
Structural modulation of the photophysical and electronic properties of pyrene-based 3D metal–organic frameworks derived from s-block metals†
Christopher N.
Coleman
,
Patrick C.
Tapping
 ,
Michael T.
Huxley
,
Tak W.
Kee
,
David M.
Huang
*,
Christian J.
Doonan
* and
Christopher J.
Sumby
*
,
Michael T.
Huxley
,
Tak W.
Kee
,
David M.
Huang
*,
Christian J.
Doonan
* and
Christopher J.
Sumby
*
Department of Chemistry and Centre for Advanced Nanomaterials, The University of Adelaide, Adelaide, South Australia 5005, Australia. E-mail: david.huang@adelaide.edu.au; christian.doonan@adelaide.edu.au; christopher.sumby@adelaide.edu.au
First published on 5th November 2020
Abstract
Materials in which charge delocalization and migration can be tuned are critical for electronic applications. Crystalline framework materials containing π-rich polycyclic aromatic moieties, such as pyrene, can provide a pathway for fast anisotropic charge transport. The extent of interchromophore interaction for structurally distinct assemblies of the π-conjugated aromatic ligand 4,4′,4′′,4′′′-(1,3,6,8-pyrenetetrayl) tetrabenzoic acid (H4TBAPy) was studied within two novel metal–organic frameworks (MOFs), Na(TBAPy)(DMF) and K(TBAPy)(DMF), via steady-state and time-resolved spectroscopic techniques. Single-crystal X-ray diffraction was used to determine the structures of K(TBAPy)(DMF) and Na(TBAPy)(DMF), which both form 3D MOFs comprising 1D rod-like SBUs surrounded by columnar stacks of TBAPy that are aligned in an eclipsed and x-shaped (staggered) geometry, respectively. Spectroscopic and computational results indicate significant chromophore interactions and potentially fast charge transport. Furthermore, distinct transient emission decay profiles are observed and are attributed to significant differences in the stacking orientation of the organic ligands in the two MOFs. Lastly, the study identifies design principles that may be exploited in the rational construction of s-block based MOFs for microelectronic and sensing applications.
Introduction
Metal–organic frameworks (MOFs) are a class of porous materials that are synthesized via a “building-block” approach from organic links and metal-based nodes.1 They have been investigated for their potential applications in catalysis,2 gas storage/separation,3 biotechnology4 and sensing.5 However, in recent years, there has been a growing interest in the synthesis of electrically active MOFs.6 One strategy that has been employed in the exploration of charge transport in both MOF, and related COF (covalent-organic framework) materials,7 is to construct networks that possess π-rich organic components.8 A design imperative of this approach is to position the organic linkers close enough together within the framework to allow for through-space charge transport.9 However, this strategy represents a significant challenge for MOFs synthesized from carboxylate-functionalized linkers as they typically react with transition metal salts to form discrete inorganic nodes that engender 3D net topologies10 in which the organic units are separated on length scales that preclude orbital overlap. An instructive example is the Zr-based MOF NU-1000, which is constructed from π-rich 4,4′,4′′,4′′′-(1,3,6,8-pyrenetetrayl) tetrabenzoic acid (TBAPy) linkers that are separated by a minimum of 10.94 Å (centroid to centroid) due to the eight-coordinate Zr6O8 secondary building units (SBUs).11 To overcome the geometrical constraints imposed by 0D nodes, 1D rod-like SBUs can be employed to facilitate close contact of the organic moieties. Although there are examples of infinite rod SBUs composed of transition metals (e.g. MIL-53 (ref. 12) and MOF-74(M)13), we sought to synthesize TBAPy-based MOFs from s-block metals which are known to form 1D SBUs. The alkali metals sodium and potassium are particularly attractive precursor elements due to their availability, low cost and ecological compatibility.14 Further, the network topologies of alkali metal MOFs are typically dominated by densely packed metal–On [n = 6–10] polyhedra, forming chains or layers, interconnected by organic linkers.15 Nevertheless, the non-directional, ionic nature of s-block carboxylate bonds renders rational design of s-block MOFs a challenging task. The net-topologies of the s-block MOFs are heavily influenced by the shape and steric bulk of the ligands, π orbital interactions, as well as solvent polarity.16 Thus, we surmised that an organic linker that has a strong preference to form π-stacked structures17 may drive the formation of a network architecture with closely spaced or “stacked” TBAPy moieties. This hypothesis is supported by recent work showing that TBAPy is capable of forming a robust hydrogen-bonded organic framework (PFC-1)18 that displays permanent porosity with a surface area of 2122 m2 g−1, and exceptional chemical stability. It is noteworthy that computational analysis on PFC-1 clearly indicates significant pyrene–pyrene π orbital interactions that are responsible for the excellent stability of this material.Previously reported MOFs based on TBAPy (NU-1000, NU-901, ROD-7)19–21 have been studied for their solid-state photophysical properties with a focus on interchromophoric interaction.22 However, in these examples the chromophore spacing is larger than optimal (<4 Å), with the closest being an 8.76 Å centroid to centroid distance in the case of ROD-7. Thus, despite the intrinsic photoactivity of TBAPy, these MOFs are insulators and provide limited capacity for exciton delocalization or charge transport. In this study we report the synthesis of two new 3D MOFs based on TBAPy organic linkers and the s-block metal ions Na+ and K+, Na(TBAPy)(DMF) and K(TBAPy)(DMF), respectively. Structure determination, by single crystal X-ray diffraction (SCXRD), reveals that in these materials the pyrene moieties are positioned <4 Å apart with distinct eclipsed and x-shaped (staggered) arrangements, allowing for significant amplification of the pyrene–pyrene electronic coupling. Thus, given the close stacking arrangement observed for Na(TBAPy)(DMF) and K(TBAPy)(DMF) we examined their fluorescence and charge-transport properties. Furthermore, Na(TBAPy)(DMF) and K(TBAPy)(DMF) offer the unique opportunity to compare how the relative alignment of the chromophores, “eclipsed” in K(TBAPy)(DMF) and a novel “x-shape” or staggered arrangement in Na(TBAPy)(DMF), influence the photophysical properties of the material. Indeed, time-resolved fluorescence spectroscopy, supported by computational analysis, suggests that energy transfer is sensitive to the orientation of the chromophores. In addition, to assess the viability of using these materials as semi-conductors, we fabricated a light-emitting diode (LED) that successfully integrates Na(TBAPy)(DMF) as a hole-transport layer.
Experimental
General details
All chemicals were obtained from commercial vendors (1,3,6,8 tetrabromopyrene, Sigma Aldrich, purity >97%; 4 ethoxycarbonylphenyl boronic acid, Boron Molecular, purity >97%) and used without further purification, unless otherwise stated. Dioxane was degassed with Ar prior to use. Tetraethyl 4,4′,4′′,4′′′-(pyrene-1,3,6,8-tetrayl) tetrabenzoate and the free carboxylic acid, H4TBAPy, were synthesized using previously reported method23 with some minor modification (see ESI† section 2). Powder X-ray diffraction data were collected on a Bruker Advance D8 diffractometer equipped with a capillary stage using Cu Kα radiation (λ = 1.5418 Å). NMR spectra were recorded on a Varian 500 MHz spectrometer operating at 23 °C and equipped with a 5 mm probe.Single crystal X-ray diffraction (SCXRD)
Diffraction data were collected for single crystals mounted on nylon loops in Paratone-N at 100 K on the MX1 beamline of the Australian Synchrotron using the BluIce software interface,24λ = 0.71073 Å. Ntot reflections were merged to Nunique (Rint quoted) after a multi-scan absorption correction (proprietary software) and used in the full matrix least squares refinements on F2, No with F > 4σ(F) being considered ‘observed’. The structure was solved using SHELXT25 and refined using SHELXL,26 interfaced through the programs X-Seed27 and Olex.28 Unless otherwise stated, anisotropic displacement parameter forms were refined for the non-hydrogen atoms; hydrogen atoms were treated with a riding model [weights: (σ2(Fo)2 + (aP)2 + (bP))−1; P = (Fo2 + 2Fc2)/3]. Neutral atom complex scattering factors were used. Additional refinement details, crystallographic data and refinement parameters are found in the ESI† (section 4, Table S2).Spectroscopy
Diffuse reflectance spectra were measured on solid samples using a Varian Cary 5000 spectrophotometer fitted with a Praying Mantis diffuse reflectance accessory. Steady-state fluorescence measurements on solid samples were conducted on a Perkin Elmer LS 55 spectrometer (slit widths: ex 2.5, em. 2.5 nm). Time-resolved fluorescence data were obtained using time-correlated single photon counting (TCSPC) on a Halcyone spectrometer sourced from Ultrafast Systems. Solid MOF samples were mounted inside 0.8 mm glass capillaries and measured in reflection mode. The 400 nm excitation light was generated by frequency doubling a portion of the output of a Ti-sapphire laser (Tsunami, Spectra Physics), with a pulse duration of ∼100 fs. The repetition rate of the laser pulses was reduced from 80 MHz to 6.7 MHz with a Pockels cell pulse-picker. Multiexponential fits of the decay kinetics were performed using a function of the form I(t) = ∑nAn![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) exp(−t/τn) convoluted with a Gaussian instrument response function of 0.65 ns. Amplitudes are normalized so that ∑n|An| = 1 (see ESI† section 3).
exp(−t/τn) convoluted with a Gaussian instrument response function of 0.65 ns. Amplitudes are normalized so that ∑n|An| = 1 (see ESI† section 3).
Quantum-chemical calculations
Density functional theory (DFT) calculations were performed with Q-Chem version 5.1.1.29 Calculations of excited electronic states used time-dependent DFT (TD-DFT), while charge-transfer calculations used constrained density functional theory (CDFT)30 and CDFT configuration interaction (CDFT-CI).31,32 All geometries were optimized at the ωB97X-D/6-31G* level,33 using DFT for ground states and TD-DFT for excited states. Single-point calculations were performed on optimized geometries at the ωB97X-D/6-31+G* level. All calculated electronic properties are at this level unless otherwise stated.Carboxylate carbon and oxygen atom positions were constrained to their positions in the experimental crystal structure (carboxylate groups from the crystal structure were terminated by hydrogens to neutralize the charge). Ground-state geometry optimizations were initiated from the experimental crystal structure, while optimizations of excited states and charge-localized states were initiated from the optimized ground-state geometry.
MOF synthesis
Crystals suitable for SCXRD were grown by slow vapor diffusion of acetone into a concentrated solution of Na(TBAPy)(DMF) in DMF/H2O (1:1). The resulting yellow rod-shaped crystals of Na(TBAPy)(acetone) were suitable for SCXRD. These were then transferred to a solution of DMF and allowed to soak for several days in order to exchange the coordinated acetone for DMF and give Na(TBAPy)(DMF). IR (νmax, cm−1): 1675 (s, CO), 1603 (m, CO), 1374 (s, C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C), 1269 (s, CO).
C), 1269 (s, CO).
C).
Preparation of LED device
An glass indium-tin oxide (ITO) glass substrate was coated with Na(TBAPy)(DMF) and a light emitting copolymer PDOF-co-MEH-PPV). Details of the device fabrication are in the ESI† (Fig. S11).Results and discussion
MOF synthesis and structures
Na(TBAPy)(DMF) and K(TBAPy)(DMF) were synthesized by reacting an aqueous suspension of H4TBAPy with sodium carbonate or potassium carbonate, respectively, under reflux for 24 hours to form the TBAPy salts (Fig. 1). Removal of water afforded pale-yellow powders that were recrystallized via slow evaporation from DMF/H2O to yield pure crystalline material (see Fig. S2 and S10†). Single crystals for an acetone-solvated form of the sodium MOF were obtained by vapor diffusion of acetone into a concentrated 1:1 DMF/H2O solution of Na(TBAPy)(DMF), followed by solvent exchange with DMF to form the DMF solvate, Na(TBAPy)(DMF) (see Fig. S4† for a structural comparison); single crystals of K(TBAPy)(DMF) were obtained by a solvothermal method.
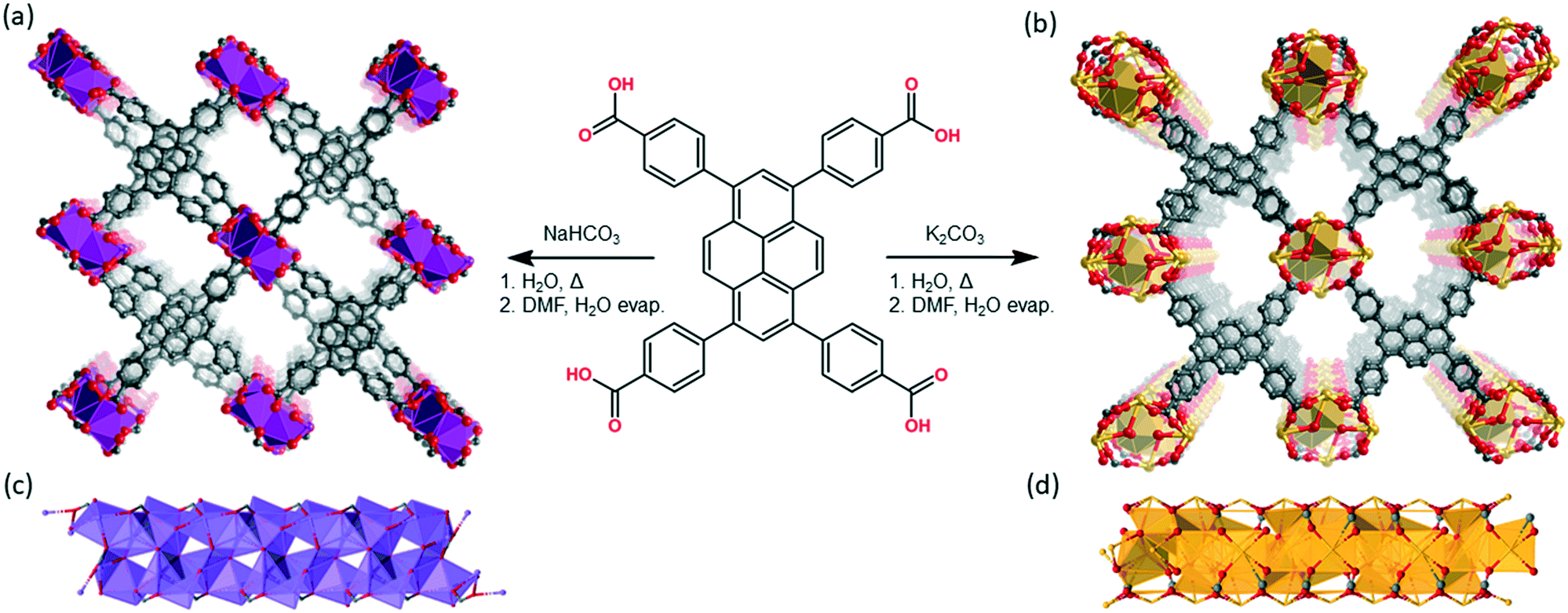 | ||
| Fig. 1 Perspective views of the structures of the TBAPy-based MOFs obtained by single crystal X-ray diffraction (hydrogen and solvent molecules omitted for clarity). The extended structures of (a) Na(TBAPy)(DMF) and (b) K(TBAPy)(DMF), and their 1D SBUs (c) and (d), respectively, are provided. The materials used for all analyses were synthesised in the two-step procedures shown. Atoms are represented as: carbon (grey), oxygen (red), sodium (purple) and potassium (orange); hydrogen atoms and the non-coordinated atoms of solvate molecules are omitted. | ||
Na(TBAPy)(DMF) crystallizes in the monoclinic space group P21/n. The ligand occupies two distinct chemical environments, with an asymmetric unit consisting of two partial ligands (one complete TBAPy molecule), four sodium atoms and two coordinated DMF molecules; this gives the formula [Na4(TBAPy)(DMF)2]. The structure of Na(TBAPy)(DMF) comprises a 1D SBU extending along the b axis (Fig. 1c), with the ligand acting as a four connecting centre. Within the linear SBU, the metal centres are present in trigonal prismatic and distorted octahedral coordination geometries. The coordination environment of the sodium SBU includes ionic bonding to oxygen atoms of carboxylate and solvent molecules, as well as bridging water molecules. The orientation by which the linkers stack is unique. As opposed to an eclipsed or “slip stacked” arrangement, the pyrene cores form an x-shape (see Fig. 1a and 4b) and the 4 benzoate groups on each ligand form two co-facial and two edge-to-face π-stacking interactions with neighbouring ligands. Furthermore, subtle structural changes can be induced for the Na-based material by exchanging the solvent and altering the crystallization methods. Powder X-ray diffraction (PXRD) (Fig. S3†) shows two distinct forms of the sodium-based MOF can be obtained, namely Na(TBAPy)(DMF) and Na(TBAPy)(acetone). Sodium is prone to coordinate a variety of solvents, and well-defined solvent molecules appear as integral features of the molecular composition, with Na(TBAPy)(acetone) featuring coordinated acetone in place of DMF but also coordinated water which subtly modifies the diameter of the SBU (see Fig. S4†) and the unit cell.
K(TBAPy)(DMF) crystallizes in the monoclinic space group P21/c. The asymmetric unit consists of two ligand molecules, nine potassium atoms, coordinated solvent (DMF and water), and a 50% occupied carbonate anion: [K9(TBAPy)2(H2O)8.25(DMF)3.25(CO3)0.5]. A salient feature of K(TBAPy)(DMF) is that, in contrast to Na(TBAPy)(DMF), the TPABY links are aligned in an eclipsed formation (Fig. 1b). This arrangement results in all carbon atoms in the stacked pyrene moieties separated by ca. 3.85 Å, with the ligands aligning in an extended H-aggregate configuration.34,35 The larger unit cell in the potassium MOF is the result of disordered solvent molecules which alter the dihedral angles on adjacent TBAPy molecules (see Fig. S9†). However, despite these crystallographic differences and in contrast to Na(TBAPy)(DMF), the linkers of K(TBAPy)(DMF) are stacked directly on top of each other with all ligands having almost identical chemical environments. This is an important consideration in the analysis of the spectral data. The SBU in K(TBAPy)(DMF) comprises 1D potassium oxide chains (Fig. 1d). The potassium ions co-ordinate with up to 7 oxygen atoms that include carboxylate groups, DMF and water molecules. PXRD data for a sample of K(TBAPy)(DMF) obtained by slow evaporation matches the structure determined by single crystal diffraction, thus confirming bulk purity (see Fig. 2).
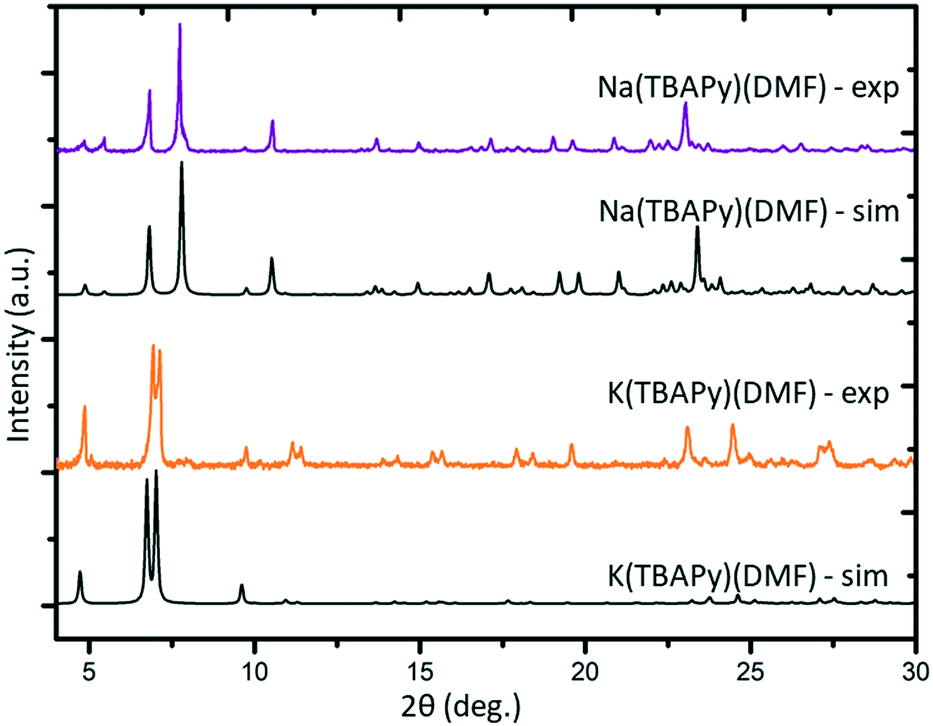 | ||
| Fig. 2 Simulated (sim) and experimental (exp) PXRD patterns of Na(TBAPy)(DMF) and K(TBAPy)(DMF). | ||
Due to the 1D rod-like SBUs that feature in Na(TBAPy)(DMF) and K(TBAPy)(DMF), close-packing of the pyrene moieties is observed. This, in theory, has the potential to facilitate charge transfer through π-orbital overlap as well as trigger noticeable interchromophoric interactions. The close packing of linkers also appears to confer some stability to the MOFs. The result is that both structures are stable and retain their crystallinity for months after being removed from solvent, provided they are kept in an environment devoid of excess moisture.
Photophysical properties
In terms of fluorescence quantum yield and excited state lifetime, pyrene ranks considerably higher than many well-known and commonly employed chromophores.36 Additionally, its spectral signature is well known to be sensitive to its environment.37 Pyrene-based MOFs have been investigated for their photophysical performance characteristics; however, Na(TBAPy)(DMF) and K(TBAPy)(DMF) provide an unprecedented opportunity to examine the effects of chromophore alignment in two close-stacked systems. Thus, to understand the nature of the chromophoric interactions we carried out steady-state and time-resolved spectroscopic studies on each MOF. We anticipated that the different stacking arrangements present in Na(TBAPy)(DMF) and K(TBAPy)(DMF) would manifest distinct emission decay profiles as a function of their topology. Previously reported spectral data for pyrene derivatives showed solvent-dependent emission peaks for the same materials.38–40 To avoid the effects of solvent polarity on the excited state energy we conducted spectroscopic studies on dried material.The high chromophore density in Na(TBAPy)(DMF) and K(TBAPy)(DMF) should result in effective photon absorption. The calculated molar density of TBAPy in each framework was 1.62 mol dm−3 for Na(TBAPy)(DMF) and 1.40 mol dm−3 for K(TBAPy)(DMF). We note that both values are considerably higher than that of the zirconium-based MOF NU-1000 (0.41 mol dm−3).39 Close positioning of photoactive ligands typically leads to nonradiative pathways that can quench fluorescence.41 However, despite the proximity of the pyrene groups in Na(TBAPy)(DMF) and K(TBAPy)(DMF), both MOFs are fluorescent in the solid state. The solid-state absorption profile features distinct peaks and troughs ranging from 225 to 450 nm (with a maximum ca. 405 nm). Despite the different relative orientations of the TBAPy ligands in the two MOFs, both show almost identical absorption profiles, as shown in Fig. 3a.
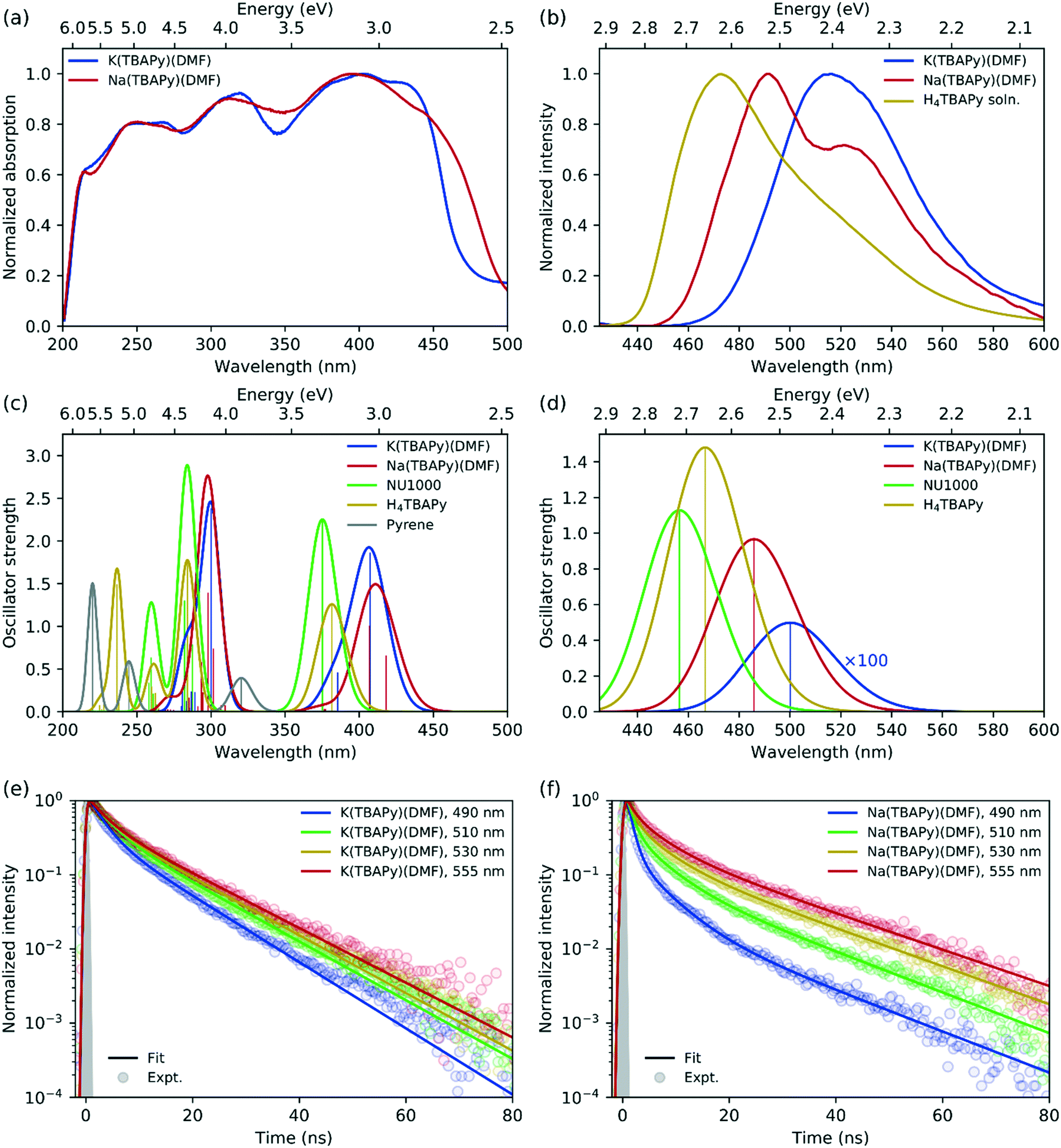 | ||
| Fig. 3 (a) Solid-state absorption spectra (measured by diffuse reflectance) of K(TBAPy)(DMF) (blue) and Na(TBAPy)(DMF) (red). (b) Emission spectra for solid-state K(TBAPy)(DMF) (blue) and K(TBAPy)(DMF) (red) using 405 nm excitation, with the H4TBAPy linker in DMF solution (yellow) for reference. Calculated (c) absorption and (d) emission spectra for the same species, with spectra for NU-1000 (green) and pyrene (grey) included for reference (computed spectra have been red-shifted by 0.4 eV and convoluted with a Gaussian of full width at half maximum (FWHM) of 0.2 eV in all cases). Time-resolved fluorescence kinetics at different detection wavelengths for (e) K(TBAPy)(DMF) and (f) Na(TBAPy)(DMF). Fits to a multiexponential decay model are shown with solid lines, with fitting parameters given in Table S1.† The experimentally determined instrument response is indicated with the grey shaded region corresponding to a Gaussian function of 0.65 ns FWHM. | ||
The solid-state emission spectra for K(TBAPy)(DMF) and Na(TBAPy)(DMF) are shown in Fig. 3b. The emission peak of K(TBAPy)(DMF) is centred at 515 nm and largely featureless but is significantly red shifted compared to that of the free H4TBAPy linker in solution. In contrast, the Na(TBAPy)(DMF) emission band exhibits distinct spectral structure, with the main peak at 490 nm and shoulder around 530 nm. The shift of the emission peaks to lower energies relative to the free H4TBAPy linker in solution is indicative of electronic coupling between the chromophore units resulting from close packing of the aromatic pyrene cores of the TBAPy linkers in the solid-state. This molecular architecture allows significant π–π interactions, and thus delocalization of the excitation. The spectral structure evident in the Na(TBAPy)(DMF) emission can be attributed to a vibronic progression. The shoulder is lower in energy than the main peak by approximately 1600 cm−1, which can be assigned to the aromatic CC bond vibration in the pyrene cores. The absence of structure in the K(TBAPy)(DMF) emission can be attributed to the distinct alignments of the pyrene units observed in the two MOFs. The packing of the linkers in K(TBAPy)(DMF) resembles that of an ideal H-aggregate,35,42 in which the eclipsed arrangement of the chromophore pairs suppresses the 0–0 vibronic transition, leaving the 0–1 peak to dominate the spectrum. The x-shaped stacking of the linkers in Na(TBAPy)(DMF) results in coupling that is intermediate between that of a J- and an H-aggregate, and thus the 0–0 and 0–1 vibronic peaks are both present. This aggregate behaviour can also account for the differences between the absorption spectra of the MOFs (Fig. 3a), which show that absorption in the Na(TBAPy)(DMF) extends further into the low-energy region, while the lowest energy 0–0 vibronic absorption peak is attenuated in K(TBAPy)(DMF).
Fluorescence decay kinetics were obtained for the two MOFs at several emission wavelengths, shown in Fig. 3e and f. The data were fit to a multiexponential decay model, with parameters given in Table S1.† For K(TBAPy)(DMF), the kinetic profiles are almost identical at all detection wavelengths. The slightly faster decay at 490 nm is at the blue edge of the absorption peak and is indicative of spectral migration to lower energies due to excited-state structural relaxation or exciton migration. Na(TBAPy)(DMF), however, displays significantly different kinetics at each detection wavelength, with noticeably faster decays at shorter wavelengths, particularly at short times (≲5–10 ns), which also indicates time-dependent spectral migration, but to a greater extent and at a faster rate than that observed in K(TBAPy)(DMF). The longer time (≳5–10 ns) decay, which occurs approximately uniformly across all wavelengths in both MOFs, can be attributed to relaxation to the ground state. The faster decays on both time scales in Na(TBAPy)(DMF) compared with K(TBAPy)(DMF) can again be attributed to the arrangement of the chromophores within the MOFs, as the H-aggregate35 nature of K(TBAPy)(DMF) is expected to result in slower emission and reduced exciton migration by resonance energy transfer.
Although pyrene dimers stacked directly on top of each other have been observed and probed previously,43 to the best of our knowledge the x-shaped or staggered configuration of pyrene present in Na(TBAPy)(DMF) (Fig. 4) has not been documented. To clarify how the configuration of ligands influences the electronic coupling and excited-state properties in the two s-block MOFs, we performed density functional theory (DFT) calculations on the two closest ligand pairs in each MOF, with the carboxylate C and O atoms fixed at their positions in the experimental SCXRD structure to mimic the coordination constraints in the MOF. For comparison, analogous calculations were performed on the closest ligand pair in NU-1000 and similar calculations were performed on the TBAPy monomer and on pyrene without geometric constraints. Absorption and emission spectra were calculated using time-dependent DFT (TD-DFT) on the optimized geometry of the electronic ground state and first excited state, respectively. Details of the calculations are given in the ESI.†
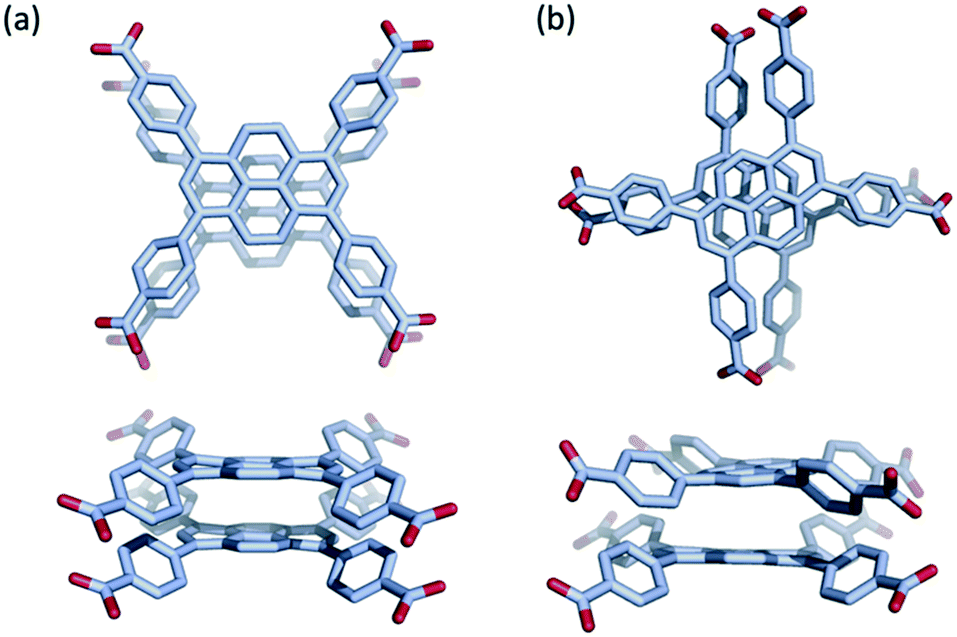 | ||
| Fig. 4 Relative orientation of the TBAPy ligand in (a) K(TBAPy)(DMF), eclipsed, and (b) Na(TBAPy)(DMF), x-shaped. The TBAPy ligands are viewed along the crystallographic b axis (top) and a axis (below). | ||
Consistent with experiment, the absorption spectra of the K(TBAPy)(DMF) and Na(TBAPy)(DMF) MOFs calculated by TD-DFT on the optimized ground-state geometries of the constrained ligand dimers have similar peak positions but are blue-shifted (by ∼0.4–0.6 eV) compared with experiment, as shown in Fig. 3c. However, the lowest energy absorption transitions are shifted to lower energy compared with those of the NU-1000 MOF and the TBAPy monomer (by ∼0.2 eV in both cases), indicating substantially stronger electronic coupling between the ligands in K(TBAPy)(DMF) and Na(TBAPy)(DMF) compared with NU-1000. The discrepancy between the calculations and experiment can be partly attributed to using a dimer model in the calculations, which neglects coupling between multiple ligands in the MOF. Also calculated is the absorption spectrum of pyrene, which is significantly blue shifted compared with the TBAPy monomer, indicating significant electron delocalization between the pyrene and phenyl groups in H4TBAPy.
The calculations show that the transition dipole moment (TDM) of the lowest energy transition of the TBAPy monomer is aligned in the plane along the long axis of the pyrene core, as shown in Fig. S13a.† The orientation of the ligands in the Na(TBAPy)(DMF) MOF dimer at an angle to one another (105°) results in an excited-state electronic coupling that is intermediate between a H and a J-type interaction, producing two almost degenerate lowest energy absorption transitions (separated by ∼0.08 eV) with significant TDMs that are almost orthogonal to one another, as shown in Fig. S13b.† On the other hand, confirming our interpretation of the spectroscopic data, the parallel orientation of the monomers in the K(TBAPy)(DMF) MOF dimer yields an H-type interaction, with the lowest energy absorption transition having negligible oscillator strength and the most intense transition being the second lowest, which has a TDM aligned in the same direction as that of the TBAPy monomer (Fig. S13c†).
Also consistent with the experiment, the emission of the Na(TBAPy)(DMF) MOF dimer is red-shifted by ∼0.4 eV with respect to absorption due to structural relaxation of the excited state (Fig. 3d), but the Stokes shift is about half that observed experimentally. This discrepancy is likely due to the strong geometric constraints placed on the carboxylate atom positions in the calculations, which neglect flexibility of the MOF. As shown in Fig. S12,† the atoms in the Na(TBAPy)(DMF) MOF dimer hardly move between the ground and excited-state geometries. The Stokes shift calculated for the TBAPy monomer, for which no geometric constraints were applied, is significantly larger, at ∼0.6 eV. This larger shift appears to be related to the larger change in the dihedral angle between the pyrene core and phenyl substituents in the excited state of the monomer compared with the MOF dimers, which leads to greater electron delocalization due to the more planar geometry. Calculations on the K(TBAPy)(DMF) MOF dimer indicate that the purely electronic S1 → S0 transition does not occur, which is consistent with the presence of H-type coupling, with emission made possible by vibronic coupling that is neglected in our purely electronic calculations.
Unlike previous reports examining the topology-dependent emissive properties of MOFs containing the TBAPy ligand,22,44 we were reluctant to classify the emission as resulting from excimer formation. Although our model overestimates how rigidly the ligands are held in position, we see no strong evidence to suggest that the experimentally observed steady-state or time-resolved emission are a result of significant geometric rearrangement of the ligands within the lattice that would be associated with excimer formation. The Stokes shift45 and peak width of the steady-state emission of both s-block MOFs are much smaller than those associated with excimer formation in pyrene, and the emission is not substantially broadened compared with the isolated TBAPy ligand, as shown in Fig. 3b. In addition, the shortest decay times measured in the time-resolved emission are an order of magnitude longer than that of excimer formation in crystalline pyrene.45 Furthermore, the calculated changes in the excited-state geometry compared with the ground-state geometry are much smaller than those calculated for pyrene excimers.43,46
Charge-transport properties
Although most MOFs are insulators with no capacity for exciton delocalization between ligands, intrinsic charge transport within a MOF is still an area of emerging interest.47 A design challenge for electrically conductive MOFs is that charge transport decreases exponentially with increased intermolecular distance between frontier orbitals.48 We have shown that the two s-block MOFs presented in this work possess a close packed arrangement of π-rich chromophores and thus may facilitate exciton delocalization and charge mobility. However, conductivity experiments on microcrystalline materials are notoriously unreliable and can differ by orders of magnitude.49 For this reason we decided to use quantum-chemical calculations to estimate the effect of the relative position and orientation of the ligands on charge-transfer rates in the s-block MOFs and a related pyrene-based MOF. Charge-transfer parameters for hole transfer in the K(TBAPy)(DMF) and Na(TBAPy)(DMF) MOFs were calculated and compared with those for NU-1000. Constrained density functional theory (CDFT) was used to localize a +1 charge on one of the monomers in each MOF dimer and CDFT-CI was used to calculate the electronic coupling Vda and reorganization energy λ for the process of transferring this +1 charge from one monomer to the other. The hole transfer rate constant kh was also calculated using Marcus theory50 according to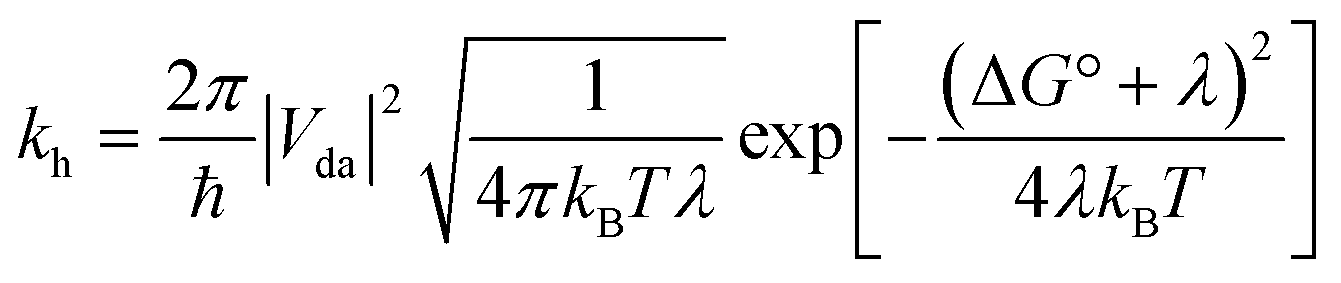 | (1) |
| MOF | Hole transfer rate constant kh (s−1) | Two closest ligands (centroid to centroid) |
|---|---|---|
| Na(TBAPy)(DMF) | 5.7 × 1012 | 3.93 Å |
| K(TBAPy)(DMF) | 1.5 × 1012 | 3.86 Å |
| NU-1000 | 3.3 × 1010 | 10.94 Å |
Given the encouraging results from quantum-chemical calculations, coupled with the well documented ability of pyrene moieties to facilitate hole transfer in several electronically active polymers and composites,51 we examined the semiconducting properties of Na(TBAPy)(DMF) by using it to construct a light emitting diode (LED). LEDs generally consist of a conductive substrate, a hole transport layer, and a light-emitting layer.52 Poly(3,4-ethylenedioxythiophene) polystyrene sulfonate (PEDOT:PSS) can be used as an effective hole transport layer53 that can prevent the LED from shorting, and transports positive charge to the active light emitting polymer layer.54 While the PEDOT:PSS layer does not itself emit any light, such hole transport layers have been found to dramatically improve the efficiency of polymer-based LEDs.55 To this end, we substituted PEDOT:PSS for Na(TBAPy)(DMF) to yield a functioning LED which emitted at 580 nm with a voltage of 4.45 V (24 mA) (see Fig. S11†). Control devices that consisted of just the conductive substrate and the light emitting layer, with no hole transport layer, failed to function. Upon introduction of an electric current, these devices immediately began to heat and did not emit any light. The functioning device represents the first time a sodium-based MOF has been used as a semiconducting hole transport layer in a functioning LED.
Conclusion
We successfully synthesized two new, stable, non-interpenetrated s-block-based MOFs, Na(TBAPy)(DMF) and K(TBAPy)(DMF), containing a π-rich photoactive organic linker. As anticipated, the non-directional, ionic metal–ligand bonds yields MOFs possessing rod-like SBUs and close proximity of the pyrene moieties. Interestingly, the pyrene chromophores adopt different relative packing arrangements (eclipsed and x-shaped) that according to computational and spectral data give rise to significantly different electronic coupling. We note that this is the first time an x-shaped (staggered) alignment of pyrene cores has been examined in the solid-state. This work shows that there is broad scope for using s-block MOFs composed of π-rich chromophores for a variety of sensing and electronic applications. Furthermore, they are ideal candidates for fundamental studies on how the level of solvation, solvent polarity, ion exchange, guest molecules, chromophore packing and alignment, and photoinduced structural changes influence the electronic properties of a material.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research was undertaken with the assistance of resources and services from the National Computational Infrastructure (NCI), which is supported by the Australian Government, and from The University of Adelaide's Phoenix High Performance Computing service. This research was undertaken in part using the MX1 beamline at the Australian Synchrotron, part of ANSTO. The time-resolved fluorescence studies were performed at a laser spectroscopic facility supported by the Australian Research Council (ARC, LE0989747) and in-house single crystal X-ray diffraction undertaken on equipment provided under ARC grant LE0989336.References
- (a) O. M. Yaghi, M. O'Keeffe, N. W. Ockwig, H. K. Chae, M. Eddaoudi and J. Kim, Nature, 2003, 423, 705–714 CrossRef CAS; (b) H. Furukawa, K. E. Cordova, M. O'Keeffe and O. M. Yaghi, Science, 2013, 341, 1230444 CrossRef.
- (a) V. Pascanu, G. González Miera, A. K. Inge and B. Martín-Matute, J. Am. Chem. Soc., 2019, 141, 7223–7234 CrossRef CAS; (b) D. Yang and B. C. Gates, ACS Catal., 2019, 9, 1779–1798 CrossRef CAS.
- (a) Z. Kang, L. Fan and D. Sun, J. Mater. Chem. A, 2017, 5, 10073–10091 RSC; (b) X. Zhao, Y. Wang, D.-S. Li, X. Bu and P. Feng, Adv. Mater., 2018, 30, 1705189 CrossRef.
- (a) K. Liang, R. Ricco, C. M. Doherty, M. J. Styles, S. Bell, N. Kirby, S. Mudie, D. Haylock, A. J. Hill, C. J. Doonan and P. Falcaro, Nat. Commun., 2015, 6, 7240 CrossRef CAS; (b) J. W. M. Osterrieth and D. Fairen-Jimenez, Biotechnol. J., 2020 DOI:10.1002/biot.202000005.
- W. P. Lustig, S. Mukherjee, N. D. Rudd, A. V. Desai, J. Li and S. K. Ghosh, Chem. Soc. Rev., 2017, 46, 3242–3285 RSC.
- L. Sun, M. G. Campbell and M. Dinca, Angew. Chem., Int. Ed., 2016, 55, 3566–3579 CrossRef CAS.
- H. Yang, S. Zhang, L. Han, Z. Zhang, Z. Xue, J. Gao, Y. Li, C. Huang, Y. Yi, H. Liu and Y. Li, ACS Appl. Mater. Interfaces, 2016, 8, 5366–5375 CrossRef CAS.
- Y. Gao, H. Liu, S. Zhang, Q. Gu, Y. Shen, Y. Ge and B. Yang, Phys. Chem. Chem. Phys., 2018, 20, 12129–12137 RSC.
- T. C. Narayan, T. Miyakai, S. Seki and M. Dincă, J. Am. Chem. Soc., 2012, 134, 12932–12935 CrossRef CAS.
- K. S. Lin, A. K. Adhikari, C. N. Ku, C. L. Chiang and H. Kuo, Int. J. Hydrogen Energy, 2012, 37, 13865–13871 CrossRef CAS.
- T. Zhang, P. Wang, Z. Gao, Y. An, C. He and C. Duan, RSC Adv., 2018, 8, 32610–32620 RSC.
- C. W. Ashling, D. N. Johnstone, R. N. Widmer, J. Hou, S. M. Collins, A. F. Sapnik, A. M. Bumstead, P. A. Midgley, P. A. Chater, D. A. Keen and T. D. Bennett, J. Am. Chem. Soc., 2019, 141, 15641–15648 CrossRef CAS.
- J. Wang, Y. Fan, H. W. Lee, C. Yi, C. Cheng, X. Zhao and M. Yang, ACS Appl. Nano Mater., 2018, 1, 3747–3753 CrossRef CAS.
- S. Krieck and M. Westerhausen, Inorganics, 2017, 5, 8–11 CrossRef.
- D. Banerjee and J. B. Parise, Cryst. Growth Des., 2011, 11, 4704–4720 CrossRef CAS.
- D. L. Reger, A. P. Leitner and M. D. Smith, Cryst. Growth Des., 2016, 16, 527–536 CrossRef CAS.
- C. G. Perkins, J. E. Warren, A. Fateeva, K. C. Stylianou, A. McLennan, K. Jelfs, D. Bradshaw and M. J. Rosseinsky, Microporous Mesoporous Mater., 2012, 157, 24–32 CrossRef CAS.
- Q. Yin, P. Zhao, R.-J. Sa, G.-C. Chen, J. Lü, T.-F. Liu and R. Cao, Angew. Chem., Int. Ed., 2018, 57, 7691–7696 CrossRef CAS.
- T. Islamoglu, K. I. Otake, P. Li, C. T. Buru, A. W. Peters, I. Akpinar, S. J. Garibay and O. K. Farha, CrystEngComm, 2018, 20, 5913–5918 RSC.
- S. J. Garibay, I. Iordanov, T. Islamoglu, J. B. DeCoste and O. K. Farha, CrystEngComm, 2018, 20, 7066–7070 RSC.
- R. J. Li, M. Li, X. P. Zhou, D. Li and M. O'Keeffe, Chem. Commun., 2014, 50, 4047–4049 RSC.
- J. Yu, J. Park, A. Van Wyk, G. Rumbles and P. Deria, J. Am. Chem. Soc., 2018, 140, 10488–10496 CrossRef CAS.
- T. C. Wang, N. A. Vermeulen, I. S. Kim, A. B. F. Martinson, J. F. Stoddart, J. T. Hupp and O. K. Farha, Nat. Protoc., 2016, 11, 149–162 CrossRef CAS.
- T. M. McPhillips, S. E. McPhillips, H. J. Chiu, A. E. Cohen, A. M. Deacon, P. J. Ellis, E. Garman, A. Gonzalez, N. K. Sauter, R. P. Phizackerley, S. M. Soltis and P. Kuhn, J. Synchrotron Radiat., 2002, 9, 401–406 CrossRef CAS.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Adv., 2015, 71, 3–8 CrossRef.
- G. M. Sheldrick, Acta Crystallogr., Sect. C: Struct. Chem., 2015, 71, 3–8 Search PubMed.
- L. J. Barbour, J. Supramol. Chem., 2001, 1, 189–191 CrossRef CAS.
- O. V. Dolomanov, L. J. Bourhis, R. J. Gildea, J. A. K. Howard and H. Puschmann, J. Appl. Crystallogr., 2009, 42, 339–341 CrossRef CAS.
- Y. Shao, Z. Gan, E. Epifanovsky, A. T. B. Gilbert, M. Wormit, J. Kussmann, A. W. Lange, A. Behn, J. Deng, X. Feng, D. Ghosh, M. Goldey, P. R. Horn, L. D. Jacobson, I. Kaliman, R. Z. Khaliullin, T. Ku
![[s with combining umlaut]](https://www.rsc.org/images/entities/char_0073_0308.gif) , A. Landau, J. Liu, E. I. Proynov, Y. M. Rhee, R. M. Richard, M. A. Rohrdanz, R. P. Steele, E. J. Sundstrom, H. L. Woodcock, P. M. Zimmerman, D. Zuev, B. Albrecht, E. Alguire, B. Austin, G. J. O. Beran, Y. A. Bernard, E. Berquist, K. Brandhorst, K. B. Bravaya, S. T. Brown, D. Casanova, C. M. Chang, Y. Chen, S. H. Chien, K. D. Closser, D. L. Crittenden, M. Diedenhofen, R. A. Distasio, H. Do, A. D. Dutoi, R. G. Edgar, S. Fatehi, L. Fusti-Molnar, A. Ghysels, A. Golubeva-Zadorozhnaya, J. Gomes, M. W. D. Hanson-Heine, P. H. P. Harbach, A. W. Hauser, E. G. Hohenstein, Z. C. Holden, T. C. Jagau, H. Ji, B. Kaduk, K. Khistyaev, J. Kim, J. Kim, R. A. King, P. Klunzinger, D. Kosenkov, T. Kowalczyk, C. M. Krauter, K. U. Lao, A. D. Laurent, K. V. Lawler, S. V. Levchenko, C. Y. Lin, F. Liu, E. Livshits, R. C. Lochan, A. Luenser, P. Manohar, S. F. Manzer, S. P. Mao, N. Mardirossian, A. V. Marenich, S. A. Maurer, N. J. Mayhall, E. Neuscamman, C. M. Oana, R. Olivares-Amaya, D. P. Oneill, J. A. Parkhill, T. M. Perrine, R. Peverati, A. Prociuk, D. R. Rehn, E. Rosta, N. J. Russ, S. M. Sharada, S. Sharma, D. W. Small, A. Sodt, T. Stein, D. Stück, Y. C. Su, A. J. W. Thom, T. Tsuchimochi, V. Vanovschi, L. Vogt, O. Vydrov, T. Wang, M. A. Watson, J. Wenzel, A. White, C. F. Williams, J. Yang, S. Yeganeh, S. R. Yost, Z. Q. You, I. Y. Zhang, X. Zhang, Y. Zhao, B. R. Brooks, G. K. L. Chan, D. M. Chipman, C. J. Cramer, W. A. Goddard, M. S. Gordon, W. J. Hehre, A. Klamt, H. F. Schaefer, M. W. Schmidt, C. D. Sherrill, D. G. Truhlar, A. Warshel, X. Xu, A. Aspuru-Guzik, R. Baer, A. T. Bell, N. A. Besley, J. Da Chai, A. Dreuw, B. D. Dunietz, T. R. Furlani, S. R. Gwaltney, C. P. Hsu, Y. Jung, J. Kong, D. S. Lambrecht, W. Liang, C. Ochsenfeld, V. A. Rassolov, L. V. Slipchenko, J. E. Subotnik, T. Van Voorhis, J. M. Herbert, A. I. Krylov, P. M. W. Gill and M. Head-Gordon, Mol. Phys., 2015, 113, 184–215 CrossRef CAS.
, A. Landau, J. Liu, E. I. Proynov, Y. M. Rhee, R. M. Richard, M. A. Rohrdanz, R. P. Steele, E. J. Sundstrom, H. L. Woodcock, P. M. Zimmerman, D. Zuev, B. Albrecht, E. Alguire, B. Austin, G. J. O. Beran, Y. A. Bernard, E. Berquist, K. Brandhorst, K. B. Bravaya, S. T. Brown, D. Casanova, C. M. Chang, Y. Chen, S. H. Chien, K. D. Closser, D. L. Crittenden, M. Diedenhofen, R. A. Distasio, H. Do, A. D. Dutoi, R. G. Edgar, S. Fatehi, L. Fusti-Molnar, A. Ghysels, A. Golubeva-Zadorozhnaya, J. Gomes, M. W. D. Hanson-Heine, P. H. P. Harbach, A. W. Hauser, E. G. Hohenstein, Z. C. Holden, T. C. Jagau, H. Ji, B. Kaduk, K. Khistyaev, J. Kim, J. Kim, R. A. King, P. Klunzinger, D. Kosenkov, T. Kowalczyk, C. M. Krauter, K. U. Lao, A. D. Laurent, K. V. Lawler, S. V. Levchenko, C. Y. Lin, F. Liu, E. Livshits, R. C. Lochan, A. Luenser, P. Manohar, S. F. Manzer, S. P. Mao, N. Mardirossian, A. V. Marenich, S. A. Maurer, N. J. Mayhall, E. Neuscamman, C. M. Oana, R. Olivares-Amaya, D. P. Oneill, J. A. Parkhill, T. M. Perrine, R. Peverati, A. Prociuk, D. R. Rehn, E. Rosta, N. J. Russ, S. M. Sharada, S. Sharma, D. W. Small, A. Sodt, T. Stein, D. Stück, Y. C. Su, A. J. W. Thom, T. Tsuchimochi, V. Vanovschi, L. Vogt, O. Vydrov, T. Wang, M. A. Watson, J. Wenzel, A. White, C. F. Williams, J. Yang, S. Yeganeh, S. R. Yost, Z. Q. You, I. Y. Zhang, X. Zhang, Y. Zhao, B. R. Brooks, G. K. L. Chan, D. M. Chipman, C. J. Cramer, W. A. Goddard, M. S. Gordon, W. J. Hehre, A. Klamt, H. F. Schaefer, M. W. Schmidt, C. D. Sherrill, D. G. Truhlar, A. Warshel, X. Xu, A. Aspuru-Guzik, R. Baer, A. T. Bell, N. A. Besley, J. Da Chai, A. Dreuw, B. D. Dunietz, T. R. Furlani, S. R. Gwaltney, C. P. Hsu, Y. Jung, J. Kong, D. S. Lambrecht, W. Liang, C. Ochsenfeld, V. A. Rassolov, L. V. Slipchenko, J. E. Subotnik, T. Van Voorhis, J. M. Herbert, A. I. Krylov, P. M. W. Gill and M. Head-Gordon, Mol. Phys., 2015, 113, 184–215 CrossRef CAS. - Q. Wu and T. Van Voorhis, Phys. Rev. A: At., Mol., Opt. Phys., 2005, 72, 7–10 Search PubMed.
- Q. Wu and T. Van Voorhis, J. Chem. Phys., 2006, 125, 164105 CrossRef.
- Q. Wu, C. L. Cheng and T. Van Voorhis, J. Chem. Phys., 2007, 127, 164119 CrossRef.
- J. Da Chai and M. Head-Gordon, Phys. Chem. Chem. Phys., 2008, 10, 6615–6620 RSC.
- T. Brixner, R. Hildner, J. Köhler, C. Lambert and F. Würthner, Adv. Energy Mater., 2017, 7, 1–33 Search PubMed.
- N. J. Hestand and F. C. Spano, Chem. Rev., 2018, 118, 7069–7163 CrossRef CAS.
- S. K. Chakraborty, Geofis. Pura Appl., 1956, 33, 17–22 CrossRef.
- R. Casier and J. Duhamel, Macromolecules, 2018, 51, 3450–3457 CrossRef CAS.
- A. Gładysiak, T. N. Nguyen, R. Bounds, A. Zacharia, G. Itskos, J. A. Reimer and K. C. Stylianou, Chem. Sci., 2019, 10, 6140–6148 RSC.
- J. H. Qin, Y. D. Huang, Y. Zhao, X. G. Yang, F. F. Li, C. Wang and L. F. Ma, Inorg. Chem., 2019, 58, 15013–15016 CrossRef CAS.
- A. Nakajima, Bull. Chem. Soc. Jpn., 1973, 46, 2602–2604 CrossRef CAS.
- A. Sharma, D. Kim, J. H. Park, S. Rakshit, J. Seong, G. H. Jeong, O. H. Kwon and M. S. Lah, Commun. Chem., 2019, 2, 39 CrossRef.
- F. C. Spano and C. Silva, Annu. Rev. Phys. Chem., 2014, 65, 477–500 CrossRef CAS.
- J. Hoche, H. C. Schmitt, A. Humeniuk, I. Fischer, R. Mitrić and M. I. S. Röhr, Phys. Chem. Chem. Phys., 2017, 19, 25002–25015 RSC.
- P. Deria, J. Yu, T. Smith and R. P. Balaraman, J. Am. Chem. Soc., 2017, 139, 5973–5983 CrossRef CAS.
- R. D. Pensack, R. J. Ashmore, A. L. Paoletta and G. D. Scholes, J. Phys. Chem. C, 2018, 122, 21004–21017 CrossRef CAS.
- J. B. Birks and L. G. Christophorou, Spectrochim. Acta, 1963, 19, 401–410 CrossRef.
- D. Sheberla, L. Sun, M. A. Blood-Forsythe, S. Er, C. R. Wade, C. K. Brozek, A. Aspuru-Guzik and M. Dincă, J. Am. Chem. Soc., 2014, 136, 8859–8862 CrossRef CAS.
- S. Patwardhan and G. C. Schatz, J. Phys. Chem. C, 2015, 119, 24238–24247 CrossRef CAS.
- L. Sun, S. S. Park, D. Sheberla and M. Dincă, J. Am. Chem. Soc., 2016, 138, 14772–14782 CrossRef CAS.
- R. A. Marcus, Annu. Rev. Phys. Chem., 1964, 15, 155–196 CrossRef CAS.
- H. Kuroda, T. Amano, I. Ikemoto and H. Akamatu, J. Am. Chem. Soc., 1967, 89, 6056–6063 CrossRef CAS.
- W. P. Lustig, Z. Shen, S. J. Teat, N. Javed, E. Velasco, D. M. O'Carroll and J. Li, Chem. Sci., 2020, 11, 1814–1824 RSC.
- C. Hou and H. Yu, J. Mater. Chem. C, 2020, 8, 4169–4180 RSC.
- J. Lu, W. Feng, G. Mei, J. Sun, C. Yan, D. Zhang, K. Lin, D. Wu, K. Wang and Z. Wei, Adv. Sci., 2020, 7, 2000689 CrossRef CAS.
- Y. K. Seo, J. W. Han, K. T. Lim, Y. H. Kim, C. W. Joo, J. Lee, N. S. Cho, S. Yu, M. H. Kang, C. Yun and B. H. Choi, Org. Electron., 2017, 42, 348–354 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Additional experimental details for MOF synthesis and characterisation; fits for time resolved fluorescence data, additional crystallographic details, information of the fabrication of the LED device and further details of the calculations. CCDC 2026823, 2026825 and 2026827. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d0ce01505a |
| This journal is © The Royal Society of Chemistry 2021 |