
Increasing the structural boundary of quasiracemate formation: 4-substituted naphthylamides†
Drew E.
Craddock
 a,
McKenzie J.
Parks
a,
Lauren A.
Taylor
a,
Benjamin L.
Wagner
b,
Michael
Ruf
c and
Kraig A.
Wheeler
*a
a,
McKenzie J.
Parks
a,
Lauren A.
Taylor
a,
Benjamin L.
Wagner
b,
Michael
Ruf
c and
Kraig A.
Wheeler
*a
aDepartment of Chemistry, Whitworth University, 300 West Hawthorne Road, Spokane, Washington, 99251, USA. E-mail: kraigwheeler@whitworth.edu
bDepartment of Chemistry, Eastern Illinois University, 600 Lincoln Avenue, Charleston, Illinois 61920, USA
cBruker AXS Inc., 5465 E. Cheryl Parkway, Madison, WI 53711, USA
First published on 17th November 2020
Abstract
Quasiracemates – materials consisting of pairs of near enantiomers – form crystalline motifs that mimic the inversion relationships observed for their racemic counterparts. Recent investigations from our group explored a family of chiral (N-benzoyl)methylbenzylamines to understand the structural boundary of cocrystallization. This investigation extends these earlier studies to include naphthylamide quasiracemates, where the molecular framework is ∼20% larger by volume than the previous diarylamides. A family of naphthylamides was prepared where the pendant functional group differs incrementally in size (i.e., H to C6H5) to give 55 possible unique pairs of racemic and quasiracemic combinations. Data collected from these materials using X-ray crystallography, thermal analysis methods and lattice energy calculations offer important insight into how a spatially larger naphthylamide molecular framework promotes greater structural variance of substituents during the pairwise assembly of quasienantiomers.
Introduction
The terms quasiracemate and quasiracemic material describe the assembly of pairs of chemically non-identical compounds of opposite handedness, e.g., (R)-X and (S)-X′.1 Because the definition of ‘non-identical’ lacks a strict set of structural descriptors, it is not surprising that the current database of known quasiracemates includes building blocks constructed from diverse molecular frameworks and functional groups.2 A notable trend that emerges from these collective investigations is that the initial design strategy for achieving quasiracemate formation often begins with pairs of structurally similar quasienantiomers. This notion of structural likeness is prevalent in the database of neutral small molecule quasiracemates,3 where for example, many entries include quasienantiomers of (R)-X/(S)-X′ that differ only by Cl/Br substitutions.4–8 While successfully pairing left- and right-handed Cl and Br components may be largely anticipated given the topological similarity of these groups, inspection of the collective entries suggests that modest variations in size (H/F (ref. 9) and O/S (ref. 10 and 11)), shape (CH3/NO2 (ref. 12)) and polarity (OCH3/Br (ref. 13)) can also be tolerated during the molecular recognition process of quasiracemate formation.Incorporating more structurally diverse sets of quasienantiomers is possible when considering other chemical classes such as organometallic,14–18 amino acid19–21 and macromolecular systems.22–26 In these cases, the contribution of the imposed structural difference to supramolecular assembly is most likely diminished in the presence of dominant intermolecular contacts or larger molecular frameworks. An indication that these systems allow for greater structural diversity of quasienantiomeric components is that the H/CH3 variation is frequently reported for organometallic27 and amino acid quairacemates19–21 – a quasienantiomeric pair not observed with neutral small organic quasiracemates. As expected, even larger structural differences are observed with protein-based quasiracemates where the contribution from the imposed structural difference is minimal compared to the overall size of the biomolecule. The significance of the H/CH3 variation can be approximated by use of group volumes.28 For example, the percent difference in substituent volumes (%ΔV) of the Cl/Br pair is 16 while %ΔV = 100 for H/CH3. This indirect measure of topological features can be used to advantage as a predictive tool for assessing quasiracemate formation.
This investigation builds on a recent study from our group that explored the structural boundaries of quasiracemate formation using a homologous family of 2-substituted diarylamides (1, Fig. 1).9 Results from processing (R)-X/(S)-X′ combinations of 1 using hot stage polarized microscopy indicated molecular recognition events by the formation of new crystalline phases. Following the success of this previous report, our attention now turns to understanding how increased crystal lattice stabilization affects the cocrystal assembly of quasiracemic components. This is achieved by constructing a larger molecular framework via the addition of a second aryl group to the original diarylamide 1 system. Such a modification results in a family of naphthylamides (2) where the molecular framework is considerably larger than 1 (%ΔV = 22). Because of the steric bulk of the naphthyl group and its potential contribution to crystal packing, it was anticipated that this structural change may allow for more variance of substituents in the pairwise assembly of quasienantiomers before molecular recognition is compromised.
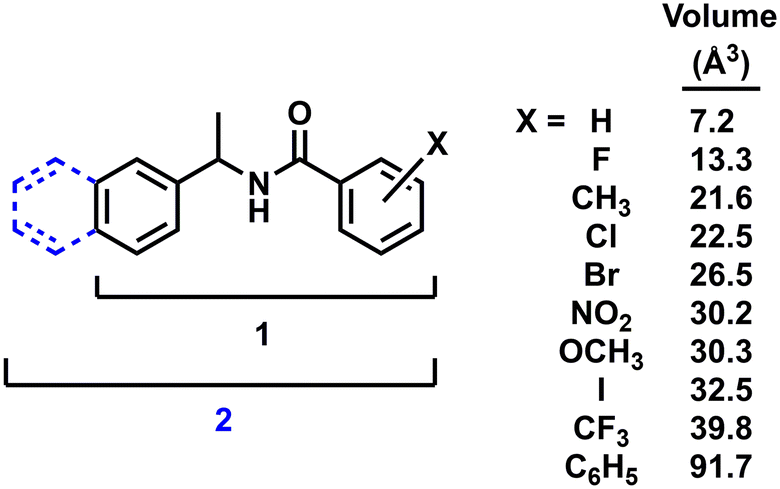 | ||
| Fig. 1 Chemical framework of diarylamide 1 and naphthylamide 2 with functional group volumes (group volume determinations from ref. 28). | ||
Results and discussion
Hot stage thermomicroscopy
Investigations into the recognition profiles of naphthylamides 2 utilized the Kofler contact fusion method29 to determine if cocrystallization would occur. The utility of this hot stage technique coupled with video recording has provided considerable insight into the modes of molecular assembly for a wide variety of materials.30,31 This method applied to the assessment of cocrystal formation involves delivering milligram quantities of two or more solid samples to a glass slide equipped with a smaller glass coverslip. Heating the system to the melting temperature of the components effectively repositions these materials under the coverslip using capillary action and immediate cooling recrystallizes these components with distinct interfaces formed between each sample. Thermal cycling of the system then either produces melting regions (eutectic regions) suggesting conglomerate formation or the generation of new crystalline phases (cocrystal formation) if recognition occurs. In the case of the 2-substituted diarylamide 1 study, processing all possible pairs of quasienantiomers (i.e., (R)-X and (S)-X′ where X/X′ = H, F, Cl, CN, CH3, NO2, Br, CF3, OCH3, I and C6H5) using the thermomicroscopy technique effectively identified the structural boundary of quasiracemate formation for this system.9 While pairing (S)-1-H and (R)-1-F quasienantiomers formed a quasiracemate, processing these same components with the other diarlyamide derivatives lacked thermal signatures indicative of cocrystallization. This outcome showed that pairing the H and F substituted diarylamides with other quasienantiomers of larger substituent volumes created a substantial topological difference, impeding cocrystallization.Given the larger molecular framework of naphthylamide 2 as compared to diarylamide 1, use of 2 as quasiracemic building blocks should be accompanied by increased crystal lattice stabilization that in turn promotes greater substituent diversity during quasiracemate formation. Because the hot stage method provides intimate details of crystal growth events, we anticipated this technique, when applied to 2, would also offer important insight to the formation of new quasiracemic crystalline phases and possibly a more flexible structural boundary capable of adopting molecular association of greater diversity.
The initial focus of this study centered on preparing a sizeable set of chiral 2-substituted naphthylamides (2) and then processing all pairs using the hot stage method similar to the diarylamide 1 investigation. Use of these materials proved to be a challenge since many combinations, including racemates, formed conglomerates from the melt. Because the observed spontaneous resolution events suggested cocrystal formation is thermodynamically less favorable than crystal growth events using only the starting components, we then turned our attention to examine the 4-substituted naphthylamides where the placement of the functional group X could promote different crystal growth behavior. As with our previous study of 1, outcomes from the thermal cycling of 4-substituted naphthylamide 2 samples indicated either one eutectic region for conglomerate mixtures or two regions, accompanied by the growth of a new crystalline phase for racemic and quasiracemic materials. Also, since heating all possible sets of enantiomers gave racemic mixtures and not conglomerates, these results show preference for the 4-substituted naphthylamide framework as a viable candidate for this investigation.
Fig. 2 depicts the hot stage micrographs for the 4-substituted (R)-2-H/(S)-2-H, (R)-2-H/(S)-2-NO2 and (R)-2-NO2/(S)-2-NO2 systems. In each case, the thermal cycling of the component pairs resulted in the growth of new racemic or quasiracemic crystalline phases. To achieve these crystal formations, molecular recognition from the melt must first occur at the interface region of the deposited R and S samples (enantiomers or quasienantiomers). These initial assemblies further propagate during the cooling portion of the experiment to give thermodynamically favored molecular alignment of (R)-X/(S)-X′. Repeating the heat–cool cycle several times increases the size of the central racemic/quasiracemic crystalline region further confirming the cocrystallization event. As additional validation, several of these new crystalline phases were examined via X-ray powder diffraction and compared to the powder patterns generated from known crystal structures of the same materials and the racemic counterparts (S3,†). It is worth noting that the H/NO2 combination did not form a quasiracemic phase when applied to diarylamide 1 nor, to our knowledge, has this functional group pairing appeared in other reports describing quasiracemates. This lack of attention to the H/NO2 pair is likely due to the sizable topological differences associated with these groups (%ΔV = 123).
 | ||
| Fig. 2 Hot stage micrographs of the 4-substituted (R)-2-H/(S)-2-H, (R)-2-H/(S)-2-NO2 and (R)-2-NO2/(S)-2-NO2 naphthylamide systems showing the formation of racemic and quasiracemic crystalline phases. | ||
Parallel processing all possible pairs of homochiral components of 2 (55 total racemic and quasiracemic combinations) using the hot stage method provided insight into the cocrystallization tendencies of the naphthylamide family. The micrographs generated by pairing enantiomeric components (Fig. 3, yellow) show distinct racemate formation. The other entries provided in Fig. 3 result from combining quasienantiomeric components ranging from H to C6H5 and show either conglomerate (red, single eutectic region) or quasiracemate formation (green, two eutectic regions). The landscape for this set of hot stage data favors successful cocrystallization events. The exceptions to these observations include six entries (F/I, NO2/CF3, NO2/OCH3, NO2/I, CF3/I and OCH3/I) along with any pairing that involves the phenyl derivative. These results are in stark contrast to the thermomicroscopy investigation of diarylamide 1, where the structural boundary of quasiracemate formation is limited to pairing H/F quasienantiomers and other sets that range at most by %ΔV = 36 (i.e., X = Cl, Me, NO2, Br, CF3 and I).
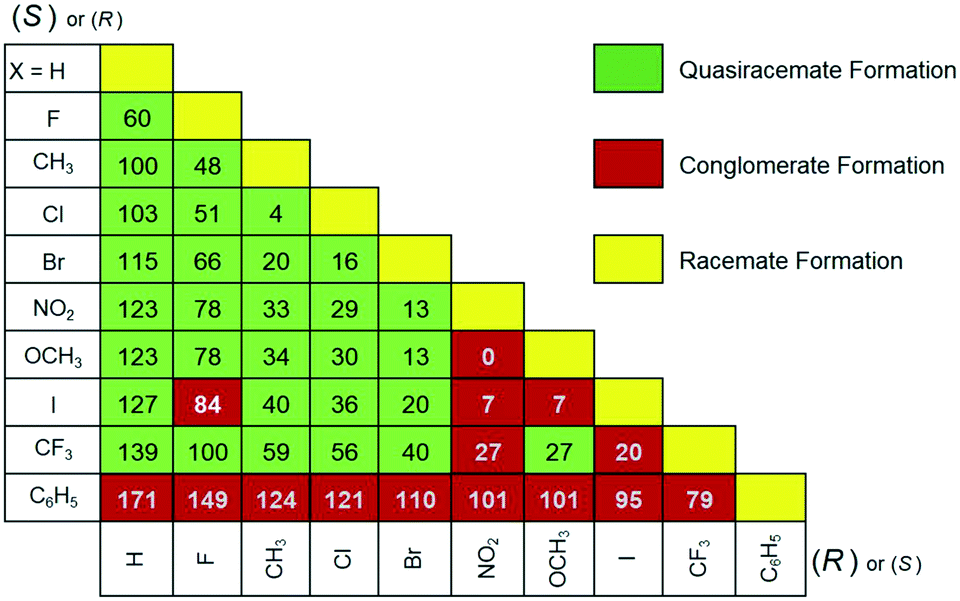 | ||
| Fig. 3 Recognition profiles of 4-substituted naphthylamides as obtained from video-assisted hot stage micrographs. The percent difference in substituent volumes (%ΔV) is also provided for each functional group pair. | ||
Crystal structure assessment
To further understand how molecular recognition occurs in the naphthylamide systems, X-ray crystallography was utilized to determine the structural trends in quasiracemate formation and capture diverse sets of previously unreported functional group pairs (Table S1†). It should be pointed out that the crystal growth studies of the naphthylamide 2 systems were often problematic, requiring multiple crystal growth attempts for each sample. This effort utilized many solvent systems and crystal growth methods with results frequently producing small (short crystal dimension typically <50 microns) poorly diffracting fibers. Samples of (±)-2-H provided the most notable challenge. After numerous attempts with crystal growth experiments and crystallographic data collections, the most satisfactory data set of (±)-2-H exhibited crystal modulation, twinning and disorder with 24 symmetry independent molecules in the asymmetric unit (Z′ = 24).As shown in Fig. 4, each naphthylamide derivative forms crystal structure motifs directed by N–H⋯O hydrogen bonds (Table S2†) and the complementary shapes of the components. With the exception of (±)-2-H (P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) , Z′ = 24) and (±)-2-NO2 (P21/n, Z′ = 1), structures corresponding to the H/F, H/NO2 and F/Br families (6 total structures) organize in triclinic space groups with two symmetry independent molecules in the asymmetric unit (Z′ = 2). The racemic entries exhibit centrosymmetric alignment in space group P with the quasiracemates organized in space group P1. As observed with other previously reported quasiracemic systems, alignment of the quasienantiomeric pairs of 2 form near inversion symmetry motifs that closely mimic those found in the related racemates. The structural similarity of these racemic and quasiracemic crystalline phases extends beyond motifs derived from only pairs of molecules to include the entire crystal landscape. Many of the triclinic structures (i.e., (±)-2-F, (±)-2-Br, (S)-2-H/(R)-2-F, (S)-2-NO2/(R)-2-H, (S)-2-F/(R)-2-Br) exhibit isostructural relationships as evident from comparable unit cell parameters, diffraction data sets and crystal packing. The propensity of these materials to form close structural relationships is even more remarkable given the diversity of substituents included with this group and that the structures of (R)-2-H/(S)-2-NO2 and (S)-2-F/(R)-2-Br display two-part disorder of the naphthyl groups.
, Z′ = 24) and (±)-2-NO2 (P21/n, Z′ = 1), structures corresponding to the H/F, H/NO2 and F/Br families (6 total structures) organize in triclinic space groups with two symmetry independent molecules in the asymmetric unit (Z′ = 2). The racemic entries exhibit centrosymmetric alignment in space group P with the quasiracemates organized in space group P1. As observed with other previously reported quasiracemic systems, alignment of the quasienantiomeric pairs of 2 form near inversion symmetry motifs that closely mimic those found in the related racemates. The structural similarity of these racemic and quasiracemic crystalline phases extends beyond motifs derived from only pairs of molecules to include the entire crystal landscape. Many of the triclinic structures (i.e., (±)-2-F, (±)-2-Br, (S)-2-H/(R)-2-F, (S)-2-NO2/(R)-2-H, (S)-2-F/(R)-2-Br) exhibit isostructural relationships as evident from comparable unit cell parameters, diffraction data sets and crystal packing. The propensity of these materials to form close structural relationships is even more remarkable given the diversity of substituents included with this group and that the structures of (R)-2-H/(S)-2-NO2 and (S)-2-F/(R)-2-Br display two-part disorder of the naphthyl groups.
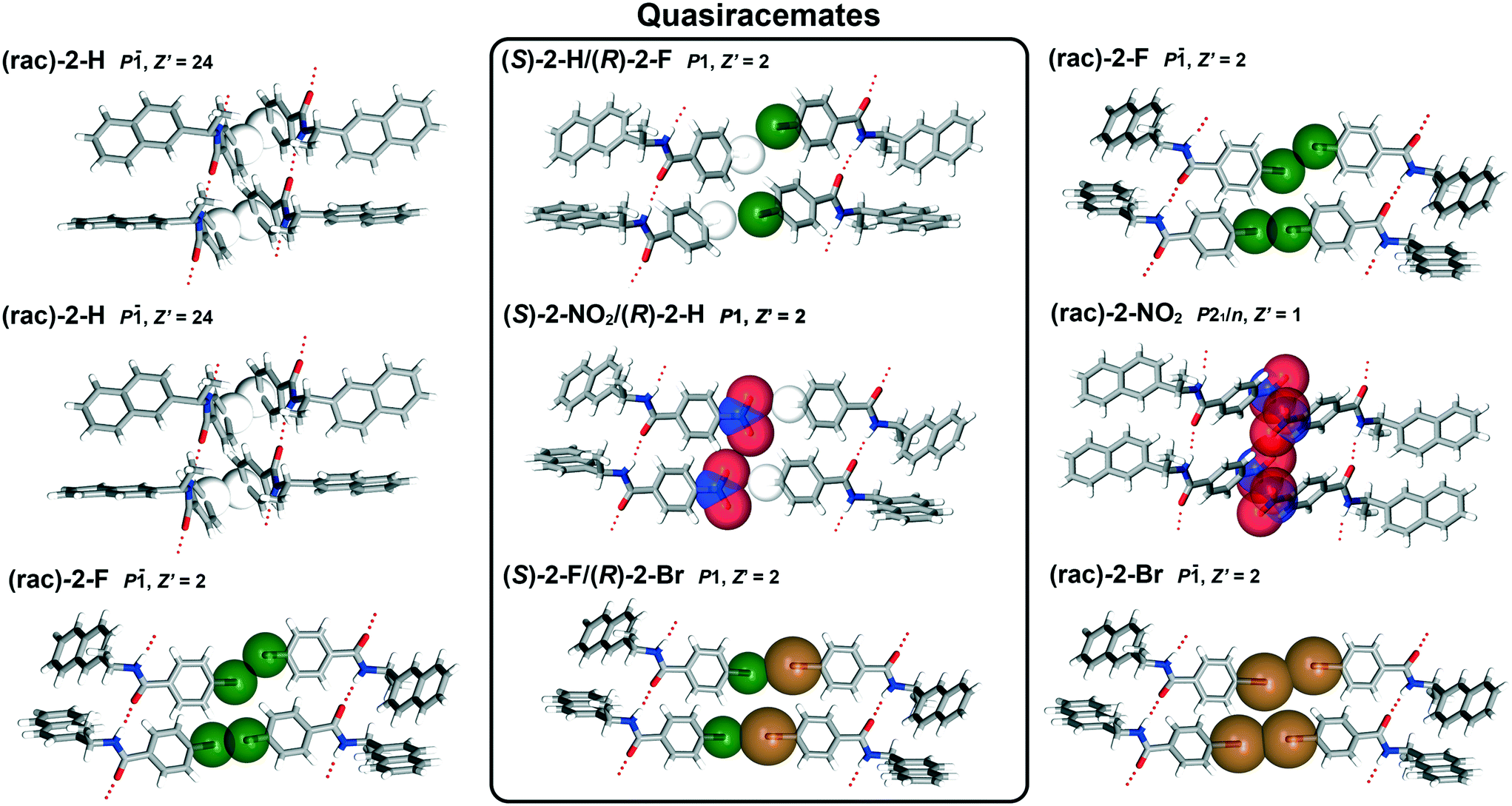 | ||
| Fig. 4 Crystal structures of the racemic and quasiracemic 4-substituted H/NO2, H/F and F/Br naphthylamides showing the attached substituents with space-filling spheres and extended motifs directed by N–H⋯O contacts. Disordered naphthyl groups of (R)-2-H/(S)-2-NO2 and (S)-2-F/(R)-2-Br are omitted for clarity. | ||
The structures presented in Fig. 4 display comparable molecular conformations with C(![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O)–N–Csp3–CAr angles ranging from 80.0 to 90.2° (the exception being 69.2 to 93.9° for (±)-2-H) and N–H⋯O contacts that either link translationally related or symmetry independent neighboring molecules. These interactions form C4 graph set motifs that extend in the crystal to give 1D hydrogen-bonded motifs.32,33 It was anticipated that the aryl groups of naphthylamide 2 would also provide noticeable crystal stabilization via close π⋯π or Caryl–H⋯π contacts. While molecules of 2 do align with aromatic groups in close proximity to each other, the positions of these molecules form motifs directed by van der Waals surfaces rather than a clear set of attractive non-bonded contacts. Excluding (±)-2-NO2 where Z′ = 1, the other six structures exhibit Z′ > 1 because of the prominent tilt angle of the naphthyl group for each symmetry unique molecule. This difference in naphthyl orientation alternates with the N–H⋯O linked molecules where the N–Csp3–CAr–CAr torsion angles are either −5 ± 5° or +120 ± 5°. When omitting the naphthyl groups, the molecules of 2 that construct the C4 structural motif closely mimic translation symmetry.
O)–N–Csp3–CAr angles ranging from 80.0 to 90.2° (the exception being 69.2 to 93.9° for (±)-2-H) and N–H⋯O contacts that either link translationally related or symmetry independent neighboring molecules. These interactions form C4 graph set motifs that extend in the crystal to give 1D hydrogen-bonded motifs.32,33 It was anticipated that the aryl groups of naphthylamide 2 would also provide noticeable crystal stabilization via close π⋯π or Caryl–H⋯π contacts. While molecules of 2 do align with aromatic groups in close proximity to each other, the positions of these molecules form motifs directed by van der Waals surfaces rather than a clear set of attractive non-bonded contacts. Excluding (±)-2-NO2 where Z′ = 1, the other six structures exhibit Z′ > 1 because of the prominent tilt angle of the naphthyl group for each symmetry unique molecule. This difference in naphthyl orientation alternates with the N–H⋯O linked molecules where the N–Csp3–CAr–CAr torsion angles are either −5 ± 5° or +120 ± 5°. When omitting the naphthyl groups, the molecules of 2 that construct the C4 structural motif closely mimic translation symmetry.
Beyond molecules linked by N–H⋯O interactions, several systems of 2 display halogen bonding. The crystal structures depicted in Fig. 4 and corresponding to (±)-2-F (F⋯F, 2.843(2) and 2.887(2) Å), (±)-2-Br (Br⋯Br, 3.2897(6) and 3.3868(7) Å) and (S)-2-F/(R)-2-Br (F⋯Br, 2.967(2) and 2.975(2) Å) show molecules related by inversion or near inversion symmetry with short X⋯X contacts. It is interesting to note that nearly identical packing patterns are observed for (S)-2-H/(R)-2-F and (S)-2-NO2/(R)-2-H; however, these structures exists without any obvious cohesive interactions involving H and F/NO2 at the same molecular interfaces. This suggests that X⋯X non-bonded contacts likely play a supportive role, but not essential for generating the packing motifs depicted in Fig. 4 for the set of isostructural materials. Additional contributors to crystal packing, such as molecular shape, must play a central role in the assembly of these naphthylamide molecules.
Crystal lattice energetics
Differential scanning calorimetry (DSC) measurements conducted on naphthylamide 2 samples offered important opportunities to assess the thermal behavior of the quasiracemic systems and their racemic counterparts. Each DSC trace provided distinct thermal signatures corresponding to the melting process of the material. For comparison, this investigation also determined crystal lattice energies using the molecular cluster approach as employed by Crystal Explorer34–36 (Gaussian16,37 B3LYP/6-31G(d,p)). This computational method starts with crystallographic data and sums the electrostatic, polarization, dispersion and repulsion contributions to determine lattice energies of the naphthylamide derivatives (2).The data provided in Table 1 supports many of the crystallographic trends identified for each naphthylamide family. For example, the set of data corresponding to the H/F family shows a close isostructural relationship between quasiracemate (R)-2-H/(S)-2-F and racemate (±)-2-F. The similarity of structural features observed for these crystal structures translates to a comparable trend in their computationally and experimentally derived lattice energies (ELatt). For the (±)-2-H entry where crystal growth was the most problematic, this data is significantly less than that for (R)-2-H/(S)-2-F and (±)-2-F where ΔELatt(Q–H) (CE) = 64.8 kJ mol−1, ΔELatt(F–H) (CE) = 78.9, ΔELatt(Q–H) (DSC) = 5.8 and ΔELatt(F–H) (DSC) = 3.6. The similar ELatt energies determined for quasiracemate (S)-2-H/(R)-2-F and (±)-2-F (ΔELatt(Q–F) (CE) = 14.1 kJ mol−1 ΔELatt(Q–F) (DSC) = 2.2) supports the notion that quasiracemic materials mimic the thermodynamically preferred centrosymmetric alignment often observed in racemates.
| Crystal lattice enthalpies, ELatt (kJ mol−1) | Density (g mL−1) | Melting point (°C) | ||
|---|---|---|---|---|
| Crystal Explorer | DSC | |||
| (±)-2-H | −92.2 | −32.7 | 1.27 | 145.9 |
| (S)-2-H/(R)-2-F | −157.0 | −38.5 | 1.32 | 166.1 |
| (±)-2-F | −171.1 | −36.3 | 1.36 | 164.6 |
| (±)-2-H | −92.2 | −32.7 | 1.27 | 145.9 |
| (R)-2-H/(S)-2-NO2 | −168.9 | −37.5 | 1.34 | 165.1 |
| (±)-2-NO2 | −178.8 | −39.4 | 1.38 | 155.9 |
| (±)-2-F | −171.1 | −36.3 | 1.36 | 164.6 |
| (S)-2-F/(R)-2-Br | −169.2 | −42.7 | 1.44 | 172.3 |
| (±)-2-Br | −172.6 | −47.2 | 1.53 | 168.7 |
Extending this study to the F/Br family again emphasizes the significance of sets of crystal structures that exhibit isostructural relationships. Quasiracemate (S)-2-F/(R)-2-Br is isostructural to both of the structurally related racemic compounds (i.e., (±)-2-F and (±)-2-Br). The melting point, crystal density and ELatt data related to these entries are quite similar and consistent with structures that exhibit closely related crystal packing. The inclusion of the H/NO2 system in the study offers a departure from the other two quasiracemates since the (±)-2-H and (±)-2-NO2 racemic structures possess fewer common structural features to (R)-2-H/(S)-2-NO2 due to differences in space group assignment, number of symmetry independent molecules (Z′) and crystal packing. Even so, the (R)-2-H/(S)-2-NO2 system is isostructural with the other quasiracemates, and as expected shows comparable ELatt energies.
Each quasiracemic crystal structure included with this study exhibits near centrosymmetric packing where the greatest departure from true symmetry occurs at the imposed structural point of difference – the H/F, H/NO2 and F/Br substitutions – rather than significant misalignment of the chemical framework. This mode of assembly effectively organizes molecules with close packing, which, in turn, maximizes crystal stabilization using the complementary shapes of the components. For racemic materials, the enantiomeric pairs are chemically identical, but in the case of quasiracemates the crystal components are chemically unique. One challenge with assembling quasiracemates relates to determining the structural boundary of quasiracemate formation and the minimal structural difference between the quasienantiomers required for successful cocrystallization. The structure of (S)-2-H/(R)-2-F was largely anticipated since other previously reported quasiracemates exist with this substitution pattern.9 Since %ΔV for H/F is 60 and the variation in size of the F/Br pair (%ΔV = 66) is not appreciably different, it was somewhat expected that other F/Br quasiracemates would be present in the literature. Their absence however may relate to the lack of use of the F/Br pair in quasiracemate design strategies rather than unsuccessful attempts by practitioners in the field. The H/NO2 pair offers quite a different deviation from the other quasiracemic systems where %ΔV = 123. Such a significant size and shape difference associated with the H and NO2 functional group pair was previously thought to be a considerable deterrent to quasiracemate formation for neutral small molecule systems. However, given the success of the hot stage and crystallographic studies assessment for (R)-2-H/(S)-2-NO2 and the lack of success with the diarylamide H/NO21, these results emphasize the importance of chemical framework selection to the recognition profile of quasienantiomeric components.
This computational investigation also provided an opportunity to evaluate the contribution of the naphthyl group in 2 to crystal stabilization. This was achieved by comparing lattice energies determined from the crystal structures of naphthylamide 2 and those of 2-substituted diarylamide 1 retrieved from the CCDC Cambridge Structural Database.38 The difference in lattice energy for the (±)-2-NO2 and (±)-1-NO2 (ELatt = −155.6 kJ mol−1, ISACOU), (±)-2-Br and (±)-1-Br (ELatt = −143.9 kJ mol−1, LUNQEQ) and (S)-2-H/(R)-2-F and (R)-1-H/(S)-1-F (ELatt = −128.8 kJ mol−1, WANLUV) systems is ΔELatt = 23.2, 28.7 and 28.2 kJ mol−1, respectively. An average ΔELatt = 26.7 kJ mol−1 of crystal lattice stabilization in favor of the naphthylamide derivatives provides considerable support that the larger naphthyl ring is more able to promote greater structural diversity of quasienantiomeric components during molecular recognition events.
Conclusions
The present study explores the role chemical framework plays on the assembly of crystalline quasiracemic materials. While the collective body of quasiracemate literature suggests larger chemical systems offer greater opportunity to cocrystallize structurally diverse quasienantiomers, to our knowledge no systemic investigation exists that provides insight into this aspect of the quasiracemate process. This study evaluated a family of naphthylamide derivatives (2) in view of previously reported diarylamides (1) where the primary structural difference between these chemical systems is a benzene group attachment. The set of naphthylamide compounds examined are ∼20% larger by volume than the diarylamides (1) and systematically differ by the attached functional groups (i.e., H to C6H5).To understand the recognition profile of the naphthylamide system (2), all possible pairs of homochiral components (55 total racemic and quasiracemic combinations) were processed using video-assisted polarized thermomicroscopy. These hot stage results draw attention to the propensity of 2 to form quasiracemates (or racemates) as evident from the growth of a new crystalline phase outlined by two eutectic regions. This work was further supported by X-ray crystallography, thermal analysis and computational methods where the collective data offers critical insight to how a larger naphthylamide system compared to diarylamide 1 tolerates greater diversity of starting components during molecular assembly.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was generously supported by the National Science Foundation (DMR1904651 and CHE1827313), MJ Murdock Charitable Trust and Whitworth University. We also thank Prof. A. L. Rheingold (Univ. of California San Diego) for key crystallographic contributions to this study.Notes and references
- A. Fredga, Bull. Soc. Chim. Fr., 1973, 1, 173–182 Search PubMed.
- Q. Zhang and D. P. Curran, Chem. – Eur. J., 2005, 11, 4866–4880 CrossRef CAS.
- S. P. Kelley, L. Fábián and C. P. Brock, Acta Crystallogr., Sect. B: Struct. Sci., 2011, 67, 79–93 CrossRef CAS.
- J. M. Spaniol and K. A. Wheeler, RSC Adv., 2016, 6, 64921–64929 RSC.
- J. T. Cross, N. A. Rossi, M. Serafin and K. A. Wheeler, CrystEngComm, 2014, 16, 7251–7258 RSC.
- A. M. Lineberry, E. T. Benjamin, R. E. Davis, W. S. Kassel and K. A. Wheeler, Cryst. Growth Des., 2008, 8, 612–619 CrossRef CAS.
- S. L. Fomulu, M. S. Hendi, R. E. Davis and K. A. Wheeler, Cryst. Growth Des., 2002, 2, 645–651 CrossRef CAS.
- F. Toda, K. Tanaka, H. Miyamoto, H. Koshima, I. Miyahara and K. Hirotsu, J. Chem. Soc., Perkin Trans. 2, 1997, 1877–1886 RSC.
- I. C. Tinsley, J. M. Spaniol and K. A. Wheeler, Chem. Commun., 2017, 53, 4601–4604 RSC.
- C. S. Frampton, K. A. Ketuly, A. Hamid, A. Hadi, J. H. Gall and D. D. MacNicol, Chem. Commun., 2013, 49, 7198–7200 RSC.
- T. Jacobs, M. W. Bredenkamp, P. H. Neethling, E. G. Rohwer and L. J. Barbour, Chem. Commun., 2010, 46, 8341–8343 RSC.
- M. S. Hendi, P. Hooter, R. E. Davis, V. M. Lynch and K. A. Wheeler, Cryst. Growth Des., 2004, 4, 95–101 CrossRef CAS.
- M. E. Breen, S. L. Tameze, W. G. Dougherty, W. S. Kassel and K. A. Wheeler, Cryst. Growth Des., 2008, 8, 3863–3870 CrossRef CAS.
- C.-P. Li, H. Zhou, Y. Sun, J.-H. Guo and M. Du, Cryst. Growth Des., 2018, 18, 4252–4256 CrossRef CAS.
- Y. Popowski, I. Goldberg and M. Kol, Chem. – Eur. J., 2016, 22, 5530–5533 CrossRef CAS.
- S. Akine, S. Hotate, T. Matsumoto and T. Nabeshima, Chem. Commun., 2011, 47, 2925–2927 RSC.
- S. Reemers and U. Englert, Inorg. Chem. Commun., 2002, 5, 829–831 CrossRef CAS.
- U. Englert, A. Haring, C. Hu and I. Kalf, Z. Anorg. Allg. Chem., 2002, 628, 1173–1179 CrossRef CAS.
- B. Dalhus and C. H. Görbitz, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 1999, 55, 1547–1555 CrossRef.
- B. Dalhus and C. H. Görbitz, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 1999, 55, 1105–1112 CrossRef.
- B. Dalhus and C. H. Görbitz, Acta Crystallogr., Sect. B: Struct. Sci., 1999, 55, 424–431 CrossRef.
- K. W. Kurgan, A. F. Kleman, C. A. Bingman, D. F. Kreitler, B. Weisblum, K. T. Forest and S. H. Gellman, J. Am. Chem. Soc., 2019, 141, 7704–7708 CrossRef CAS.
- B. Yan, L. Ye, W. Xu and L. Liu, Bioorg. Med. Chem., 2017, 25, 4953–4965 CrossRef CAS.
- K. Mandal, B. Dhayalan, M. Avital-Shmilovici, A. Tokmakoff and S. B. H. Kent, ChemBioChem, 2016, 17, 421–425 CrossRef CAS.
- D. F. Kreitler, D. E. Mortenson, K. T. Forest and S. H. Gellman, J. Am. Chem. Soc., 2016, 138, 6498–6505 CrossRef CAS.
- S. Gao, M. Pan, Y. Zheng, Y. Huang, Q. Zheng, D. Sun, L. Lu, X. Tan, X. Tan, H. Lan, J. Wang, T. Wang, J. Wang and L. Liu, J. Am. Chem. Soc., 2016, 138, 14497–14502 CrossRef CAS.
- B. Calmuschi, M. Alesi and U. Englert, Dalton Trans., 2004, 1852–1857 RSC.
- Y. H. Zhao, M. H. Abraham and A. M. Zissimos, J. Org. Chem., 2003, 68, 7368–7373 CrossRef CAS.
- L. Kofler and A. Kofler, Thermal micromethods for the study of organic compounds and their mixtures, Wagner, Innsbrook, Austria, 1952 Search PubMed.
- A. Kumar, P. Singh and A. Nanda, Appl. Microsc., 2020, 50, 12 CrossRef.
- R. Teng, L. Wang, M. Chen, W. Fang, Z. Gao, Y. Chai, P. Zhao and Y. Bao, J. Mol. Struct., 2020, 1217, 128432 CrossRef CAS.
- J. Bernstein, R. E. Davis, L. Shimoni and N.-L. Chang, Angew. Chem., Int. Ed. Engl., 1995, 34, 1555–1573 CrossRef CAS.
- M. C. Etter, J. C. MacDonald and J. Bernstein, Acta Crystallogr., Sect. B: Struct. Sci., 1990, 46, 256–262 CrossRef.
- C. F. Mackenzie, P. R. Spackman, D. Jayatilaka and M. A. Spackman, IUCrJ, 2017, 4, 575–587 CrossRef CAS.
- M. A. Spackman and D. Jayatilaka, CrystEngComm, 2009, 11, 19–32 RSC.
- S. P. Thomas, P. R. Spackman, D. Jayatilaka and M. A. Spackman, J. Chem. Theory Comput., 2018, 14, 1614–1623 CrossRef CAS.
- M. J. Frisch, et al., GAUSSIAN16, Gaussian, Inc., Wallingford, CT, USA, 2016, http://www.gaussiancom Search PubMed.
- C. R. Groom, I. J. Bruno, M. P. Lightfoot and S. C. Ward, Acta Crystallogr., Sect. B: Struct. Sci., Cryst. Eng. Mater., 2016, 72, 171–179 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Synthetic procedures, NMR spectroscopic data, X-ray crystallographic assessments (powder and single-crystal), hydrogen-bond tables, differential scanning calorimetry and computational methods. CCDC 2019789–2019794 and 2026035. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d0ce01331e |
| This journal is © The Royal Society of Chemistry 2021 |