipemidic acid, one oxalate dianion, and five water molecules (Fig. S3†). The intermolecular hydrogen bonds in the structure of 3·5H2O (Table 3) form a two-dimensional network that propagates in the ab plane (Fig. 4a). This network includes the binary motif C22(7)[R24(8)] of chains that are formed by bonds vii, ix, xi and xiv and propagate along crystallographic axis a. Additional motifs within the layer include ternary motifs R66(16) (vii, xi, xiii) and R66(18) (vii, xiii, xiv) and quaternary motifs R88(20) (iii, iv, vii, xii) and R88(22) (iii, iv, ix, xii). Both symmetrically independent cationic moieties interact with the two-dimensional hydrogen-bonded network by a pair of charge-assisted hydrogen bonds with the anion using one donor site (N101–H104 for bonds x and xviii, N201–H204 for bonds xvi and xvii), that give rise to two R21(5) patterns and by a hydrogen bond with a water molecule using the other donor site (N101–H102 for bond v and N201–H202 for bond vi). Additionally, the individual hydrogen-bonded layers are interconnected by hydrogen bond xv which forms chains C34(19) propagating along c together with bonds iii, ix and xvi. Notably, only one of the symmetrically independent cations participates in this motif. Furthermore, π–π stacking interaction of pyridopyrimidine systems of centrosymmetrically related cation moieties (rings C101–C102–C103–N103–C107–N104–C104–N105–C105–C106 and C201–C202–C203–N203–C207–N204–C204–N205–C205–C206, distance between centroids 3.4784(7) Å, 3.3552(6) Å respectively, distance between planes 3.3492(4) Å, 3.3144(4) Å respectively) also interconnects the adjacent hydrogen-bonded layers (Fig. 4b).
| Bond | D–H⋯A | Equivalent position code | Distance H⋯A, [Å] | Distance D⋯A, [Å] | Angle DHA, [°] |
|---|---|---|---|---|---|
| i | O201–H201⋯O203 | x, y, z | 1.58(2) | 2.507(2) | 165(2) |
| ii | O101–H101⋯O103 | x, y, z | 1.59(2) | 2.559(2) | 165(2) |
| iii | O5W–H5B⋯O4W | −x + 1, −y + 1, −z + 1 | 1.76(3) | 2.715(2) | 174(2) |
| iv | O1W–H1B⋯O5W | x, y, z | 1.77(2) | 2.719(2) | 176(2) |
| v | N101–H102⋯O3W | x + 1, y − 1, z | 1.82(2) | 2.723(2) | 169(2) |
| vi | N201–H202⋯O1W | x − 1, y, z | 1.84(2) | 2.717(2) | 172(2) |
| vii | O4W–H4A⋯O2 | −x, −y + 1, −z + 1 | 1.89(2) | 2.751(2) | 160(2) |
| viii | O3W–H3A⋯O2W | x, y + 1, z | 1.89(3) | 2.806(2) | 168(2) |
| ix | O4W–H4B⋯O3 | −x + 1, −y + 1, −z + 1 | 1.91(2) | 2.752(2) | 174(3) |
| x | N101–H104⋯O2 | −x + 1, −y, −z + 1 | 1.94(2) | 2.803(2) | 153(2) |
| xi | O2W–H2A⋯O3 | x − 1, y, z − 1 | 1.94(2) | 2.818(2) | 165(2) |
| xii | O1W–H1A⋯O1 | −x + 1, −y + 1, −z + 2 | 1.94(2) | 2.790(2) | 168(2) |
| xiii | O3W–H3B⋯O4 | −x + 1, −y + 1, −z + 1 | 1.95(2) | 2.755(2) | 179(3) |
| xiv | O2W–H2B⋯O2 | x, y, z − 1 | 2.02(2) | 2.841(2) | 164(2) |
| xv | O5W–H5A⋯O201 | −x + 2, −y + 1, −z + 1 | 2.11(3) | 2.911(2) | 157(2) |
| xvi | N201–H204⋯O3 | −x + 1, −y + 1, −z + 2 | 2.13(2) | 2.862(2) | 141(2) |
| xvii | N201–H204⋯O1 | −x + 1, −y + 1, −z + 2 | 2.20(2) | 2.871(2) | 133(2) |
| xviii | N101–H104⋯O4 | −x + 1, −y, −z + 1 | 2.33(2) | 2.937(2) | 123(1) |
 | ||
| Fig. 4 (a) Hydrogen bonded network in the structure of 3·5H2O viewed along c (cations were omitted for clarity); (b) view of the π–π stacking (green dotted line) in the structure of 3·5H2O. | ||
The asymmetric units of 4S and 4R consist of one protonated pipemidic acid and one camphorsulfonate anion (Fig. S4†). The crystal packing in the pair of enantiomers 4S and 4R gives rise to only two charge-assisted hydrogen bonds between the secondary ammonium moiety of the cation and the sulfonate group of the anion (Table 4). The bonds constitute motif C22(6) of infinite chains that propagate along crystallographic axis a (Fig. 5). Additionally, three symmetrically related chains are created by applying the three crystallographic two-fold screw axes on this motif.
| Bond | D–H⋯A | Equivalent position code | Distance H⋯A, [Å] | Distance D⋯A, [Å] | Angle DHA, [°] |
|---|---|---|---|---|---|
| i | O1–H1⋯O3 | x, y, z | 1.59(6) [1.67(6)] | 2.523(5) [2.531(5)] | 166(6) [162(6)] |
| ii | N1–H4⋯O22 | x − 1, y, z | 1.95(9) [2.05(6)] | 2.847(6) [2.852(6)] | 151(7) [155(5)] |
| iii | N1–H2⋯O21 | x, y, z | 1.97(6) [1.90(6)] | 2.796(6) [2.795(6)] | 162(5) [167(5)] |
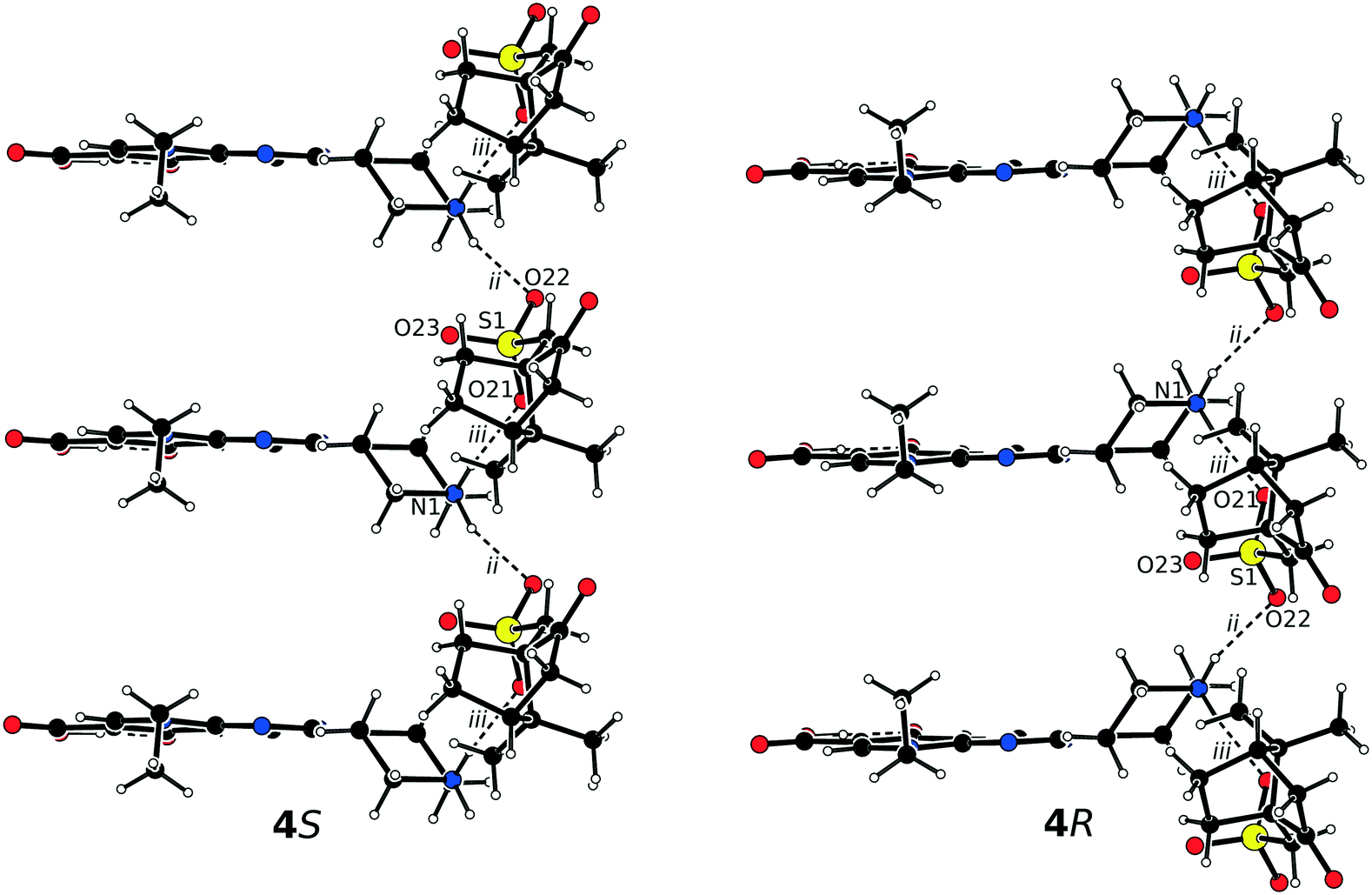 | ||
| Fig. 5 Hydrogen bonded chains in the structure of 4S (left) and 4R (right) viewed along c. | ||
These 5 salts were obtained as pure phases in very high yields, as it can be seen by the comparison of the simulated and experimental diffractograms (Fig. S6–S10†). Furthermore, these compounds have shown to be stable on shelf for at least 15 months (Fig. S13–S17†).
Regarding the thermal stability, both hydrated forms (1·2H2O and 3·5H2O) are the least stable, with the water molecules being released before 130 °C. TGA data of 1·2H2O and 3·5H2O reveal mass losses of 8.30% and 10.96%, respectively, corresponding to the two and five water molecules (calc. 8.67% and 11.44%, respectively) present in the structures at room temperature. The other three structures are stable until melting and decomposition, which occur at higher temperatures (>250 °C). Table 5 summarizes the DSC/TGA data for the five salts reported herein. All this data is supported by hot-stage microscopy observations (Fig. S24–S28†).
| Compound | Mass loss before melting/decomposition (temperature range) | Melting point/decomposition (onset) |
|---|---|---|
| 1·2H2O | 8.3% (<130 °C) | 200 °C |
| 2 | 0% | 160 °C |
| 3·5H2O | 10.96% (<130 °C) | 250 °C |
| 4 S | 0% | 220 °C |
| 4 R | 0% | 200 °C |
Metal complex and BioMOF
In complex 5, pipemidic acid assumes a zwitterionic role, with the protonation of its secondary amine site and the deprotonation of the carboxylic moiety, and the nitrate ions working as counterions (Fig. S5†). The piperazine ring of the protonated pipemidic moiety assumes a chair conformation with the Cremer–Pople θ angle63 value approaching 180° (177.9(5) °) and the puckering amplitude Q (ref. 63) of 0.566(5)°. The ethyl group again adopts a staggered conformation and points in the same direction as the piperazine ring with respect to the pyridopyrimidine plane. The carboxylate group still lies in the plane of the aromatic pyridopyrimidine system. However, the aforementioned O–H⋯O intramolecular hydrogen bond involving the keto and carboxylic groups (graph set62 descriptor S(6)) present in the previously described salt structures, is replaced by the coordination to the metal site in 5, with both the keto and carboxylate groups coordinating to Cu2+ (Fig. 6). The Cu center resides on an inversion center coordinating to two crystallographically dependent pipemidic acid zwitterions by the carbonyl and carboxylate moieties (1.891(3) and 1.918(3) Å, respectively), fulfilling a square planar geometry (coordination number 4), with O–Cu–O angles of 86.45(12), 92.55(12) and 180.0°. Further bond distances and angles are given in Table S6.† | ||
| Fig. 6 Representation of coordination of pipemidic acid to Cu(II) (ellipsoids are set at 30% probability level). | ||
The protonated secondary amine site of the piperazine ring of the pipemidic moiety participates as the donor in three charge-assisted hydrogen bonds (Table 6): i) connecting with the OCOO− that is not coordinated to Cu, and ii and iii) interacting with the nitrate counterion. These contacts give rise to a 1-D hydrogen bonded framework that aligns in the ac plane (Fig. 7).
| Bond | D–H⋯A | Equivalent position code | Distance H⋯A, [Å] | Distance D⋯A, [Å] | Angle DHA, [°] |
|---|---|---|---|---|---|
| i | N1–H2⋯O2 | x, y, z | 1.85(3) | 2.720(5) | 167(3) |
| ii | N1–H4⋯O5 | x, y, z | 2.15(3) | 2.982(6) | 154(3) |
| iv | N1–H4⋯O6 | 1 + x, −1 + y, z | 2.06(4) | 2.842(6) | 145(3) |
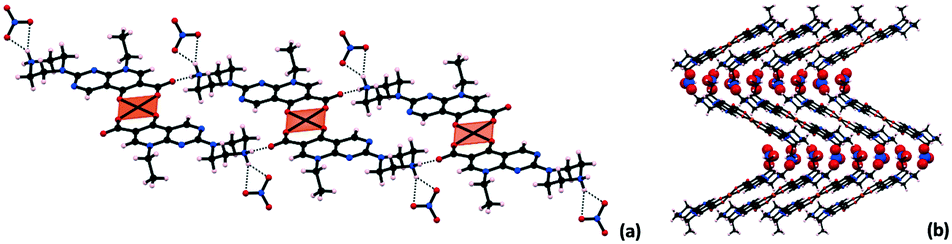 | ||
| Fig. 7 (a) Packing in a view along b depicting the 1D hydrogen bonded framework (polyhedral representation of Cu for better visualization); (b) packing in a view along a (nitrate ions are represented in space fill for better visualization). | ||
The structural analysis of 5 was complemented by FTIR data (Fig. S30†). It is known that the stretching vibration of the CO of carboxylic acid appears in the range of 1700–1725 cm−1. In the solid infrared spectrum of compound 5 this band is not present, thus this functional group is in the carboxylate form. The asymmetric and symmetric stretching vibrations of O–C–O are assigned to be in the range of 1650–1510 cm−1 and 1400–1280 cm−1, appearing in this case at 1570 cm−1 and 1400 cm−1.64,65 The interactions of the 4-oxo and the 3-carboxylate quinolone groups with the metallic ion can be noted in the range of 1800–1300 cm−1.65,66 Additionally, it can be noted that the stretching vibration of CN (1690–1640 cm−1) and of N–H (3500–3100 cm−1) are kept, confirming that the coordination is not established via nitrogen atoms.
Complex 5 was obtained as pure phase by LAG in very high yields, as proved by the comparison of the simulated and experimental diffractograms (Fig. S11†). Complex 5 is stable for at least 15 months on shelf (Fig. S18†). Additionally, its thermal stability, inferred from hot-stage microscopy (HSM), suggests that it is stable up 220 °C (Fig. S29†).
Regarding the Ag-MOF, 6, its crystal structure was previously reported, being already deposited at CSD under the refcode PICWUV.52 A similar structure is also disclosed at CSD, under the refcode GIWNOR.51 The asymmetric unit of these structures encloses two deprotonated pipemidic acids, two crystallographically independent silver cations, and 3.5 hydration water molecules. The piperazine rings of the protonated pipemidic moieties maintain the chair conformation with the Cremer–Pople θ angle63 values approaching 180° (163.7(6) and 171.8(6)°) and the puckering amplitudes Q (ref. 63) of 0.570(6) and 0.566(6)°. One of the silver sites assumes a distorted square pyramidal geometry (coordination number 5) coordinating in the equatorial positions to two pipemidic acid anions via the carbonyl and carboxylate moieties (Ag–O 2.349–2.515 Å; O–Ag–O 72.48–102.48°), and, in the axial position, to other pipemidic acid via the piperazine ring (Ag–N 2.316 Å; O–Ag–N 86.92–132.90°). The other silver cation bonds to three pipemidic acids via the carboxylate (Ag–O 2.363–2.425 Å) and to a fourth PA via the piperazine ring (Ag–N 2.301 Å) exhibiting a distorted tetrahedral geometry (coordination number 4) with the bond angles ranging from 85.98 to 118.37°. There are no direct hydrogen bonds between pipemidic acid, all involving water molecules. The overall arrangement gives rise to a 3D metal–organic framework, in which the water clusters lie on the pores of the structure (Fig. 8).
 | ||
| Fig. 8 View along a of the 3D metal–organic framework 6. | ||
The synthesis of this BioMOF by mechanochemistry is one more proof that this technique presents indeed major advantages. This compound had previously been synthesized by solvothermal methods, requiring over 6 days, temperature of 155 °C, and requiring the use of NaHCO3 and CH3COOH.52 We present here an alternative via manual grinding for 3 minutes, at room temperature, using minor amounts of aqueous ammonia and water.
With all these examples (1–6), it is possible to see that charge-assisted hydrogen bonds are the main driving force of the supramolecular assemblies both in the salts and in the metal-coordinated compounds reported herein. The presence of the charge is known to impact the geometric and energetic parameters of the hydrogen bonds, contributing for their stabilization and thus for the stabilization of the compounds.
Experimental
Reagents
The following reagents were used without further purification: pipemidic acid (Sigma-Aldrich, Germany), glycolic acid (Sigma-Aldrich, Germany), oxalic acid (Sigma-Aldrich, Germany), camphorsulfonic acid (TCI, Ireland), copper nitrate trihydrate (Merck, Germany), silver oxide (Alfa Aesar, Germany), aqueous ammonia 25% (Merck, Germany), and methanol (Carlo Erba, France).Synthesis of the salts
![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) with four water molecules per unit cell.
with four water molecules per unit cell.
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 molar ratio mixture of pipemidic (348.3 mg, 1.15 mmol) and oxalic (51.6 mg, 0.57 mmol) acids was placed into a 25 mL stainless steel cylinder together with two stainless steel balls (7 mm in diameter, 1.377 g methanol (40 μL; η = 0.1 μL mg−1)), the cylinder was sealed and the mixture was milled at the shaker ball mill at 30 Hz for 15 minutes. The product of milling was then left in a slurry in 2 ml of water overnight to achieve a pure bulk. Single crystals suitable for X-ray diffraction analysis (3·5H2O, dimensions 0.20 mm × 0.24 mm × 0.30 mm) were obtained from recrystallization in water (20 mg in 25 ml, dissolved after heating to 55 °C). The resulting solvate crystallizes in the space group P with ten water molecules per unit cell.
:1 molar ratio mixture of pipemidic (348.3 mg, 1.15 mmol) and oxalic (51.6 mg, 0.57 mmol) acids was placed into a 25 mL stainless steel cylinder together with two stainless steel balls (7 mm in diameter, 1.377 g methanol (40 μL; η = 0.1 μL mg−1)), the cylinder was sealed and the mixture was milled at the shaker ball mill at 30 Hz for 15 minutes. The product of milling was then left in a slurry in 2 ml of water overnight to achieve a pure bulk. Single crystals suitable for X-ray diffraction analysis (3·5H2O, dimensions 0.20 mm × 0.24 mm × 0.30 mm) were obtained from recrystallization in water (20 mg in 25 ml, dissolved after heating to 55 °C). The resulting solvate crystallizes in the space group P with ten water molecules per unit cell.
Synthesis of the metal complex and MOF
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/b_char_2009.gif) :Cu complex (5).
Complex 5 was obtained via manual grinding of 0.25 mmol of copper nitrate trihydrate (60.4 mg) and 0.50 mmol of pipemidic acid (151.6 mg), for 5 minutes, with water (100 μL; η = 0.47 μL mg−1) and aqueous ammonia (50 μL, 0.668 mmol). Aqueous ammonia was used to promote the deprotonation of pipemidic acid, and a slight excess was necessary to offset its high volatility. Single crystals suitable for X-ray diffraction analysis were obtained from the recrystallization in water at room temperature, by slow evaporation of the solvent.
:1 mixture of pipemidic acid (50.8 mg, 0.2 mmol) and silver oxide (23.2 mg, 0.1 mmol) was manually ground with water (50 μL; η = 0.68 μL mg−1) and aqueous ammonia (50 μL, 0.668 mmol) for 3 minutes. Also here aqueous ammonia was used to promote the deprotonation of pipemidic acid, and an excess was necessary to offset its high volatility. Single crystals suitable for X-ray diffraction analysis were obtained from recrystallization in a mixture of water/aqueous ammonia, with the same unit cell parameters as reported in the bibliography.52
:Cu complex (5).
Complex 5 was obtained via manual grinding of 0.25 mmol of copper nitrate trihydrate (60.4 mg) and 0.50 mmol of pipemidic acid (151.6 mg), for 5 minutes, with water (100 μL; η = 0.47 μL mg−1) and aqueous ammonia (50 μL, 0.668 mmol). Aqueous ammonia was used to promote the deprotonation of pipemidic acid, and a slight excess was necessary to offset its high volatility. Single crystals suitable for X-ray diffraction analysis were obtained from the recrystallization in water at room temperature, by slow evaporation of the solvent.
:1 mixture of pipemidic acid (50.8 mg, 0.2 mmol) and silver oxide (23.2 mg, 0.1 mmol) was manually ground with water (50 μL; η = 0.68 μL mg−1) and aqueous ammonia (50 μL, 0.668 mmol) for 3 minutes. Also here aqueous ammonia was used to promote the deprotonation of pipemidic acid, and an excess was necessary to offset its high volatility. Single crystals suitable for X-ray diffraction analysis were obtained from recrystallization in a mixture of water/aqueous ammonia, with the same unit cell parameters as reported in the bibliography.52
General characterization
:100 in weight).
Single crystal X-ray diffraction studies (SCXRD)
Crystals suitable for single X-ray diffraction studies were mounted on a loop with Fomblin© protective oil. Data were collected either on a Bruker AXS-KAPPA APEX II or on a Bruker AXS-KAPPA D8 – QUEST diffractometer, both with graphite-monochromated radiation (Mo Kα, λ = 0.71073 Å). X-ray generators were operated at 50 kV and 30 mA and APEX2 and APEX368 programs monitored the data collections. Data were corrected for Lorentzian polarization and absorption effects using SAINT69 and SADABS70 programs.
The structures 1·2H2O, 2, 3·5H2O, 4S and 4R were solved by direct methods with SHELXS-201371 and refined by full-matrix least-squares against F2 using SHELXL-2018.72 The CH, CH2 and CH3 hydrogen atoms were located on the difference electron density maps and refined as riding atoms with their Uiso(H) fixed to a multiple of Ueq of their bonding carbon atom (Uiso(H) = 1.2Ueq(C) for CH and CH2 groups and Uiso(H) = 1.5Ueq(C) for the CH3 groups). All acidic hydrogens were located on the difference electron density maps and refined freely.
For the structure 5, SHELXT 2014/473 was used for structure solution and SHELXL 2014/772 was used for full matrix least-squares refinement on F2. These programs are included in the WINGX-Version 2014.174 program package. A full-matrix least-squares refinement was used for the non-hydrogen atoms with anisotropic thermal parameters. The hydrogens of carbons were inserted in idealized positions and allowed to refine in the parent carbon atom. The hydrogen atoms connected to nitrogen were located from the electron density map and the distances were restrained. MERCURY 2020.167 and PLATON75 were used for packing diagrams. ORTEP-376 plots of all the compounds are presented in ESI.† All geometric calculations were performed using PLATON software.75 The values are rounded with respect to their estimated standard deviations.
Table 7 summarizes data collection and refinement details. Crystallographic data of complexes 1 to 5 were deposited at the Cambridge Crystallographic Data Centre (CCDC 2031938–2031943).
| Structure | 1·2H2O | 2 | 3·5H2O | 4 S | 4 R | 5 |
|---|---|---|---|---|---|---|
| a R 1 = ∑‖Fo| − |Fc‖/∑ |Fo|. b wR2 = [∑[w(Fo2 − Fc2)2]/∑[w(Fo2)2]]1/2. | ||||||
| Formula | C16H25N5O8 | C16H19N5O7 | C30H46N10O15 | C24H33N5O7S | C24H33N5O7S | C28H34CuN10O6•2(NO3) |
| M | 415.41 | 393.36 | 786.77 | 535.61 | 535.61 | 794.22 |
| Crystal system | Triclinic | Monoclinic | Triclinic | Orthorhombic | Orthorhombic | Monoclinic |
| Space group |
P |
P21/c |
P |
P212121 | P212121 | P21/c |
| T [K] | 150(2) | 150(2) | 150(2) | 150(2) | 296(2) | 296(2) |
| a [Å] | 7.2922(3) | 9.8463(8) | 7.5920(3) | 6.7041(8) | 6.7095(4) | 12.1073(9) |
| b [Å] | 7.8666(3) | 9.3601(7) | 15.4924(7) | 11.8458(10) | 11.8584(6) | 6.2472(5) |
| c [Å] | 17.1901(6) | 18.1965(12) | 16.2129(8) | 30.358(4) | 30.4373(15) | 21.3210(15) |
| α [°] | 81.064(2) | 90 | 72.946(3) | 90 | 90 | 90 |
| β [°] | 79.634(2) | 91.888(3) | 83.283(2) | 90 | 90 | 94.374(4) |
| γ [°] | 80.387(2) | 90 | 81.684(2) | 90 | 90 | 90 |
| V [Å3] | 948.38(6) | 1676.1(2) | 1798.30(14) | 2410.9(5) | 2421.7(2) | 1608.0(2) |
| Z | 2 | 4 | 2 | 4 | 4 | 2 |
| F(000) | 440 | 824 | 832 | 1136 | 1136 | 822 |
| μ(Mo, Kα) [mm−1] | 0.118 | 0.124 | 0.118 | 0.191 | 0.191 | 0.765 |
| Collected reflections | 27174 |
15376 |
31823 |
10172 |
12850 |
27329 |
| Unique reflections | 7545 | 3938 | 8306 | 4452 | 5472 | 3272 |
| R int [%] | 6.80 | 5.75 | 6.12 | 7.20 | 9.05 | 14.79 |
| θ max [°] | 33.813 | 27.772 | 27.595 | 25.457 | 27.471 | 26.445 |
| Number of parameters | 294 | 269 | 560 | 346 | 346 | 247 |
| R 1,a wR2b [I ≥ 2σ(I)] | 0.0509, 0.1234 | 0.0430, 0.1100 | 0.0472, 0.1066 | 0.0569, 0.1036 | 0.0611, 0.1163 | 0.0571, 0.1171 |
| GOF on F2 | 0.945 | 0.931 | 0.960 | 0.952 | 0.960 | 1.003 |
Conclusions
The main structural features of five new molecular salts of pipemidic acid with glycolic, oxalic and (R)- and (S)-camphorsulfonic acids are discussed herein. The charge assisted hydrogen bonds established between the protonated piperazine ring of pipemidic acid and the deprotonated acid (either carboxylic or sulfonic) moiety of the coformer are the most relevant determining the three dimensional assembly of these multicomponent forms. An extended hydrogen bonding network mediated by the solvating water molecules is observed in the hydrated molecular salts. Albeit the five crystalline solids are stable on shelf for over 15 months, the presence of water in compounds 1·2H2O and 3·5H2O is responsible for their lower thermal stability when compared with the three anhydrous forms.A Cu(II) complex (5) is also unveiled herein. The coordination to the square planar Cu(II) centre is established via the keto and carboxylate groups of pipemidic acid. Hydrogen bonds between neighbouring pipemidic acid moieties and between pipemidic acid and the nitrate anions give rise to a 1D hydrogen-bonded network. Similarly to what was observed for the molecular salts, also complex 5 is stable on shelf for at least 15 months and it is stable until temperatures above 200 °C.
Mechanochemistry is in the basis of the development of these crystalline forms, and the synthesis of the previously reported Ag-MOF (6) by liquid-assisted grinding is the confirmation of mechanochemistry value, with a solution synthetic procedure of over 6 days, at high temperature, being instead, accomplished in a few minutes, at room temperature. Our results make it evident, once again, that mechanochemistry is an excellent sustainable, efficient and fast tool for the discovery of new crystal forms of old drugs. This technique offers great potential to be further explored for several other diverse applications.
Author contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript. All authors contributed equally.Funding sources
Fundação para a Ciência e a Tecnologia (FCT, Portugal) (projects UIDB/00100/2020, UIDP/00100/2020, UID/QUI/00100/2019 and PTDC/QUI-OUT/30988/2017, and contract under DL No. 57/2016 regulation) and FEDER, Portugal 2020 and Lisboa 2020 are acknowledged for funding (project LISBOA-01-0145-FEDER-030988).Conflicts of interest
There are no conflicts to declare.Acknowledgements
Fundação para a Ciência e a Tecnologia (FCT, Portugal) (projects UIDB/00100/2020, UIDP/00100/2020, UID/QUI/00100/2019 and PTDC/QUI-OUT/30988/2017, and contracts under DL No. 57/2016 regulation and CEECIND2018) and FEDER, Portugal 2020 and Lisboa 2020 are acknowledged for funding (project LISBOA-01-0145-FEDER-030988). Dr. Auguste Fernandes is acknowledged for the DSC/TGA and FTIR data.References
- G.-B. Fernando, Chem. Int., 2019, 41, 12–17 Search PubMed.
- J. L. Howard, Q. Cao and D. L. Browne, Chem. Sci., 2018, 9, 3080–3094 RSC.
- V. André, A. R. F. da Silva, A. Fernandes, R. Frade, C. Garcia, P. Rijo, A. M. M. Antunes, J. Rocha and M. T. Duarte, ACS Appl. Bio Mater., 2019, 2, 2347–2354 CrossRef.
- T. Friščić, J. Mater. Chem., 2010, 20, 7599–7605 RSC.
- T. Friščić, C. Mottillo and H. M. Titi, Angew. Chem., Int. Ed., 2020, 59, 1018–1029 CrossRef.
- J.-L. Do and T. Friščić, ACS Cent. Sci., 2017, 3, 13–19 CrossRef CAS.
- V. André, S. Quaresma, J. L. F. da Silva and M. T. Duarte, Beilstein J. Org. Chem., 2017, 13, 2416–2427 CrossRef.
- S. Karki, T. Friščić, W. Jones and W. D. S. Motherwell, Mol. Pharmaceutics, 2007, 4, 347–354 CrossRef CAS.
- D. Hasa, G. S. Rauber, D. Voinovich and W. Jones, Angew. Chem., Int. Ed., 2015, 54, 7371–7375 CrossRef CAS.
- V. Andre, A. Hardeman, I. Halasz, R. S. Stein, G. J. Jackson, D. G. Reid, M. J. Duer, C. Curfs, M. Teresa Duarte and T. Friscic, Angew. Chem., Int. Ed., 2011, 50, 7858–7861 CrossRef CAS.
- D. Hasa, E. Carlino and W. Jones, Cryst. Growth Des., 2016, 16, 1772–1779 CrossRef CAS.
- D. Braga, L. Maini and F. Grepioni, Chem. Soc. Rev., 2013, 42, 7638–7648 RSC.
- A. Delori, T. Friščić and W. Jones, CrystEngComm, 2012, 14, 2350–2362 RSC.
- N. Kumari, B. Bhattacharya, P. Roy, A. A. L. Michalchuk, F. Emmerling and A. Ghosh, Cryst. Growth Des., 2019, 19, 6482–6492 CrossRef CAS.
- R. Shaikh, R. Singh, G. M. Walker and D. M. Croker, Trends Pharmacol. Sci., 2018, 39, 1033–1048 CrossRef CAS.
- C. Maheshwari, V. Andre, S. Reddy, L. Roy, T. Duarte and N. Rodrigez-Hornedo, CrystEngComm, 2012, 14, 4801–4811 RSC.
- S. Salehi, S. M. M. Moghaddam, M. Tarin and A. S. Saljooghi, Phys. Chem. Res., 2020, 8, 91–110 Search PubMed.
- Q. H. Tran and T. T. Doan, New J. Chem., 2020, 44, 13036–13045 RSC.
- S. Abdolmaleki, M. Ghadermazi, M. Ashengroph, A. Saffari and S. M. Sabzkohi, Inorg. Chim. Acta, 2018, 480, 70–82 CrossRef CAS.
- A. S. McCalmont, A. Ruiz, M. C. Lagunas, W. T. Al-Jamal and D. E. Crawford, ACS Sustainable Chem. Eng., 2020, 8, 15243–15249 CrossRef CAS.
- I. Sović, S. Lukin, E. Meštrović, I. Halasz, A. Porcheddu, F. Delogu, P. C. Ricci, F. Caron, T. Perilli, A. Dogan and E. Colacino, ACS Omega, 2020, 5, 28663–28672 CrossRef.
- S. Rojas, T. Devic and P. Horcajada, J. Mater. Chem. B, 2017, 5, 2560–2573 RSC.
- R. A. A. van Oosterom and E. G. Hartman, Vet. Q., 1986, 8, 2–5 CrossRef CAS.
- K. Hirai, A. Ito, Y. Abe, S. Suzue, T. Irikura, M. Inoue and S. Mitsuhashi, Antimicrob. Agents Chemother., 1981, 19, 188–189 CrossRef CAS.
- M. Lavorgna, R. Iacovino, C. Russo, C. Di Donato, C. Piscitelli and M. Isidori, Int. J. Mol. Sci., 2019, 20, 416–430 CrossRef.
- M. I. Andersson and A. P. MacGowan, J. Antimicrob. Chemother., 2003, 51, 1–11 CrossRef CAS.
- V. Uivarosi, Molecules, 2013, 18, 11153–11197 CrossRef CAS.
- A. Ahmed and M. Daneshtalab, J. Pharm. Pharm. Sci., 2011, 15, 52–72 Search PubMed.
- C. Sissi and M. Palumbo, Curr. Med. Chem.: Anti-Cancer Agents, 2003, 3, 439–450 CrossRef CAS.
- Y. Pommier, E. Leo, H. Zhang and C. Marchand, Chem. Biol., 2010, 17, 421–433 CrossRef CAS.
- A. Fabrega, S. Madurga, E. Giralt and J. Vila, Microb. Biotechnol., 2009, 2, 40–61 CrossRef CAS.
- P. Ball, J. Antimicrob. Chemother., 2000, 46, 17–24 CrossRef CAS.
- T. D. M. Pham, Z. M. Ziora and M. A. T. Blaskovich, MedChemComm, 2019, 10, 1719–1739 RSC.
- P. C. Appelbaum and P. A. Hunter, Int. J. Antimicrob. Agents, 2000, 16, 5–15 CrossRef CAS.
- K. J. Aldred, R. J. Kerns and N. Osheroff, Biochemistry, 2014, 53, 1565–1574 CrossRef CAS.
- I. Turel, Coord. Chem. Rev., 2002, 232, 27–47 CrossRef CAS.
- R. Singh, A. Debnath, D. T. Masram and D. Rathore, Res. J. Chem. Sci., 2013, 3, 83–94 CAS.
- V. Andre, F. Galego and M. Martins, Cryst. Growth Des., 2018, 18, 2067–2081 CrossRef CAS.
- P. C. Alves, P. Rijo, C. Bravo, A. M. M. Antunes and V. André, Molecules, 2020, 25, 2374–2387 CrossRef CAS.
- C. R. Groom, I. J. Bruno, M. P. Lightfoot and S. C. Ward, Acta Crystallogr., Sect. B: Struct. Sci., Cryst. Eng. Mater., 2016, 72, 171–179 CrossRef CAS.
- C. R. Groom and F. Allen, Angew. Chem., Int. Ed., 2014, 53, 662–671 CrossRef CAS.
- A. Sakon, A. Sekine and H. Uekusa, Cryst. Growth Des., 2016, 16, 4635–4645 CrossRef CAS.
- R. Ceolin, F. Clanet, G. Ghemard, C. Souleau and P. Khodadad, J. Appl. Crystallogr., 1979, 12, 612–612 CrossRef CAS.
- I. Fonseca, S. Martínez-Carrera and S. García-Blanco, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 1986, 42, 1618–1621 CrossRef.
- H. K. Fun, K. Chinnakali, I. A. Razak, S. Z. Zhan, C. J. Hu and Q. Meng, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 1999, 55, 766–768 CrossRef.
- G.-J. Zhang, J.-H. He, S.-W. Yan, Z.-L. Ye and G.-H. Xin, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2011, 67, o1011–o1012 CrossRef CAS.
- L. Yang, D. Tao, X. Yang, Y. Li and Y. Guo, Chem. Pharm. Bull., 2003, 51, 494–498 CrossRef CAS.
- E. K. Efthimiadou, Y. Sanakis, N. Katsaros, A. Karaliota and G. Psomas, Polyhedron, 2007, 26, 1148–1158 CrossRef CAS.
- X. Cao, G. E. Xing and Y. Zhang, J. Mol. Struct., 2016, 1123, 133–137 CrossRef CAS.
- J.-Q. Sha, X. Li, H.-B. Qiu, Y.-H. Zhang and H. Yan, Inorg. Chim. Acta, 2012, 383, 178–184 CrossRef CAS.
- C.-M. Xue, S.-X. Li, L. Zhang, J.-Q. Sha, T.-Y. Zheng, Q.-N. Zhang and L. Li, J. Inorg. Organomet. Polym. Mater., 2013, 23, 1468–1476 CrossRef CAS.
- M.-T. Li, J.-W. Sun, J.-Q. Sha, H.-B. Wu, E.-L. Zhang and T.-Y. Zheng, J. Mol. Struct., 2013, 1045, 29–34 CrossRef CAS.
- L.-N. Duan, Q.-Q. Dang, C.-Y. Han and X.-M. Zhang, Dalton Trans., 2015, 44, 1800–1804 RSC.
- J.-H. He, D.-R. Xiao, S.-W. Yan, D.-Z. Sun, H.-Y. Chen, X. Wang, J. Yang, Z.-L. Ye, R. Yuan and E.-B. Wang, Solid State Sci., 2012, 14, 1203–1210 CrossRef CAS.
- Z.-L. Ye, G.-H. Xin, F.-T. Zhang and D.-R. Xiao, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2013, 69, m127 CrossRef CAS.
- W. Xu, D.-S. Zhu, X.-D. Song and Z. An, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2009, 65, m1223 CrossRef CAS.
- J. Huang, W.-P. Hu and Z. An, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2008, 64, m547 CrossRef CAS.
- L. Zhu and T. Zhou, Z. Kristallogr. - New Cryst. Struct., 2016, 231, 447–449 CAS.
- J.-H. He, D.-Z. Sun, D.-R. Xiao, S.-W. Yan, H.-Y. Chen, X. Wang, J. Yang and E.-B. Wang, Polyhedron, 2012, 42, 24–29 CrossRef CAS.
- D.-R. Xiao, J. H. He, D. Z. Sun, H. Y. Chen, S. W. Yan, X. Wang, J. Yang, R. Yuan and E. B. Wang, Eur. J. Inorg. Chem., 2012, 2012, 1783–1789 CrossRef CAS.
- D.-Z. Sun, G.-J. Zhang, H.-Y. Chen, J.-H. He and S.-W. Yan, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2011, 67, m388 CrossRef CAS.
- J. Bernstein, R. E. Davis, L. Shimoni and N.-L. Chang, Angew. Chem., Int. Ed. Engl., 1995, 34, 1555–1573 CrossRef CAS.
- D. Cremer and J. A. Pople, J. Am. Chem. Soc., 1975, 97, 1354–1358 CrossRef CAS.
- G. B. Deacon and R. J. Phillips, Coord. Chem. Rev., 1980, 33, 227–250 CrossRef CAS.
- B. Szymanska, D. Skrzypek, D. Kovala-Demertzi, M. Staninska and M. A. Demertzis, Spectrochim. Acta, Part A, 2006, 63, 518–523 CrossRef.
- Z.-F. Chen, B.-Q. Li, Y.-R. Xie, R.-G. Xiong, X.-Z. You and X.-L. Feng, Inorg. Chem. Commun., 2001, 4, 346–349 CrossRef CAS.
- C. F. Macrae, I. J. Bruno, J. A. Chisholm, P. R. Edgington, P. McCabe, E. Pidcock, L. Rodriguez-Monge, R. Taylor, J. van de Streek and P. A. Wood, J. Appl. Crystallogr., 2008, 41, 466–470 CrossRef CAS.
- Bruker, Bruker Analytical Systems, Madison, WI, 2016 Search PubMed.
- Bruker, Bruker Analytical Systems, Madison, WI, 2014 Search PubMed.
- Bruker, Bruker Analytical Systems, Madison, WI, 2014 Search PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Crystallogr., 2008, 64, 112–122 CrossRef CAS.
- G. M. Sheldrick, Acta Crystallogr., Sect. C: Struct. Chem., 2015, 71, 3–8 Search PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Adv., 2015, 71, 3–8 CrossRef.
- L. Farrugia, J. Appl. Crystallogr., 1999, 32, 837–838 CrossRef CAS.
- A. L. Spek, Inorg. Chim. Acta, 2018, 470, 232–237 CrossRef CAS.
- C. L. Barnes, J. Appl. Crystallogr., 1997, 30, 568–568 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Structural data and DSC/TGA details. CCDC 2031938–2031943 for compounds 1 to 5. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d0ce01533d |
| This journal is © The Royal Society of Chemistry 2021 |