
Multicomponent crystals of baclofen with acids and bases—conformational flexibility and synthon versatility†
Ramokone Junia
Malapile
 a,
Kudzanai
Nyamayaro
a,
Luigi R.
Nassimbeni
b and
Nikoletta B.
Báthori
*a
a,
Kudzanai
Nyamayaro
a,
Luigi R.
Nassimbeni
b and
Nikoletta B.
Báthori
*a
aDepartment of Chemistry, Cape Peninsula University of Technology, P. O. Box 652, Cape Town, 8000, South Africa. E-mail: bathorin@cput.ac.za; Fax: +27 21 460 3854; Tel: +27 21 460 8354
bDepartment of Chemistry, University of Cape Town, Rondebosch 7701, South Africa
First published on 11th November 2020
Abstract
Baclofen (BAC, (R/S)-4-amino-3-(4-chlorophenyl)butanoic acid), was used to form multicomponent crystals (MCCs) with coformers (CFs) of different acidic strength and molecular size. The crystallisation of BAC with oxalic acid (OXA), salicylic acid (SAL), phenoxyacetic acid (POA), 2,4-dichlorophenoxyacetic acid (2,4ClPOA), 4-picoline (4PIC) and 3,4-lutidine (3,4LUT) yielded seven MCCs. The bulk materials were analysed by thermoanalytical methods (thermogravimetry and differential scanning calorimetry), powder X-ray diffraction analysis and Fourier transform infrared spectrometry. Liquid assisted grinding was also applied to form the MCCs. X-ray analysis of the single crystal structures revealed that BAC exists in zwitterionic or cationic form, and the MCCs represent a large variety of crystal classes, such as salts, salt solvates, cocrystal salt solvates or solvates with different stoichiometry. It was noted that BAC appeared in many different conformations in the MCCs, but comparative crystal packing analysis revealed similarities in the packing arrangement of the MCCs. This structural feature can be used for crystal engineering purposes when MCCs of BAC are designed.
Introduction
The solid state modification of pharmaceuticals via the application of crystal engineering principles1 is a sustainable way to form new solid forms2 with improved physicochemical or solid state properties of a selected active pharmaceutical ingredient (API) without altering its molecular structure.3 Crystal engineering includes the manipulation of supramolecular synthons4 and the molecular conformations5 of the API and the coformer (CF). The first approach is a well-understood process, and there are experimental and theoretical methods6 to achieve success, while the second requirement—influencing the conformation of a given molecule in the solid state—is particularly challenging.In this work, we focus on baclofen (BAC), (R/S)-4-amino-3-(4-chlorophenyl)butanoic acid (Chart 1), a GABAB receptor agonist, used in the treatment of cerebral palsy,7 neuralgia,8 spinal cord injuries9 and addictive behaviours.10 Recently, BAC was repurposed for the treatment of early-onset alcoholism11 and also showed promising results in vitro when applied in combination with acamprosate in Alzheimer's disease therapy.12 BAC is chiral, and its R enantiomer is the eutomer whereas the S enantiomer is the distomer without known side-effects. The available formulations contain the racemic modification,13 but on a few occasions, successful optical resolution was carried out.14 BAC is a γ-amino acid and the co-occurrence of the carboxylic acid/carboxylate (pKa = 3.87)15 and the primary amine/ammonium (pKa = 9.62)15 functional groups promote the formation of well-known robust supramolecular synthons. BAC is slightly water-soluble (4.3 mg ml−1, pH 7.6)16 thus, our interest is in its supramolecular modification.
 | ||
| Chart 1 Experimental flow chart (BAC: baclofen, (R/S)-4-amino-3-(4-chlorophenyl)butanoic acid, OXA: oxalic acid, SAL: salicylic acid, POA: phenoxyacetic acid, 2,4ClPOA: 2,4-dichlorophenoxyacetic acid, 4PIC: 4-picoline or 4-methylpyridine, 3,4LUT: 3,4-lutidine or 3,4-dimethylpyridine). | ||
In our previous study,17 BAC was exposed to a series of monocarboxylic acids with different sizes of hydrophobic substituents (benzoic acid, p-toluic acid, 1-hydroxy-2-naphthoic acid and p-toluene sulfonic acid). The packing of these crystals was similar, viz. the molecules formed alternating polar and aromatic layers. The similar packings were formed despite the differing fine details of the hydrogen bonds in the polar layer and the significant difference in the size of the coformers. We also noted that BAC has sufficient molecular flexibility to accommodate the selected coformers of variable molecular size.
In this work, we extend the crystalline landscape of BAC by cocrystallising with acidic coformers of increasing size and varying polarity, such as salicylic acid (SAL), phenoxyacetic acid (POA) and 2,4-dichlorophenoxyacetic acid (2,4ClPOA). SAL was selected because of the previous successful cocrystallisation with the structurally similar benzoic acid. POA and its halogenated form, 2,4ClPOA are plant growth retardants/herbicides, thus the reason of using them as coformers is because of structural considerations and not their chemical suitability. Based on our previous studies,17 we speculated that increasing the number of the chloro substituents on the aromatic ring of the carboxylic acid coformer the formation of Cl⋯Cl interactions in the aromatic layer will be encouraged, while the overall layered structure of the crystal should be maintained. Therefore these crystallisations tested crystal engineering principles, i.e. fine-tuning the intermolecular interactions in the aromatic layer. Based on the pKa values of BAC and previous successful cocrystallisations of the API with acids it can be concluded that BAC is a good proton acceptor (base). However, this raises the question of whether BAC can act as an acid when cocrystallised with a basic coformer. Therefore BAC was exposed to two basic N-heterocyclic coformers, 3,4-lutidine (3,4LUT) and 4-picoline (4PIC).
The seven MCCs were studied by single crystal X-ray diffraction and the bulk materials were analysed by thermoanalytical methods, powder X-ray diffraction analysis and Fourier transform infrared spectrometry. Liquid assisted grinding was also used to seek environmentally friendly methods for the synthesis of the MCCs. Conformation analysis of the BAC moieties and comparative crystal packing analysis of the MCCs were carried out.
Experimental details
Details of the crystallisation conditions, single crystal data collections and refinements,18 as well as details of hydrogen bonds for all the discussed crystal structures and their visualisation are given in the ESI,† Tables S1–S17, with the bulk property analysis and the results of the grinding experiments (PXRD, TGA, DSC and FTIR, Fig. A1–A34). The classification of the multicomponent crystals is based on the recent work of de Gelder et al.,19 which defines three main classes (salt, solvate and cocrystal) and seven subclasses of the MCCs (true solvate, true salt, true cocrystal, salt solvate, cocrystal solvate, cocrystal salt and cocrystal salt solvate).Results and discussion
Baclofen was cocrystallised with 36 acidic and 17 basic coformers, but only six coformers resulted in MCC formation (success rate: 11%). The crystal structures of baclofen with oxalic acid (OXA), salicylic acid (SAL), phenoxyacetic acid (POA), 2,4-dichlorophenoxyacetic acid (2,4ClPOA), 4-picoline (4PIC) and 3,4-lutidine (3,4LUT) are presented. The cocrystallisations were carried out by the slow evaporation crystallisation (SEC) method. Racemic baclofen was used for the crystallisations, and the asymmetric units were selected to contain the R enantiomer for easy comparison with already published results.(BAC+)(OXA−)·w
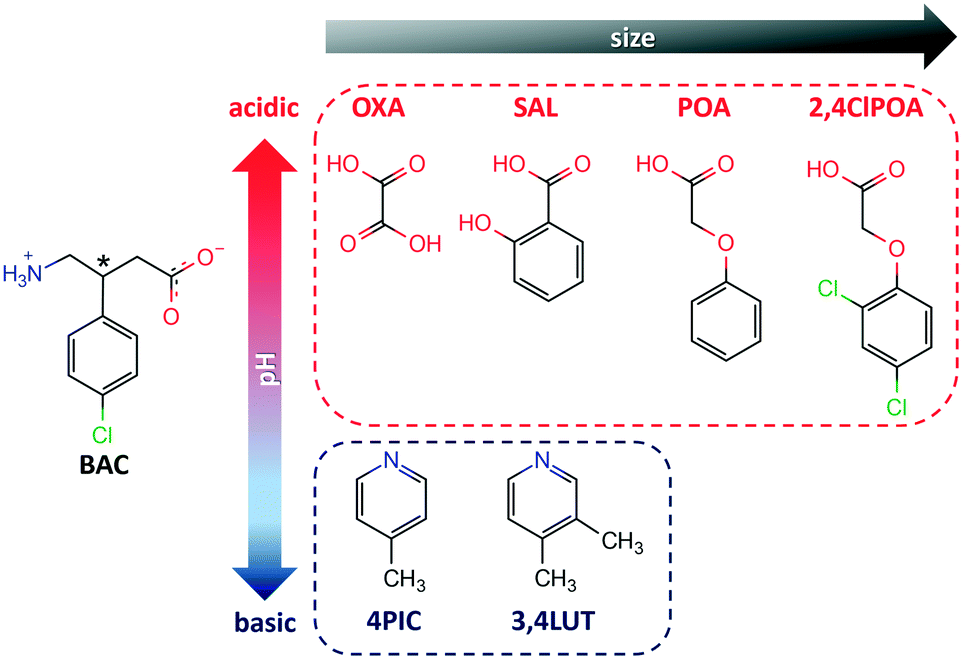
Baclofen (BAC) was previously cocrystallised with oxalic acid (OXA) from ethanol solvent (CSD code: LUSXOO)17 the asymmetric unit (ASU) contained one molecule of BAC and half a molecule of OXA, (BAC+)(½OXA2−) (P21/c, no. 14). Both of the carboxylic acid protons were transferred to the BAC moiety; thus the crystal is a salt (Fig. S1†). The cocrystallisation was repeated for experimental training purposes under the same conditions, but surprisingly, a different stoichiometric ratio of the constituents was obtained. BAC cocrystallised with two half oxalic acids, where one of the OXA is neutral, and the other is double deprotonated (OXA2−) and the BAC is protonated (BAC+). Also, one molecule of water is included in the ASU (Fig. 1a). The crystal can be described as a cocrystal salt solvate (hydrate), (BAC+)(½OXA2−)·½OXA·H2O (P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) , no. 2). For simplicity, this crystal will be referred to as (BAC+)(OXA−)·w from now onwards. The oxalic acids form hydrogen bonded chains in an alternating manner of OXA⋯(OXA2−)⋯OXA via O13–H13⋯O15 (2.464(2) Å, 171°) in [001] direction. The parallel acid chains are linked together via water molecules forming R42(8) supramolecular synthon via the O17–H17A⋯O12 (2.782(7) Å, 169°) and O17–H17B⋯O12 (2.862(4) Å, 162°) hydrogen bonds (Fig. 1b, yellow). The amino groups of the BAC molecules are incorporated into this ionic layer and form hydrogen bonds with the neighbouring water (N1–H1B⋯O17, 2.790(7) Å, 171°) and two oxalic acid molecules (N1–H1A⋯O16, 2.836(7) Å, 167° and N1–H1C⋯O16, 2.898(3) Å, 153°) and the latter forms a R42(8) graph set (Fig. 1b, green). Two BAC molecules from adjacent ionic layers form carboxylic acid dimers by O1–H1⋯O2 (2.659(9) Å, 178°) hydrogen bonds and these BAC dimers interact with neighbouring dimers via C8–H8B⋯O1 (3.544(3) Å, 171°) hydrogen bonds. Both of these interactions can be described by the R22(8) graph set (Fig. 1c, with blue and purple, respectively). It is interesting to note that in the structure of (BAC+)(½OXA2−) there are no carboxylic acid dimers between the BAC molecules, but the BACs are interacting via –COOH⋯NH3+ interactions in a head-to-tail manner (the carboxylic acid group defined as the head and the amino group as the tail). The OXA molecules do not form hydrogen bonds to each other but are bridging between adjacent hydrogen bonded BAC chains. The resultant two hydrogen bonding motifs can be described with the R33(11) and the R44(22) graph sets (Fig. S1b,† blue and green, respectively). The other difference noted between the two oxalic acid MCCs is the conformation of the BAC molecules. The crystal packing for both MCCs is layered. The packing for (BAC+)(½OXA2−) has chlorine atoms of BAC which are facing each other in a herringbone pattern (Fig. 1d and S1c†).
, no. 2). For simplicity, this crystal will be referred to as (BAC+)(OXA−)·w from now onwards. The oxalic acids form hydrogen bonded chains in an alternating manner of OXA⋯(OXA2−)⋯OXA via O13–H13⋯O15 (2.464(2) Å, 171°) in [001] direction. The parallel acid chains are linked together via water molecules forming R42(8) supramolecular synthon via the O17–H17A⋯O12 (2.782(7) Å, 169°) and O17–H17B⋯O12 (2.862(4) Å, 162°) hydrogen bonds (Fig. 1b, yellow). The amino groups of the BAC molecules are incorporated into this ionic layer and form hydrogen bonds with the neighbouring water (N1–H1B⋯O17, 2.790(7) Å, 171°) and two oxalic acid molecules (N1–H1A⋯O16, 2.836(7) Å, 167° and N1–H1C⋯O16, 2.898(3) Å, 153°) and the latter forms a R42(8) graph set (Fig. 1b, green). Two BAC molecules from adjacent ionic layers form carboxylic acid dimers by O1–H1⋯O2 (2.659(9) Å, 178°) hydrogen bonds and these BAC dimers interact with neighbouring dimers via C8–H8B⋯O1 (3.544(3) Å, 171°) hydrogen bonds. Both of these interactions can be described by the R22(8) graph set (Fig. 1c, with blue and purple, respectively). It is interesting to note that in the structure of (BAC+)(½OXA2−) there are no carboxylic acid dimers between the BAC molecules, but the BACs are interacting via –COOH⋯NH3+ interactions in a head-to-tail manner (the carboxylic acid group defined as the head and the amino group as the tail). The OXA molecules do not form hydrogen bonds to each other but are bridging between adjacent hydrogen bonded BAC chains. The resultant two hydrogen bonding motifs can be described with the R33(11) and the R44(22) graph sets (Fig. S1b,† blue and green, respectively). The other difference noted between the two oxalic acid MCCs is the conformation of the BAC molecules. The crystal packing for both MCCs is layered. The packing for (BAC+)(½OXA2−) has chlorine atoms of BAC which are facing each other in a herringbone pattern (Fig. 1d and S1c†).
 | ||
| Fig. 1 The ASU of (BAC+)(OXA−)·w (a); halves of the OXA that are not part of the ASU are faded for clarity. The ionic layer formed by oxalic acid and water molecules with the amino groups that anchor BAC molecules (b). Interactions between the adjacent BAC molecules (c) and the layered crystal packing viewed down [100] direction (d) (OXAs are yellow, water molecules are blue). | ||
BAC·SAL·w
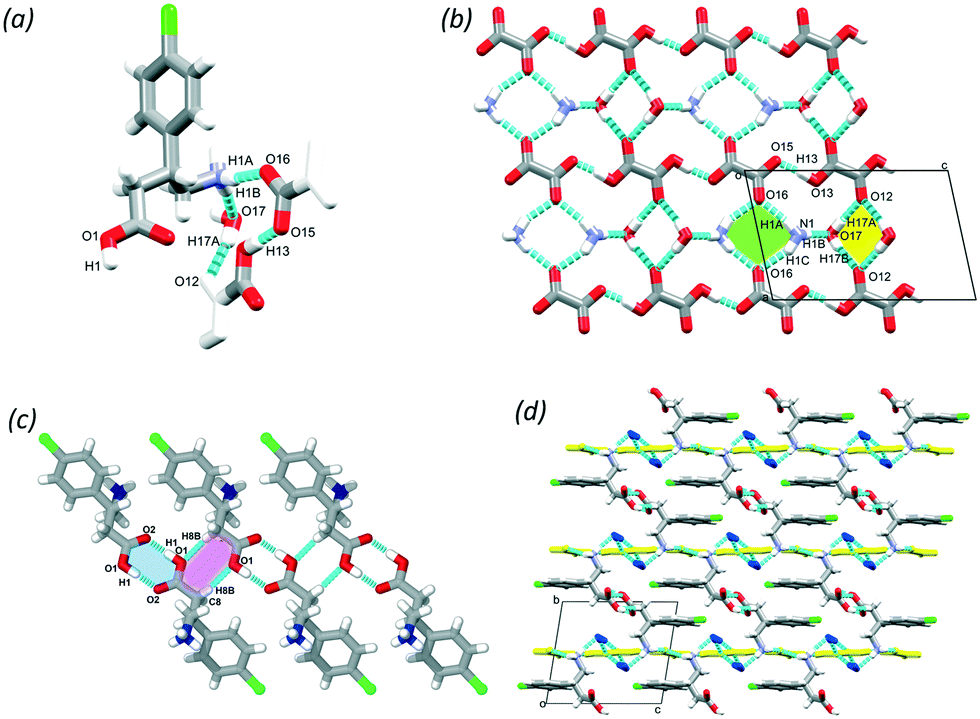
BAC was previously cocrystallised with benzoic acid (BA)17 that is generally regarded as safe (GRAS) substance in a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio and resulted in a crystal with two molecules of BAC in zwitterionic form and three molecules of neutral BA in the ASU (P21/c, no. 14). The crystallisation of BAC with salicylic acid (SAL), was a logical extension of this work. Cocrystallisation of BAC with SAL in 1:1 ratio from ethanol resulted in a crystal that contains a zwitterionic BAC, a neutral SAL and a water molecule in 1:1:1 ratio (BAC·SAL·w) in the ASU (P21/c, no. 14), and can be classified as a cocrystal solvate (hydrate) (Fig. 2). The backbone of the BAC molecule is disordered in 90:10 ratio. A supramolecular unit, defined as a tetramer, can be identified based on the hydrogen bonding between two BAC molecules of opposite chirality, two SAL and two water molecules and may be described with the R24(8) graph set (Fig. 2a, yellow). The tetramers interact via hydrogen bonds that are formed between neighbouring BAC units (N1–H1B⋯O2, 2.774(7) Å, 175° and C7–H7A⋯O2, 2.898(3) Å, 153°) and the SAL and adjacent BAC molecules (O18–H18⋯O21, 2.541(5) Å, 175° and O21–H21A⋯O1, 2.695(8) Å, 156°). The crystal has a layered structure, built from alternating hydrophilic and aromatic regions (Fig. 2b, blue and green, respectively). There are Cl⋯π interactions in the aromatic layer that are formed between the BAC molecules and the adjacent aromatic rings of the SAL molecules (Cl1⋯C14, 3.44 Å, 163°, Fig. 2c).
:1 ratio and resulted in a crystal with two molecules of BAC in zwitterionic form and three molecules of neutral BA in the ASU (P21/c, no. 14). The crystallisation of BAC with salicylic acid (SAL), was a logical extension of this work. Cocrystallisation of BAC with SAL in 1:1 ratio from ethanol resulted in a crystal that contains a zwitterionic BAC, a neutral SAL and a water molecule in 1:1:1 ratio (BAC·SAL·w) in the ASU (P21/c, no. 14), and can be classified as a cocrystal solvate (hydrate) (Fig. 2). The backbone of the BAC molecule is disordered in 90:10 ratio. A supramolecular unit, defined as a tetramer, can be identified based on the hydrogen bonding between two BAC molecules of opposite chirality, two SAL and two water molecules and may be described with the R24(8) graph set (Fig. 2a, yellow). The tetramers interact via hydrogen bonds that are formed between neighbouring BAC units (N1–H1B⋯O2, 2.774(7) Å, 175° and C7–H7A⋯O2, 2.898(3) Å, 153°) and the SAL and adjacent BAC molecules (O18–H18⋯O21, 2.541(5) Å, 175° and O21–H21A⋯O1, 2.695(8) Å, 156°). The crystal has a layered structure, built from alternating hydrophilic and aromatic regions (Fig. 2b, blue and green, respectively). There are Cl⋯π interactions in the aromatic layer that are formed between the BAC molecules and the adjacent aromatic rings of the SAL molecules (Cl1⋯C14, 3.44 Å, 163°, Fig. 2c).
 | ||
| Fig. 2 The tetramer formed in BAC·SAL·w (a) from two BAC, two SAL and two water molecules creating a hydrogen bonded ring (yellow), the alternating hydrophilic and aromatic layers (blue and green, respectively) (b) and the observed Cl⋯π interaction in the aromatic layer (c). | ||
(BAC+)(POA−), (BAC+)(2,4ClPOA−) and (BAC+)2(2,4ClPOA−)·w
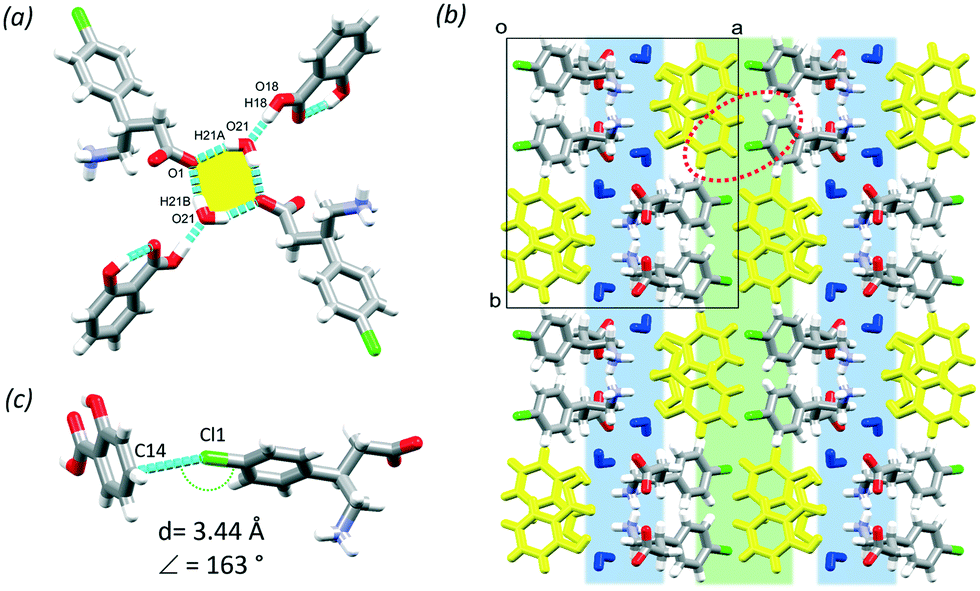
BAC was cocrystallised in 1:1 mole ratio with POA from methanol and resulted in a crystal with one molecule of BAC and one POA in the ASU (P, no. 2). The POA protonated the BAC and interacts via the charge assisted O1–H1⋯O4 (2.603(2) Å, 159°) hydrogen bond. The crystal (BAC+)(POA−) is classified as a true salt. A supramolecular unit forms from two BAC+ and two POA− and is held together by the O1–H1⋯O4 (2.603(2) Å, 159°), N1–H1A⋯O4 (2.742(7) Å, 151°), C10–H10A⋯O2 (3.462(7) Å, 168°) hydrogen bonds and their symmetry generated counterparts (Fig. 3a, yellow). The protonated amino group of the BAC hydrogen bonds to three POA ions that are parallel and eventually causes the crystal packing to have hydrophilic and aromatic layers (Fig. 3b, blue and green, respectively); the former formed by the charge assisted hydrogen bonds between the carboxylic acid, carboxylate and amino functional groups, and the latter is created by the alternating aromatic rings of BAC and POA. Cl⋯Cl close contacts were identified in the aromatic layer that are formed between two BAC molecules (Cl1⋯Cl1, 3.57 Å, 155°, Fig. 3c).
 | ||
| Fig. 3 The supramolecular unit of (BAC+)(POA−) (a), the hydrophilic (blue) and aromatic layers (green) (b) and the Cl⋯Cl interactions in the aromatic layer (c). | ||
BAC was cocrystallised in 1:1 mole ratio with 2,4ClPOA using 1:1 isopropanol:water solvent. The MCC has one molecule of BAC and one molecule of 2,4ClPOA in the ASU (P21/c, no. 14). Similar to (BAC+)(POA−), the 2,4ClPOA protonates the BAC and interacts via the charge assisted O1–H1⋯O5 (2.476(6) Å, 171°) hydrogen bond. The crystal (BAC+)(2,4ClPOA−) is classified as a true salt. A similar supramolecular unit that was discussed in (BAC+)(POA−) can be described: two BAC+ and two 2,4ClPOA− are held together by the O1–H1⋯O5 (2.476(6) Å, 171°), N1–H1A⋯O4 (2.847(2) Å, 154°), N1–H1B⋯O4 (2.830(2) Å, 151°), N1–H1C⋯O2 (2.735(8) Å, 140°) hydrogen bonds (Fig. 4a, yellow). These supramolecular units interact with adjacent ones via C6–H6⋯O5 (3.535(1) Å, 161°), C8–H8B⋯O5 (3.470(3) Å, 152°) and C10–H10A⋯O5 (3.267(4) Å, 153°) interactions. The crystal packing is also very similar to (BAC+)(POA−) and resembles its layered structure with alternating hydrophilic and aromatic layers (Fig. 4b, blue and green, respectively). It is important to note that the crystallisation was successful from a crystal engineering perspective too. The BAC interacted with the carboxylic moiety as was observed in the POA and previous structures of BAC MCCs with aromatic carboxylic acids.17 Thus the Cl substituent in the para-position of the aromatic ring of 2,4ClPOA was directed towards the aromatic layer, subsequently increasing the number of Cl substituents in that region of the crystal. Interestingly, there are only Cl⋯π interactions formed between BAC and 2,4ClPOA molecules in the aromatic layer (Cl1⋯C13, 3.38 Å, 139°, Fig. 4c).
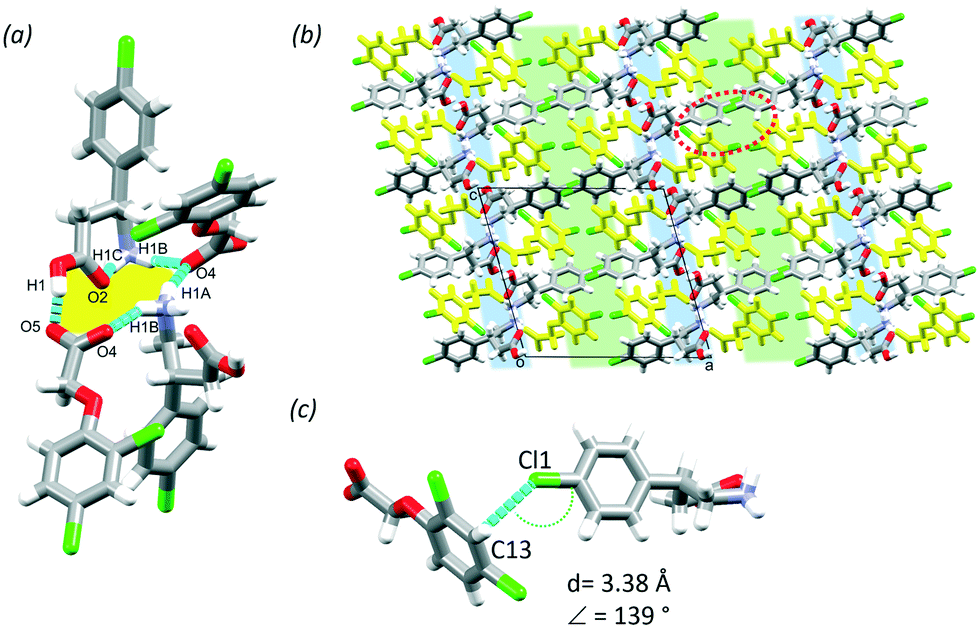 | ||
| Fig. 4 The supramolecular unit of (BAC+)(2,4ClPOA−) (a), hydrophilic (blue) and aromatic layers (green) in the crystal (b) and Cl⋯π interactions in the aromatic layer (c). | ||
BAC was also cocrystallised in 1:2 mole ratio with 2,4ClPOA in 1:1 isopropanol/water and yielded crystals with one molecule of BAC, two molecules of 2,4ClPOA and one water in the ASU (P, no. 2). The BAC is protonated by one of the 2,4ClPOA, the other 2,4ClPOA is neutral (Fig. 5a); thus the crystal may be depicted as (BAC+)(2,4ClPOA−)·2,4ClPOA·w, a cocrystal salt solvate (hydrate). For simplicity, it will be referred to as (BAC+)2(2,4ClPOA−)·w. The BAC+ forms a hydrogen bond with the neutral 2,4ClPOA (O1–H1⋯O8, 2.648(1) Å, 173°) and interacts with the deprotonated (2,4ClPOA−) via a bridging water (O9–H9A⋯O2, 2.936(9) Å, 170° and O9–H9B⋯O4, 2.960(7) Å, 147°). A larger supramolecular unit can be identified and its hydrogen bonding can be described with the R88(28) graph set (Fig. 5b, yellow). These units interact with neighbouring ones by anchoring them through hydrogen bonds via their NH3+ groups (N1–H1A⋯O5 (2.868(4) Å, 145°), N1–H1B⋯O4 (2.763(6) Å, 158°) and N1–H1C⋯O2 (2.902(3) Å, 159°). The crystal packing is very similar to the previously discussed layered structures, having alternating hydrophilic and aromatic layers (Fig. 5c, blue and green, respectively). It is interesting to note that although there are two symmetrically independent 2,4ClPOA moieties in the crystal and they differ in their protonation state, their structural role is very similar. The two 2,4ClPOA moieties form centrosymmetric tetramers via C25–H25A⋯O7 (3.499(3) Å, 151°) and O7–H7A⋯O5 (2.447(2) Å, 177°) hydrogen bonds and their symmetry-related counterparts, in that their aromatic rings are in an offset face-to-face arrangement (ring distance is 3.59 Å and their angle is 2.28°, Fig. 5d and e). It is important to note that (1) despite the inclusion of the water and the different stoichiometry, the nature of the crystal stayed layered, and (2) despite the number of chlorine functional groups of the molecules, there are no interactions formed by these moieties that fulfil the geometrical requirements to be noted as any form of halogen bond.
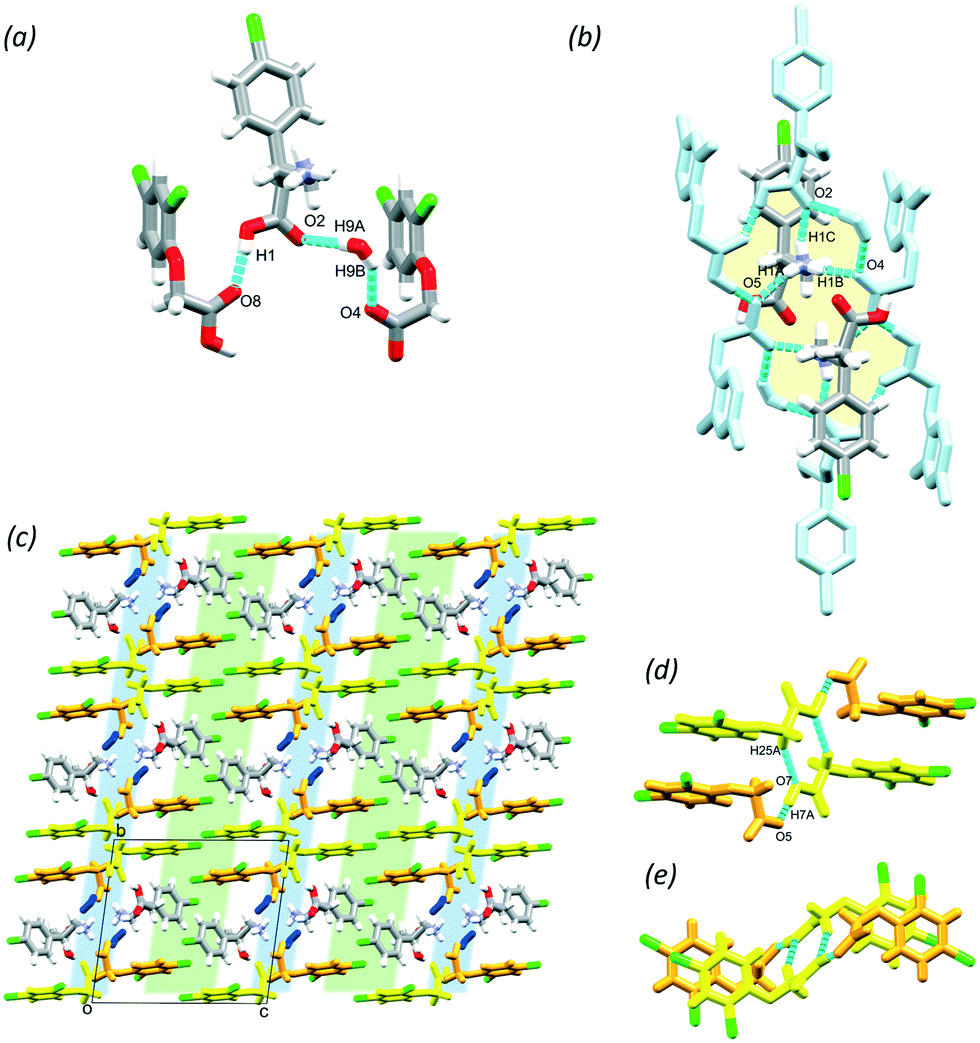 | ||
| Fig. 5 ASU (a), supramolecular unit (b) and packing interactions that result in the layered structure in (BAC+)2(2,4ClPOA−)·w (c) with the interactions between the acids (side view-d, top view-e). | ||
2BAC(4PIC)·w and 2BAC(3,4LUT)·w
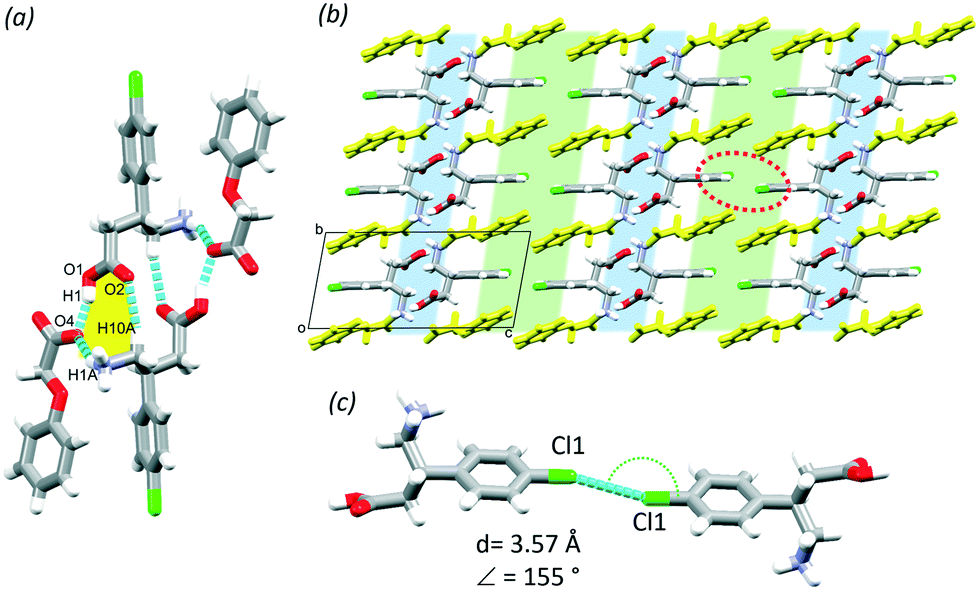
In this section, the crystal structures formed from BAC and two basic N-heterocyclic coformers, 3,4-lutidine (3,4LUT) and 4-picoline (4PIC) are discussed.The ASU of the crystal obtained with 4PIC have two BAC, one 4PIC and one water molecule (2BAC(4PIC)·w, P, no. 2). The ASU of 2BAC(3,4LUT)·w has the same stoichiometry and the same space group symmetry. In both MCCs, both BAC molecules are in zwitterionic form and no protonation of the base was noted. The crystals can be classified as cocrystal solvates (hydrates). There is a notable difference between the conformations of the two BAC molecules in the ASU of 2BAC(4PIC)·w. The amino acid backbone is linear in Mol A but bent in Mol B (Fig. 6a). The two BAC molecules hydrogen bond in a head-to-tail manner (N1A–H1B⋯O1B, 2.728(3) Å, 178°) and the 4PIC molecule cleave between them through N1A–H1C⋯N11 (2.906(1) Å, 162°) and C16–H16⋯O2B (3.427(7) Å, 157°) hydrogen bonds. The water molecule is stabilised by BAC (Mol B) accepting C8B–H8D⋯O18 (3.443(4) Å, 160°) and N1B–H1D⋯O18 (2.824(2) Å, 155°) hydrogen bonds. The ASUs of 2BAC(4PIC)·w and 2BAC(3,4LUT)·w are isostructural (RMS = 0.0576 Å) (Fig. 6b) but minor differences are noted in the position of the base. The main hydrogen bonds form two supramolecular synthons that can be described by R22(12) and R46(24) graph sets (Fig. 6c, blue and green, respectively). The water molecules are bridging between the parallel BAC chains.
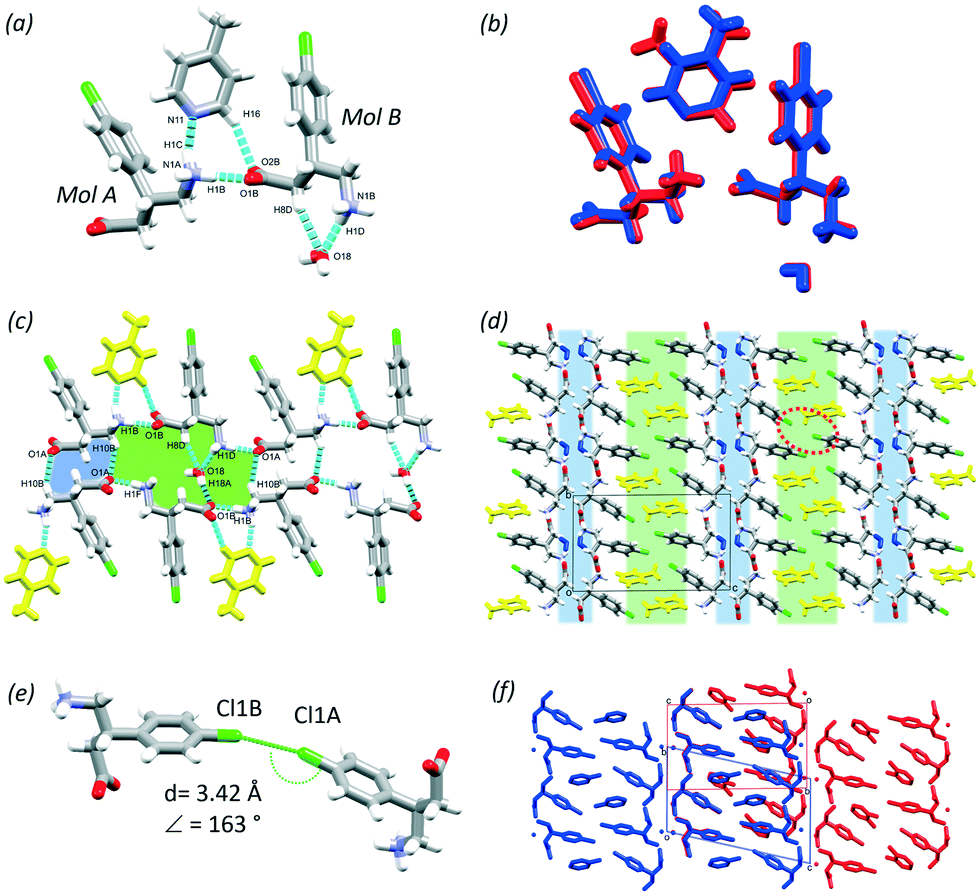 | ||
| Fig. 6 ASU of 2BAC(4PIC)·w (a) and when superimposed with the ASU of 2BAC(3,4LUT)·w (b); hydrogen bonds in 2BAC(4PIC)·w (c); hydrophilic (blue) and aromatic layers (green) in 2BAC(4PIC)·w (water molecules are blue, 4PICs are yellow) (d), the observed Cl⋯Cl interaction in the aromatic layers (e) and the superimposed structures (f). | ||
The crystal packing is layered, similar to the previously discussed structures, having alternating hydrophilic and aromatic layers (Fig. 6d, blue and green, respectively). There are Cl⋯Cl interactions in the aromatic layer that are formed between two symmetrically independent BAC molecules (Cl1A⋯Cl1B, 3.42 Å, 163°, Fig. 6e). This interaction was observed in 2BAC(3,4LUT)·w too with slightly different descriptors (Cl1A⋯Cl1B, 3.46 Å, 141°).
It is interesting to note that although the ASUs of 2BAC(4PIC)·w and 2BAC(3,4LUT)·w are very similar, the subtle difference between the position of the bases leads to a more pronounced difference between the crystal structures. The superimposition of the two structures reveals that there is a subtle shift between the adjacent aromatic layers in 2BAC(3,4LUT)·w compared to 2BAC(4PIC)·w (Fig. 6f).
Bulk property analysis (PXRD, TGA, DSC, FTIR)
PXRD analysis was used to confirm if the single crystal selected for analysis (SC) is a true representation of the bulk crystals and whether the cocrystallisation was successful when repeated with LAG (for experimental details see the ESI†). This was true for only (BAC+)(OXA−)·w and (BAC+)(POA−), where the XRD patterns of the bulk material agree well with the generated pattern of the single crystal structure (SC) and also show good agreement with the XRD pattern of the LAG products (see all XRD data in the ESI†). The XRD pattern of the bulk material agrees with the generated pattern from the single crystal for compound (BAC+)(2,4ClPOA−). The generated XRD patterns for 2BAC(4PIC)·w and 2BAC(3,4LUT)·w resemble the bulk, but minor differences can be noted. These can be attributed to the partial desolvation of the crystals at the time of the analysis. The LAG products do not show similarity with the single crystal structures; thus, LAG is not suitable for preparing these compounds. The crystal selected for data collection and the bulk is significantly different for BAC·SAL·w and (BAC+)2(2,4ClPOA−)·w. For BAC·SAL·w, the LAG product seems to be the same as the bulk material and agrees well with the pattern of SAL, one of the starting materials. The single crystal structure of (BAC+)2(2,4ClPOA−)·w does not represent the bulk of the crystals. The pattern of the bulk is similar to the 1:1 salt, thus it is very likely that the crystallisation of the 1:2 cocrystal salt hydrate resulted in the formation of a mixture of crystals with different stoichiometry.
The results of the thermogravimetric analysis (TGA) and differential scanning calorimetry (DSC) for the bulk materials obtained via the solvent evaporation crystallisation (SEC) and by liquid assisted grinding (LAG) are summarised in Tables S14 and S15,† respectively, and the thermograms are presented in the ESI.† The (BAC+)(OXA−)·w has a three-step decomposition. The first step (5%) is due to dehydration, the second step is related to the loss of one oxalic acid (28%), and the last step is the final decomposition of the crystal. The material obtained via LAG shows similar behaviour. Congruent melting occurs in BAC·SAL·w, (BAC+)(POA−), (BAC+)(2,4ClPOA−) and (BAC+)2(2,4ClPOA−)·w where the decomposition of the MCC happens in a single step. The Tonset in the presented (BAC+)(2,4ClPOA−) and (BAC+)2(2,4ClPOA−)·w are very similar for the material obtained from the LAG and supports the conclusion drawn from the XRD results. The melting points for (BAC+)(OXA−)·w and BAC·SAL·w obtained via LAG are lower than the melting point of the bulk crystals from SEC, and this can be credited to impurities. LAG experiments often do not mean 100% conversion of the starting materials to the final products and thus contamination by the starting materials lowers the melting point of the product. The crystals of 2BAC(4PIC)·w and 2BAC(3,4LUT)·w obtained via SEC show very similar thermal decomposition where the first mass loss (20% and 22%, respectively) occurs due to the simultaneous release of one mole of water and one mole of 4PIC or 3,4LUT, and this is followed by the total decomposition. Preparation of these MCCs with LAG was unsuccessful; the TGA shows no sign of included solvent, and this was supported by the PXRD and FTIR results, where the patterns of the LAG agreed with the pure BAC. The melting points of the (BAC+)(OXA−)·w, (BAC+)(POA−), (BAC+)(2,4ClPOA−) and (BAC+)2(2,4ClPOA−)·w lie between the melting point of BAC and the respective solid coformer, however, the melting point of for BAC·SAL·w is lower than its starting materials. Generally, the Tonset for MCCs obtained by LAG is lower than the crystals obtained by SEC and this is also an indication of the presence of excess starting material.
The crystallisation experiments were also monitored by FTIR spectrometry. BAC is zwitterionic under the applied crystallisation conditions (see Table S16† for pKa values for BAC and CFs), therefore the analysis focuses on the stretching frequencies of the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O, C–O, O–H and N–H bonds (the summary of the frequencies are in Table S17, spectra are in the ESI†). From the structures of (BAC+)(OXA−)·w, (BAC+)(POA−), (BAC+)(2,4ClPOA−) and (BAC+)2(2,4ClPOA−)·w, it was observed that the BAC moiety has been protonated on the COO− functional group. The CO and O–H bands are observed in the wavelength regions of 1710–1680 cm−1 and 3300–2400 cm−1, respectively. Furthermore, BAC·SAL·w, 2BAC(4PIC)·w and 2BAC(3,4LUT)·w are in the zwitterionic form; bands in the 3500–3200 cm−1 and 1550–1450 cm−1 regions suggest the presence of the NH3+ and COO− functional groups. The spectra for (BAC+)(2,4ClPOA−) and (BAC+)2(2,4ClPOA−)·w for LAG do not show the expected broad O–H band.
O, C–O, O–H and N–H bonds (the summary of the frequencies are in Table S17, spectra are in the ESI†). From the structures of (BAC+)(OXA−)·w, (BAC+)(POA−), (BAC+)(2,4ClPOA−) and (BAC+)2(2,4ClPOA−)·w, it was observed that the BAC moiety has been protonated on the COO− functional group. The CO and O–H bands are observed in the wavelength regions of 1710–1680 cm−1 and 3300–2400 cm−1, respectively. Furthermore, BAC·SAL·w, 2BAC(4PIC)·w and 2BAC(3,4LUT)·w are in the zwitterionic form; bands in the 3500–3200 cm−1 and 1550–1450 cm−1 regions suggest the presence of the NH3+ and COO− functional groups. The spectra for (BAC+)(2,4ClPOA−) and (BAC+)2(2,4ClPOA−)·w for LAG do not show the expected broad O–H band.
Conformation analysis of the BAC moieties in the MCCs
The conformational variety of BAC was noted during the discussion of the MCC structures and a comparative analysis was carried out. BAC has four rotatable bonds and the following four torsion angles can describe its conformational flexibility: τ1 indicates the rotation of the –COOH/–COO− moiety, τ2 describes the rotation of the –CH2–COOH/–CH2–COO− moiety, τ3 used to measure the movement of the –CH2–NH2/–CH2–NH3+ group, and the rotation of the aromatic ring relative to the amino acid functionality is defined by τ4 (Fig. 7). CSD-Discovery software was used20 to identify the conformers that are the most probable to form in the solid state. The knowledge-based conformer generation resulted in 23 lowest energy conformers (Fig. 7), and their superimposition revealed that (1) the amine/amino moieties prefer two positions, (2) the carboxylic/carboxylate moieties cluster around three positions and (3) the aromatic ring can take up almost any position. By not taking into account the aromatic ring rotation and focusing only on the amino acid backbone, analysis of the 23 conformers revealed that there are six main conformers, and based on the τi (Cacid–Cα⋯Cγ–N) improper torsion angle values these six conformers can be classified further using the syn/anti and clinal/periplanar descriptors. The complexity of the BAC moiety requires to use all five torsion angles and create subsets of the typical conformers to distinguish between them. The torsion angle values of the BAC moieties in the current MCCs are summarised in Table 1. The seven described MCCs contain nine molecules of BAC in their ASU showing conformational variety: two of them has the sp-I conformation when the backbone is linear, one moiety has the sp-II conformation with the U-shape backbone, and six of them can be classified as ac-I or ac-II, where the backbone is bent. This conformational variety reflects the observed conformations in the already known 13 BAC crystals (containing 16 BAC moieties) found in the CSD (Table S18†): the majority have ac conformation (ac-I: 7, ac-II: 4) and five moieties are in the sp-I conformation. It is interesting to note that the crystal (BAC+)(2,4ClPOA−) represents a new low energy conformer (sp-II) of BAC found in the solid state.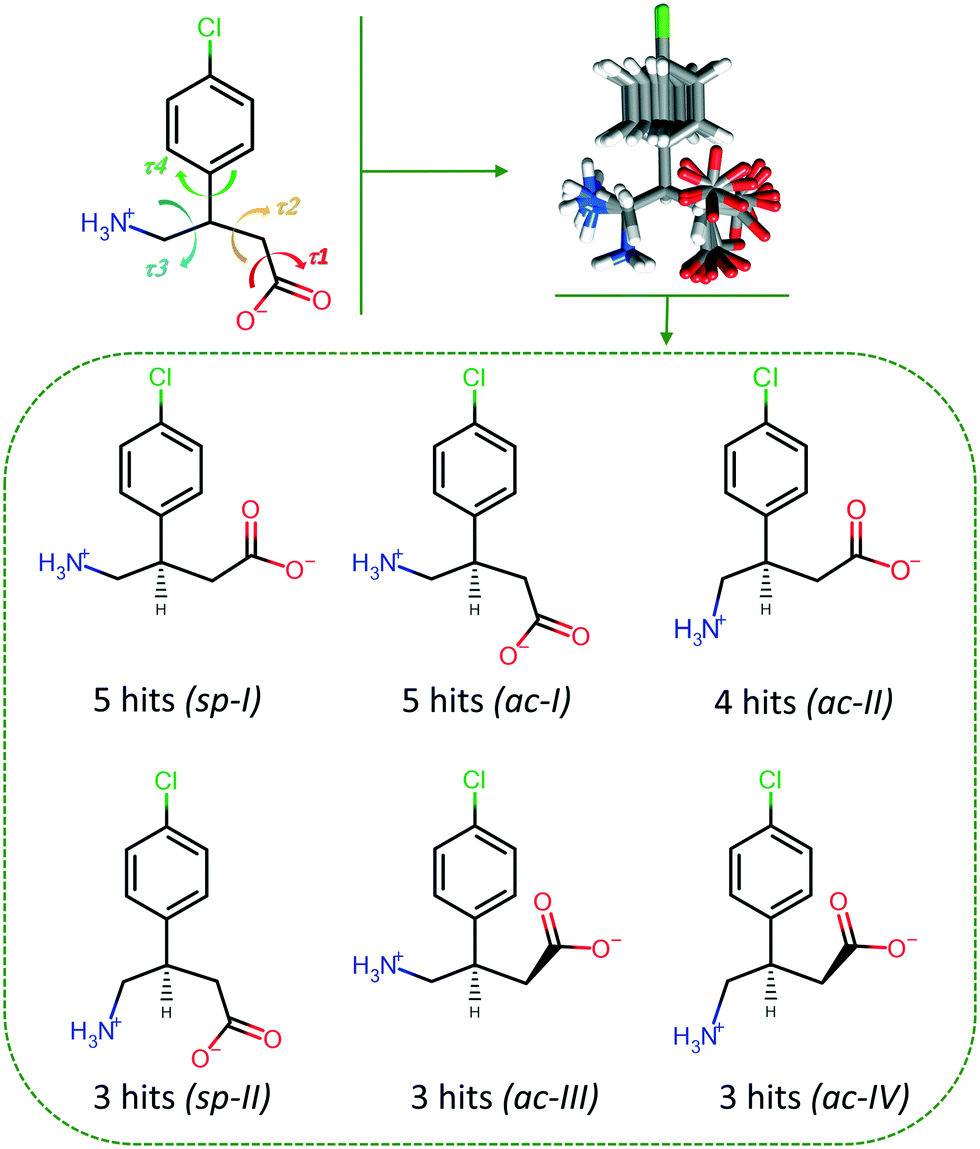 | ||
| Fig. 7 Graphical explanation of the torsion angles used to describe the four rotatable bonds of BAC and the superimposed molecules (23) of the generated lowest energy conformers. Below are the main conformers and their occurrence in the 23 generated lowest energy conformers. | ||
| MCC | τ 1 (O*–C9–C8–C7) | τ 2 (C9–C8–C7–C1) | τ 3 (C1–C7–C10–N1) | τ 4 (C2–C1–C7–C10) | τ i (Cacid–Cα⋯Cγ–N) | Conformation |
|---|---|---|---|---|---|---|
| (BAC+)(OXA−)·w | 12.58 | −157.43 | −58.67 | −118.15 | −108.06 | ac-I |
| BAC·SAL·w | 53.16 | −173.75 | 63.62 | −106.80 | −111.79 | ac-I |
| (BAC+)(POA−) | 54.18 | −172.91 | 62.18 | −144.03 | −119.22 | ac-I |
| (BAC+)(2,4ClPOA−) | −22.29 | −158.36 | 171.14 | −123.72 | 9.46 | sp-II |
| (BAC+)2(2,4ClPOA−)·w | 62.49 | −161.95 | 64.47 | −120.48 | −102.83 | ac-I |
| 2BAC(4PIC)·w (Mol A) | −55.54 | −49.75 | 54.51 | −114.02 | 7.28 | sp-I |
| (Mol B) | −55.72 | −60.56 | 175.03 | −114.18 | 120.50 | ac-II |
| 2BAC(3,4LUT)·w (Mol A) | −55.52 | −49.80 | 57.50 | −110.98 | 12.28 | sp-I |
| (Mol B) | −52.74 | −60.68 | 176.22 | 116.01 | 120.84 | ac-II |
Comparative crystal packing analysis of the MCCs
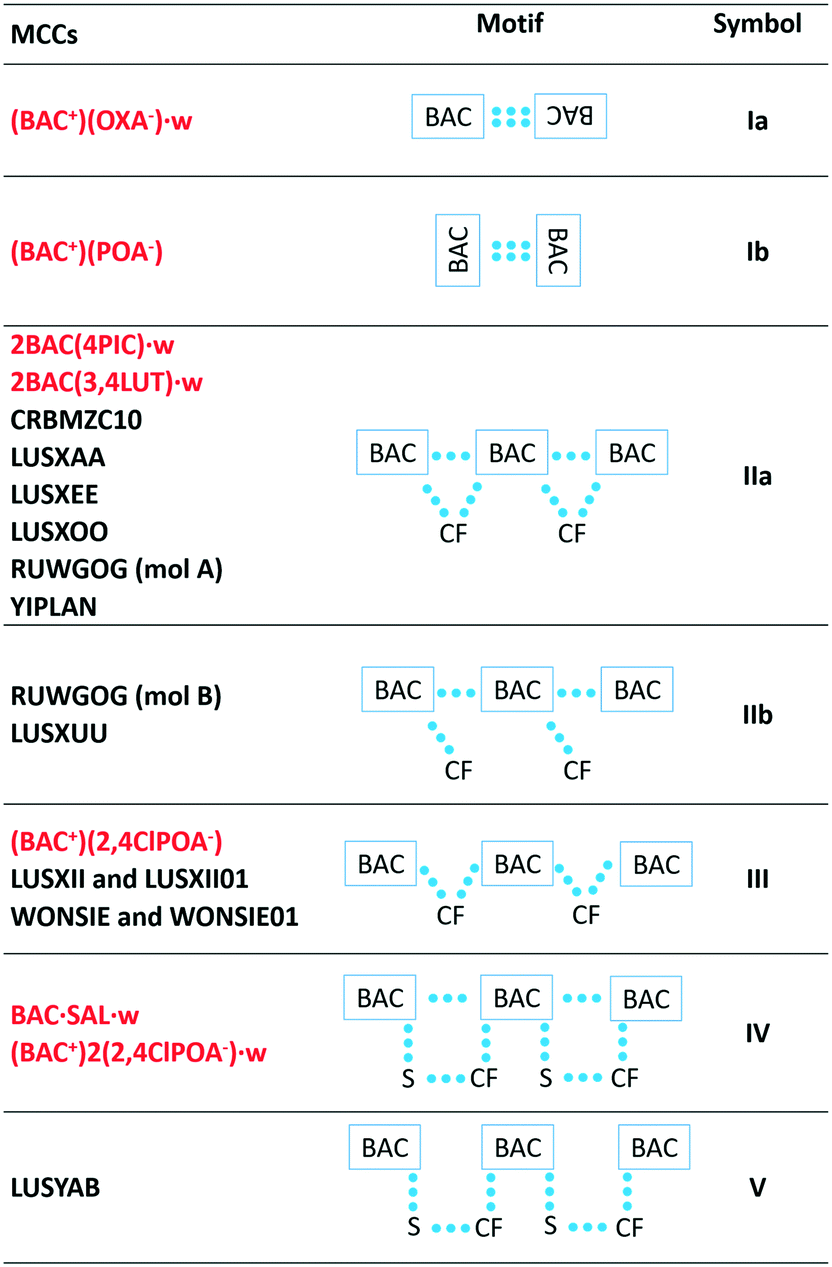
The packing of all the MCCs, except for the oxalate salt, resembles the packing observed in pure hydrophobic α-amino acids,21 namely the hydrogen bonded layers formed by the polar functional groups of the BAC and the coformers are alternating with the hydrophobic aromatic layers. In contrast, the interactions in the polar layers show great variety (Table 2). The BAC moieties form centrosymmetric dimers in two crystals, (BAC+)2(OXA−)·w and (BAC+)(POA−) in a different manner. In the (BAC+)(OXA−)·w they interact via forming the well-known carboxylic acid dimers (depicted as Ia), and in (BAC+)(POA−) they interact via –CH⋯carbonyl interactions (Ib). In the most often occurring packing the BAC moieties hydrogen bond to one another in a head-to-tail manner and the coformer (CF) cleaves itself between the two neighbouring BAC molecules (IIa). This motif is noted in the crystals of 2BAC(4PIC)·w and 2BAC(3,4LUT)·w, and six other already known crystals. In (BAC+)(2,4ClPOA−), and other two published crystals, the lack of interactions was noted between the adjacent BAC moieties (III). In BAC·SAL·w and (BAC+)2(2,4ClPOA−)·w crystals, the head-to-tail arrangement of the neighbouring BAC molecules is based on hydrogen bonds, and the coformer and the solvent bridging between the BAC molecules (IV). Motif V is similar to IV but lacks the hydrogen bonds between the BACs. In summary, the most typical packing motif of BAC MCCs is the formation of the hydrogen bonded BAC chains in a head-to-tail manner. The motif can be disrupted by the CF occasionally, but this cannot be related to the size or the polarity of the CF.
|
Conclusions
Baclofen (BAC, (R/S)-4-amino-3-(4-chlorophenyl)butanoic acid) was the centre of interest of this work. BAC has low aqueous solubility and thus its efficacy is compromised. From previous research, it is known that BAC can form MCCs with CFs of different acid strength and molecular size. The project aimed to extend the landscape of the MCCs of BAC (1) by cocrystallising the API with halogenated aromatic carboxylic acids and also (2) evaluate whether BAC can act as an acid when crystallising with basic CFs. Cocrystallisations with 36 acidic and 17 basic CFs were carried out by solvent evaporation crystallisation and yielded only seven MCCs that contained BAC and the respective CFs. The single crystal results were supported by the analysis of the bulk material with TGA, DSC, PXRD and FTIR spectrometry. Liquid assisted grinding was also used to form the MCCs, and the products were analysed similarly to the bulk compounds. The overall conclusion of the bulk property analysis is that the chemical composition of the bulk material agrees well with the single crystal for compounds (BAC+)(OXA−)·w, (BAC+)(POA−), (BAC+)(2,4ClPOA−), but discrepancies between the bulk and the single crystal structure were noted for BAC·SAL·w and (BAC+)2(2,4ClPOA−)·w, 2BAC(4PIC)·w and 2BAC(3,4LUT)·w; and only for the preparation of (BAC+)(OXA−)·w and (BAC+)(POA−) compounds was the LAG found to be a suitable method.X-ray diffraction analysis of the single crystal structures revealed that (1) BAC exists in zwitterionic or cationic form in these crystals, and (2) the MCCs represent a large variety of different crystal classes, such as salts, solvates, salt solvates, or cocrystal salt solvates with (3) differing stoichiometry. It was also noted that BAC appeared in many different conformations and the most often occurring conformations are the anti-clinal (ac-I and II) and the syn-periplanar (sp-I). The first appearance of one of the low energy conformers (sp-II) was observed in (BAC+)(2,4ClPOA−).
Comparative crystal packing analysis revealed that the hydrogen bonded BAC chains in a head-to-tail manner is the most typical packing motif of BAC MCCs and the packing arrangement of all the MCCs—with the exception of the oxalate salt—resembles the packing observed in pure hydrophobic α-amino acids, where the hydrogen bonded layers formed by the polar functional groups of the BAC and the CFs alternating with the hydrophobic aromatic layers.
In conclusion, it was proved that BAC could form MCCs with both acidic and basic CFs, and the robustness of the hydrogen bonding between the adjacent BAC molecules aids the formation of the alternating hydrophilic and hydrophobic layers in the crystal structures and opens further opportunities for fine-tuning the interactions in the latter.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The financial assistance from the National Research Foundation (NRF Pretoria, South Africa, Unique Grant Numbers 105969, 115091 and 105242) towards this research is hereby acknowledged. Opinions expressed and conclusions arrived at are those of the authors and are not necessarily to be attributed to the NRF. RJM acknowledges postgraduate funding received from CPUT.Notes and references
- (a) R. Pepinsky, Phys. Rev., 1955, 100, 971 CAS; (b) G. M. J. Schmidt, Pure Appl. Chem., 1971, 27, 647 CAS; (c) G. R. Desiraju, Crystal Engineering: The design of Organic Solids, Elsevier, Amsterdam, 1989 Search PubMed; (d) E. R. T. Tiekink, J. J. Vittal and M. Zaworotko, Organic crystal engineering: frontiers in crystal engineering, Wiley, Hoboken, N.J., 2010 CrossRef.
- (a) Multi-Component Crystals, Synthesis, Concepts, Function, ed. E. Tiekink, J. Zukerman-Schpector and S. Aitipamula, et al., De Gruyter, Berlin, Boston, 2017 Search PubMed; (b) D. Braga and F. Grepioni, Making crystals by design: methods, techniques and applications, Wiley-VCH, Weinheim, 2007 Search PubMed; (c) C. B. Aakeröy and A. S. Sinha, Co-crystals: Preparation, Characterization and Applications, Royal Society of Chemistry, Cambridge, 2018 RSC.
- (a) Pharmaceutical Salts and Co-crystals, ed. J. Wouters and L. Quéré, RSC Drug Discovery Series No. 16, Royal Society of Chemistry, 2012 Search PubMed; (b) G. Bolla and A. Nangia, Chem. Commun., 2016, 52, 8342–8360 RSC; (c) M. A. E. Yousef and V. R. Vangala, Cryst. Growth Des., 2019, 19, 7420–7438 CrossRef CAS; (d) M. Karimi-Jafari, L. Padrela, G. M. Walker and D. M. Croker, Cryst. Growth Des., 2018, 18(10), 6370–6387 CrossRef CAS; (e) O. N. Kavanagh, D. M. Croker, G. M. Walker and M. J. Zaworotko, Drug Discovery Today, 2019, 24(3), 796–804 CrossRef CAS.
- (a) G. R. Desiraju, Angew. Chem., Int. Ed. Engl., 1995, 21, 2311–2327 CrossRef; (b) A. Nangia and G. R. Desiraju, Supramolecular Synthons and Pattern Recognition, in Design of Organic Solids. Topics in Current Chemistry, ed. E. Weber, et al., Springer, Berlin, Heidelberg, 1998, vol. 198 Search PubMed; (c) A. K. Nangia and G. R. Desiraju, Angew. Chem., Int. Ed., 2019, 58, 4100–4107 CrossRef CAS; (d) P. Bombicz, T. Gruber, C. Fischer, E. Weber and A. Kálmán, CrystEngComm, 2014, 16, 3646–3654 RSC.
- (a) S. Long and T. Li, Cryst. Growth Des., 2009, 9(12), 4993–4997 CrossRef CAS; (b) P. Chen, Z. Zhang, S. Parkin, P. Zhou, K. Cheng, C. Li, F. Yu and S. Long, RSC Adv., 2016, 6, 81101–81109 RSC; (c) H. P. G. Thompson and G. M. Day, Chem. Sci., 2014, 5, 3173–3182 RSC; (d) S. E. Wright, M. J. Bryant and A. J. Cruz-Cabeza, CrystEngComm, 2020, 22, 7217–7228 RSC.
- (a) S. A. Ross, D. A. Lamprou and D. Douroumis, Chem. Commun., 2016, 52, 8772 RSC; (b) M. K. Corpinot and D.-K. Bučar, Cryst. Growth Des., 2019, 19(2), 1426–1453 CrossRef CAS; (c) N. Sarkar, J. Mitra, M. Vittengl, L. Berndt and C. B. Aakeröy, CrystEngComm, 2020, 22, 6776–6779 RSC; (d) L. E. Hatcher, A. J. Burgess, P. Paynea and C. C. Wilson, CrystEngComm, 2020, 22, 7475–7489 RSC.
- (a) A. L. Albright, J. Child Neurol., 1996, 11(2), 77–83 CrossRef CAS; (b) K. Kolaski and L. R. Logan, NeuroRehabilitation, 2007, 22, 383–395 Search PubMed.
- (a) G. H. Fromm, C. F. Terence and A. S. Chattha, Ann. Neurol., 1984, 15(3), 240–244 CrossRef CAS.
- A. Dario and G. Tomei, Drug Saf., 2004, 27(11), 799–818 CrossRef CAS.
- (a) S. M. Assadi, R. Radgoodarzi and S. A. Ahmadi-Abrahi, BMC Psychiatry, 2003, 3, 16 CrossRef; (b) R. L. Corwin, J. Boan, K. F. Peters and J. S. Ulbrecht, Behav. Pharmacol., 2012, 23(5–6), 616–625 CrossRef CAS; (c) L. A. Berner, M. E. Bocarsly, B. G. Hoebel and N. M. Avena, Behav. Pharmacol., 2009, 20, 631–634 CrossRef CAS; (d) S. C. Heinrichs, K. A. Leite-Morris, R. J. Carey and G. B. Kaplan, Behav. Brain Res., 2010, 207, 353–359 CrossRef CAS.
- (a) G. Addolorato, F. Caputo, E. Capristo, M. Domenicali, M. Bernardi, L. Janiri, R. Agabio, G. Colombo, G. L. Gessa and G. Gasbarrini, Alcohol Alcohol., 2002, 37(5), 504–508 CrossRef CAS; (b) R. de Beaurepaire, Front. Psychiatry, 2012, 3, 103 CAS; (c) J. Pinot, L. Rigal, B. Granger, S. Sidorkiewicz and P. Jaury, Front. Psychiatry, 2018, 9, 486 CrossRef; (d) S. Guerzoni, L. Pellesi, L. A. Pini and F. Caputo, Pharmacol. Res., 2018, 133, 65–76 CrossRef CAS.
- I. Chumakov, S. Nabirotchkin, N. Cholet, A. Milet, A. Boucard, D. Toulorge, Y. Pereira, E. Graudens, S. Traoré, J. Foucquier, M. Guedj, E. Vial, N. Callizot, R. Steinschneider, T. Maurice, V. Bertrand, C. Scart-Grès, R. Hajj and D. Cohen, Sci. Rep., 2015, 5, 7608 CrossRef CAS.
- W. Maniukiewicz, M. Oracz and L. Sieron, J. Mol. Struct., 2016, 1123, 271–275 CrossRef CAS.
- (a) M. R. Caira, R. Clauss, L. R. Nassimbeni, J. L. Scott and A. F. Wildervanck, J. Chem. Soc., Perkin Trans. 2, 1997, 763–768 RSC; (b) E. N. Córdova-Villanueva, C. Rodríguez-Ruiz, O. Sánchez-Guadarrama, J. Rivera-Islas, D. Herrera-Ruiz, H. Morales-Rojas and H. Höpfl, Cryst. Growth Des., 2018, 18, 7356–7367 CrossRef; (c) F.-X. Gendron, J. Mahieux, M. Sanselme and G. Coquerel, Cryst. Growth Des., 2019, 19, 4793–4801 CrossRef CAS.
- Marvin was used for drawing, displaying and characterizing chemical structures, substructures and reactions. Calculator Plugins were used for structure property prediction and calculation, Marvin 16.4.25.0, 2016, ChemAxon, (http://www.chemaxon.com) Search PubMed.
- Analytical Profiles of Drug Substances, ed. K. Florey, Academic Press, Orlando, FL, 1985, vol. 14, pp. 527–548 Search PubMed.
- N. B. Báthori and O. E. Y. Kilinkissa, CrystEngComm, 2015, 17(43), 8264–8272 RSC.
- (a) Bruker, SAINT-Plus (including XPREP), Version 7.12, Bruker AXS Inc, Madison, Wisconsin, USA, 2004 Search PubMed; (b) L. Krause, R. Herbst-Irmer, G. M. Sheldrick and D. Stalke, SADABS-2016/2 (Bruker AXS), J. Appl. Crystallogr., 2015, 48, 3–10 CrossRef CAS; (c) Bruker, XPREP, Version 6.14, Bruker AXS Inc, Madison, Wisconsin, USA, 2003 Search PubMed; (d) G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Adv., 2015, 71, 3–8 CrossRef; (e) L. J. Barbour, J. Appl. Crystallogr., 2020, 53, 1141 CrossRef CAS; (f) C. F. Macrae, I. J. Bruno, J. A. Chisholm, P. R. Edgington, P. McCabe, E. Pidcock, L. Rodriguez-Monge, R. Taylor, J. van de Streek and P. A. Wood, Mercury CSD 4.2.0 - New Features for the Visualization and Investigation of Crystal Structures, J. Appl. Crystallogr., 2008, 41, 466–470 CrossRef CAS.
- H. Grothe, E. Meekes, J. H. Vlieg, R. ter Horst and R. de Gelder, Cryst. Growth Des., 2016, 16, 3237–3243 CrossRef.
- J. C. Cole, O. Korb, P. McCabe, M. G. Read and R. Taylor, J. Chem. Inf. Model., 2018, 58, 615–629 CrossRef CAS.
- C. H. Görbitz, K. Vestli and R. A. Orlando, Acta Crystallogr., Sect. B: Struct. Sci., 2009, 65, 393–400 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available: Details of the crystallisation conditions, single crystal data collections and refinements, details of hydrogen bonds (Tables S1–S17), results of the bulk property analysis and the grinding experiments (PXRD, TGA, DSC and FTIR, Fig. A1–A34). The structures were deposited in the CCDC (deposition numbers 2031575–2031581). For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d0ce01522a |
| This journal is © The Royal Society of Chemistry 2021 |