
Photochromism and photoresponsive luminescence in naphthalenediimide coordination polymers with high thermostability†
Le-Tian
Zhang
a,
Bin
Xia
a,
Xia
Zhang
b,
Sha
Lu
a,
Xian-Xian
Zhou
a,
Quan-Wen
Li
 *c and
Qing-Lun
Wang
*a
*c and
Qing-Lun
Wang
*a
aCollege of Chemistry, Key Laboratory of Advanced Energy Materials Chemistry (Ministry of Education), Nankai University, Tianjin 300071, P. R. China. E-mail: wangql@nankai.edu.cn
bSchool of Chemical Engineering and Technology, Hebei University of Technology, Tianjin, 300130, P. R. China
cSchool of Materials Science and Engineering, National Institute for Advanced Materials, Tianjin Key Laboratory of Metal and Molecule-Based Material Chemistry, Nankai University, Tianjin 300350, P. R. China. E-mail: liqw@nankai.edu.cn
First published on 20th October 2020
Abstract
Two novel photochromic complexes [Zn2(bpdc)2(m-DPNDI)2]·H2O (1) and [Cd(bpdc)(m-DPNDI)]·H2O (2) (H2bpdc = 4,4′-diphenic acid, m-DPNDI = N,N′-bis(3-pyridyl)-1,4,5,8-naphthalenetetracarboxydiimide) were prepared through a solvothermal method. In complexes 1 and 2, the selection of electron-deficient ligand m-DPNDI and electron-rich ligand bpdc2− is aimed at forming a complete donor–acceptor pair and establishing a suitable electron transfer pathway to realize photoinduced electron transfer. Both complexes exhibited interesting photochromic and photoresponsive luminescence behaviours, which was verified by EPR, XRD and IR measurements. Furthermore, the existence of interpenetrating networks and abundant π⋯π interactions in 1 makes the framework intact at below 460 °C.
Introduction
Photochromic compounds can change their colours and some other physical properties between two different states before and after illumination.1 Such properties are of great interest in applications for sensors,2 molecular switches3–5 and information storage.6–8 In the past few years, the number of photochromic species rapidly increased according to the assembly of metal cations and photochromic ligands.9,10 Among them, electron transfer photochromic compounds are special, which always differ dramatically in electron states with photoisomers rather than molecular structures. The negligible change of the structure results in smaller chemical stress, which is beneficial to the reversibility and max cycle times.11 In particular, naphthalenediimide (NDI) derivates are widely studied due to good electron affinity, charge carrier mobility, and excellent thermal and oxidative stability.12 Furthermore, the functionalization of NDI can be realized easily by an amidation reaction between 1,4,5,8-naphthalenetetracarboxylic acid dianhydride (NDA) and different primary amines. However, since Kelly et al. developed a series of NDI derivatives including N,N′-bis(4-pyridyl)-1,4,5,8-naphthalenetetracarboxydiimide (DPNDI) ligands,13,14 only a few complexes were reported with N-heterocyclic substitutional NDI ligands, while most other complexes were constructed with carboxylic NDI ligands. As is known, N-donor ligands present better affinity with low valence metal ions according to the hard and soft of acids and bases (HSAB) principle.15,16 The strong coordination effect enhances the difficulty of ligand dissociation and would increase the stability. However, N-heterocyclic rings are lacking electron donor capability. The lone pair in nitrogen atoms coordinated with a metal will further reduce this capability.17 Hence, introduction of auxiliary electron-rich ligands is required to constitute an electron donor–acceptor pair.Herein, we choose N,N′-bis(3-pyridyl)-1,4,5,8-naphthalenetetracarboxydiimide (m-DPNDI) as an electron acceptor and 4,4′-diphenic acid (H2bpdc) as an electron donor to construct two novel photochromic coordination polymers, [Zn2(bpdc)2(m-DPNDI)2]·H2O (1) and [Cd(bpdc)(m-DPNDI)]·H2O (2), respectively. The photoinduced electron transfer from donors to acceptors not only influences the absorption bands, but also influences the transition of fluorescence emission. Therefore, the compounds can present colour changes and photoresponsive luminescence behaviours. Furthermore, the interpenetrating network and strong π⋯π interaction in the structure provide high stability for compound 1. The switchable photoresponsive behavior in color and fluorescence gives both compounds good potential in molecular switches, information storage, anti-counterfeiting marks, etc.18
Experimental
Materials and methods
All reagents and solvents were of reagent grade quality and purchased from commercial sources without further purification.The FT-IR spectra were recorded using the powder samples on a Bruker TENSOR II FT-IR spectrophotometer in the 4000–400 cm−1 region. UV-vis absorption spectra were recorded in the reflectance mode at room temperature on a Persee TU1901 UV-vis spectrophotometer with an integrating sphere attachment and BaSO4 pellets as the background. C, H, and N elemental analyses were performed by using an Elementar Vario EL Cube analyzer. Powder X-ray diffraction (PXRD) patterns were collected on a Rigaku D/max-2500 diffractometer using Cu-Kα radiation (λ = 1.5406 Å) at room temperature. Variable temperature patterns were collected with a heating accessory in an Ar atmosphere. Thermogravimetric (TG) analysis was conducted on a NETZSCH TG209 analyzer with a ramp rate of 10 °C min−1 in an N2 atmosphere. The simulated PXRD patterns were derived from the free Mercury software. The electron paramagnetic resonance (EPR) spectra were recorded on a Bruker EMX-6/1 spectrometer with a 9.832 GHz magnetic field in the X band at room temperature. A 300 W xenon lamp (PLS-SXE 300C) system equipped with an infrared filter as the light source was used to illuminate the samples (c.a. 1.1 W cm−2 detected with a light intensity meter) for achieving various spectra, and the distance between these samples and the Xe lamp was 30 cm to reduce the photothermal effect.
Crystallography
Single crystal X-ray diffraction (SXCRD) data were collected on a computer controlled Rigaku XtalAB PRO MM007 DW diffractometer with graphite-monochromated Cu-Kα radiation with a radiation wavelength of 1.54184 Å. Lorentz polarization and absorption corrections were applied. The crystal structure data were solved by direct methods using the SHELXS program of the SHELXTL package and refined by full-matrix least-squares methods with SHELXL.19 The non-hydrogen atoms were located by successive difference Fourier syntheses and refined with anisotropic thermal parameters on F2. Hydrogen atoms were generated theoretically at specific atoms and refined isotropically with fixed thermal factors. The SQUEEZE method was conducted by Platon software to remove the residual electron density of free solvent molecules in the structure of 1.20 CCDC reference numbers 2026863 and 2026864 are for 1 and 2, respectively. The refinement details and selected bond lengths and angles are given in Tables 1 and S1,† respectively.| Complex | 1 | 2 |
|---|---|---|
| Empirical formula | C76H42N8O17Zn2 | C38H22CdN4O9 |
| Formula weight | 1469.96 | 790.99 |
| Temperature/K | 120.00(10) | 120.00(10) |
| Crystal system | Triclinic | Orthorhombic |
| Space group |
P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) |
Pbca |
| a/Å | 14.1670(2) | 18.6379(2) |
| b/Å | 14.5777(2) | 11.3482 (1) |
| c/Å | 17.0380(2) | 28.6405(3) |
| α/° | 89.140 (1) | 90 |
| β/° | 79.848 (1) | 90 |
| γ/° | 62.998(1) | 90 |
| Volume/Å3 | 3077.58(8) | 6057.66(11) |
| Z | 2 | 8 |
| D (g cm−3) | 1.586 | 1.735 |
| μ/mm−1 | 1.689 | 6.386 |
| F(000) | 1500 | 3184 |
| GOF on F2 | 1.041 | 1.073 |
| Final R indices [I ≥ 2σ(I)] | R 1 = 0.0391 | R 1 = 0.0259 |
| wR2 = 0.1126 | wR2 = 0.0684 | |
| Final R indices [all data] | R 1 = 0.0412 | R 1 = 0.0277 |
| wR2 = 0.1144 | wR2 = 0.0694 |
Synthesis
Phase purity of the bulk samples of crystalline compounds 1 and 2 was checked by PXRD measurements (Fig. S1†). FT-IR spectra of both compounds are given in Fig. S2.†
Results and discussion
Description of the crystal structure
SCXRD analysis revealed that complex 1 crystallizes in the triclinic space group P (no. 2). The asymmetric unit includes two zinc(II) cations, two bpdc2− ions, two m-DPNDI molecules and a free water molecule (Fig. 1a). Each Zn1 centre is four coordinated with two pyridyl N atoms (N7 and N8) of different m-DPNDI ligands and two carboxylic O atoms (O4 and O5) of two bpdc2− ligands. Therein, the coordination bond lengths of both Zn⋯O bonds are nearly 1.95 Å while that of Zn⋯N bonds are a bit longer (Zn1⋯N7, 2.05 Å; Zn1⋯N8, 2.04 Å), which results in a distorted tetrahedral [ZnN2O2] geometry. Another crystallographic independent zinc cation (Zn2) is in a similar [ZnN2O2] coordination environment to the Zn1 cation and only differs slightly in bond length and angles (Table S1†). Both m-DPNDI and bpdc2− ligands crosslink the neighbouring zinc cations to form a hexagonal ring at the bc plane (Fig. 1b). Each ring fuses with other six rings by sharing the edges of the ligands and vertices of the metal centres and construct a 2D hcb network. Each 2D layer connects with another layer (Fig. 1c). Two m-DPNDI ligands link two neighbouring zinc cations and leave a box at the ab plane, which is penetrated by bpdc2− of another 2D layer according to the π⋯π interaction between aromatic rings of bpdc2− and two NDI parts (Fig. 1d). The distances of those two interactions are 3.59 Å and 3.61 Å, respectively. The two interpenetrating layers can be considered as a set, which packs with other sets in a staggered arrangement through the π⋯π stacking (3.52 Å) at the a-axis (Fig. 1e). The sets pack in ABAB form and finally result in a 2-fold interpenetrating hcb topology in space (Fig. 1f).
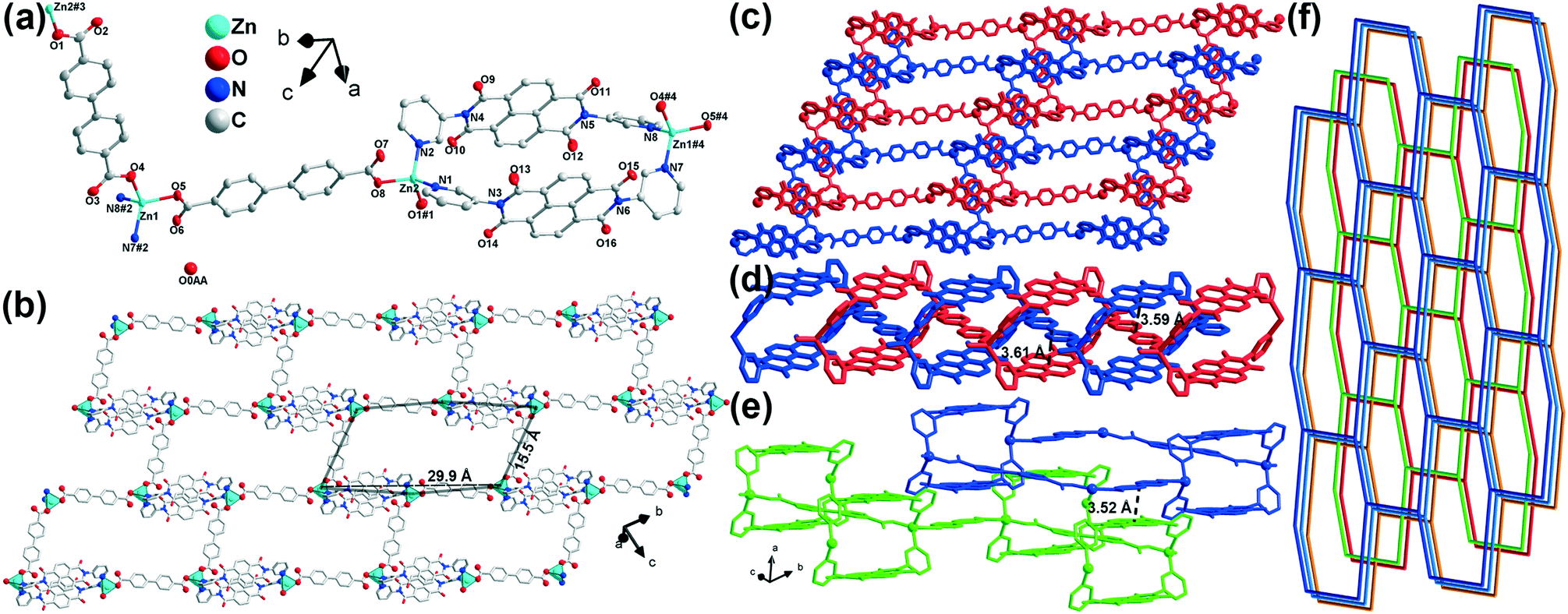 | ||
| Fig. 1 Structure of 1. H atoms are omitted for clarity. (a) Coordination environments of the metal ion. ORTEP diagrams were drawn at the 50% level. Symmetry code: #1 x + 1, y − 2, z; #2 x − 1, y + 2, z + 1; #3 x − 1, y + 2, z; #4 x + 1, y − 2, z − 1. (b) 2D layer structure. (c) 2-Fold interpenetrating networks. (d and e) π⋯π interactions between bpdc2− and m-DPNDI in interpenetrating layers and two neighbouring layers, respectively. (f) Topology of 1. | ||
Compound 2 crystallizes in the orthorhombic space group Pbca (no. 61). The structure solution of diffraction data indicates the overall 1D-chain structure of 2. Each asymmetric unit contains a cadmium(II) cation, an m-DPNDI molecule, a coordinated water molecule and a bpdc2− ligand (Fig. 2a). The Cd centre lies in a seven-coordinated pentagonal bipyramid [CdN2O5] coordination geometry. Therein, four O atoms (O6, O7, O8, and O9) from bpdc2− and an aqua O atom (O5) occupy the coordination sites at the equatorial plane while pyridyl N atoms of m-DPNDI occupy the vertices. Four coordination bond lengths of Cd⋯O (carboxylic) range roughly from 2.4 Å to 2.5 Å and the other three are all nearly 2.3 Å (Table S1†). Bpdc2− and m-DPNDI serve as crosslinkers and connect neighbouring cations in the same direction, which extends at the c axis to construct a zig-zag 1D chain structure (Fig. 2b). Two adjacent chains pack in space through a strong offset face-to-face π⋯π interaction between bpdc2− and m-DPNDI from the respective chain with a distance of 3.39 Å (Fig. 2c).
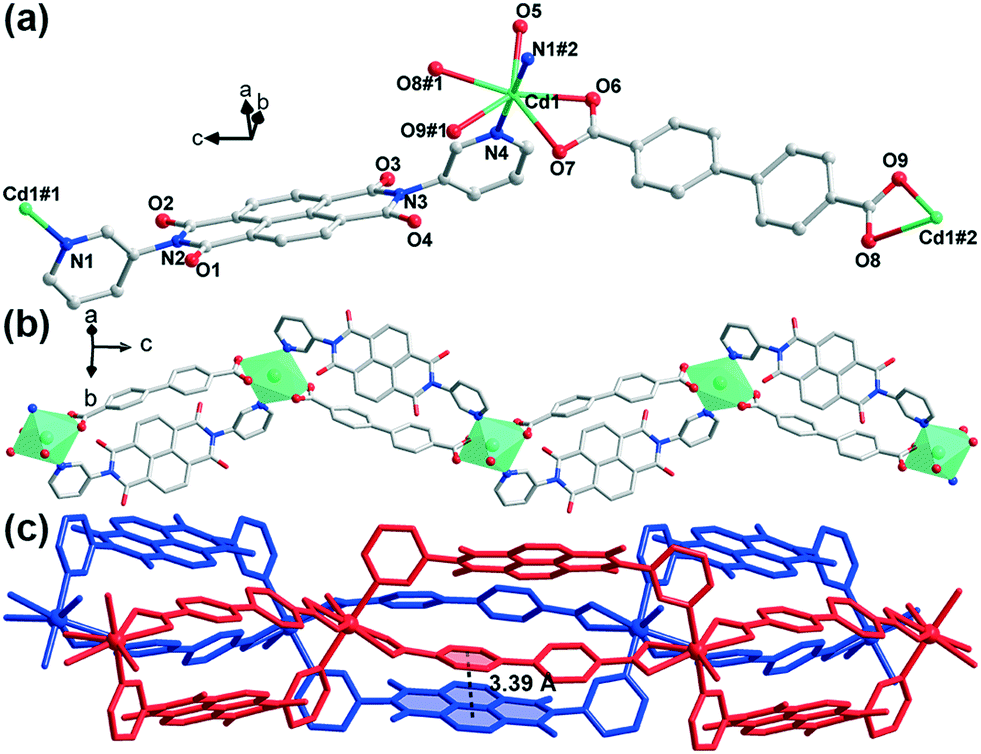 | ||
| Fig. 2 Structure of 2. H atoms are omitted for clarity. (a) Coordination environments of the metal ion. ORTEP diagrams were drawn at the 50% level. Symmetry code: #1 −x + 3/2, −y + 1, z + 1/2; #2 −x + 3/2, −y + 1, z − 1/2. (b) 1D chain structure. (c) π⋯π interactions between bpdc2− and m-DPNDI. | ||
Thermostability
The 2D framework of compound 1 presents an excellent thermal stability. TG analysis of compound 1 after solvent exchange with CH2Cl2 reveals that there is almost no weight loss before 460 °C (Fig. S3†). Upon further heating to 530 °C, a weight loss of 33.9% was observed, indicating that the organic ligands broke down rapidly which can be attributed to the thermolysis of bpdc2− (exp. value: 33.0%). As the temperature further increased, m-DPNDI gradually decomposed. Variable temperature PXRD studies were conducted to further verify its thermal stability.‡ The solvent peak at the (−4 −1 2) crystal plane disappeared at above 100 °C, which corresponds to the vaporization temperature of water (Fig. 3). The main peak at 25.4° belongs to the (4 2 0) crystal plane, which is assigned to bpdc2− aromatic rings (Fig. S4†). With the increase of temperature, this peak shifts to a smaller degree and reaches 25.1° at 460 °C, which indicates larger distances of π⋯π interaction between bpdc2− and m-DPNDI rings. No other obvious variation was found in the XRD patterns of 1 until 460 °C, which indicates that the skeleton of 1 can remain intact below this temperature. The 2D framework collapsed gradually upon further heating and no peak of 1 was found at 500 °C. The result of variable temperature PXRD is quite consistent with that of TG analysis and both measurements confirm the relatively good thermostability of 1. As a kind of π acid, m-DPNDI have strong affinity with electron-rich bpdc2− rings.12 The abundant strong π⋯π interactions between these aromatic components in the crystal structure would be responsible for the high thermostability of 1. Furthermore, the interpenetrating structure in 1 also makes contributions to the integrity of the whole framework.16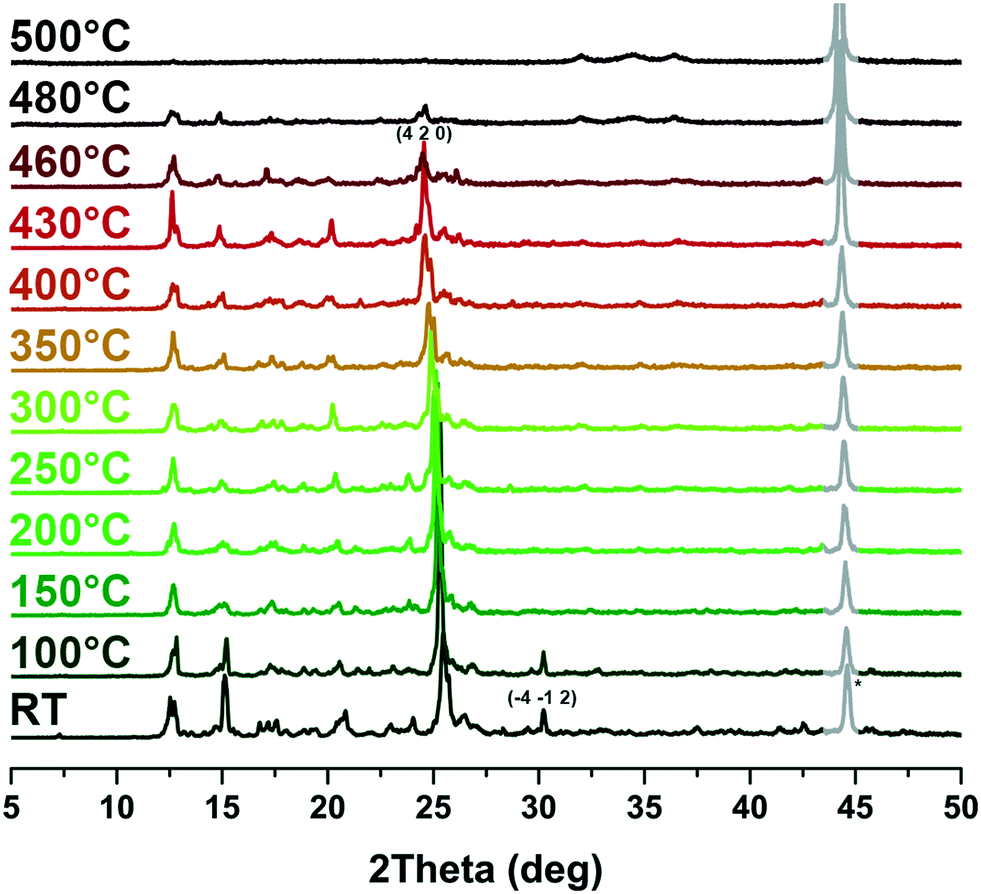 | ||
| Fig. 3 Variable temperature PXRD patterns of 1. The irremovable peak (*) of the background is expressed in gray lines. | ||
TG analysis of compound 2 was also carried out. Due to the 1D structure of 2, solvent exchange was not conducted. A continuous weight loss (9.9%) until 170 °C was first observed, which was caused by coordinated water and unsolved free solvent molecules (DMF, H2O) in the lattice. Other components started pyrolysis at 285 °C.
Photochromism
Both compounds can present photochromic behaviours. As shown in Fig. 4a, compound 1 shows strong absorption before 400 nm, which can be assigned to the superposition of π → π* and n → π* transitions of the bpdc2− ligands and m-DPNDI ligands, which is consistent with the results reported for other NDI based compounds.22 The weak absorbance at 400–500 nm arises from the π⋯π stacking of chromophores in m-DPNDI, which is clearly found in the crystal structure. In comparison with the absorption spectra of the irradiated samples, a small amount of molecule irradiated by ambient light would be responsible for the other absorption bands. After irradiation with a xenon lamp, the absorbance of compound 1 increased at the full range measured and five new absorption bands at 350, 511, 615, 740 and 821 nm were found by comparing the spectra before and after irradiation (Fig. S5†). Meanwhile, its colour changed from yellow to olive green. The sample colour can be recovered in a dark environment for a day or by heating at 100 °C in air for 30 minutes. Cycle experiments for compound 1 were carried out by monitoring the absorbance at 615 nm of the same samples. The result shows the good reversibility of its photochromic behaviours and can be recycled at least 7 times (Fig. 4c).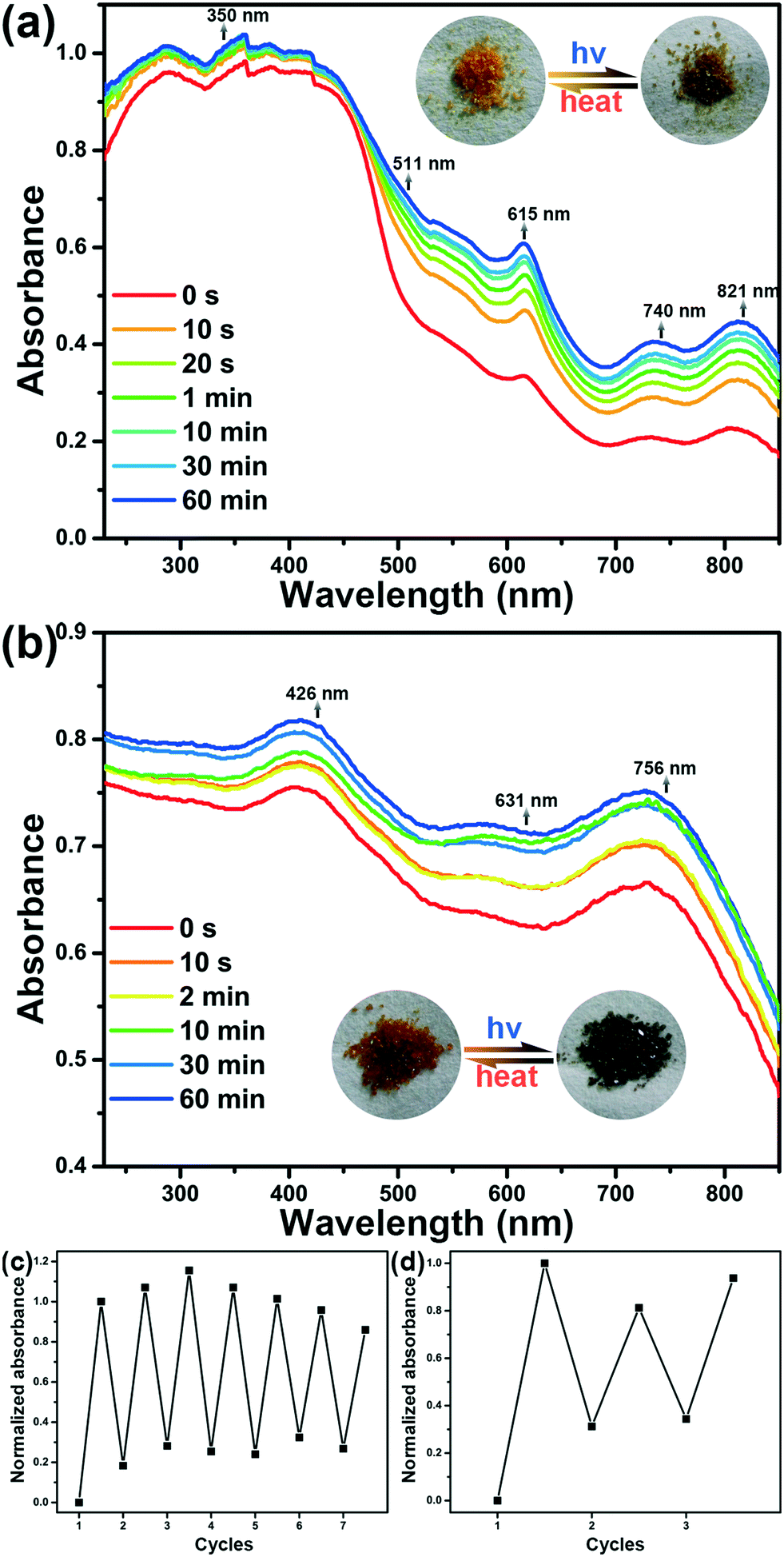 | ||
| Fig. 4 Time-dependent UV-vis diffuse reflectance spectra of 1 (a) and 2 (b) upon UV-visible light irradiation. Cycle experiment results of compounds 1 (c) and 2 (d). | ||
Irradiation on the orange crystals of 2 will change their colour to dark green and can be reverted to their original colour by annealing the irradiated samples at 100 °C for 45 minutes or placing them in the dark for a day. The increased absorption of compound 2 at full ranges was observed after irradiation, which is consistent with the colour changes detected with the naked eye (Fig. 4b). Three new absorption bands at 426, 631, and 756 nm were found, respectively. The colour change of 2 can be recycled for at least 3 times by monitoring absorbance at 756 nm (Fig. 4d).
Photoresponsive luminescence
The emission spectra of compounds 1 and 2 are shown in Fig. 5. Fluorescence intensity of both compounds can be affected by light irradiation. Before irradiation, similar broad structureless emission bands were found centred at 450 and 445 nm for compounds 1 and 2, respectively, under an excitation of 223 nm. The broad emission bands are characteristic of ligand-to-metal charge transfer (LMCT) transition, indicating that the emission in both compounds probably raised from the transition between the pyridyl groups of m-DPNDI and metal cations.23 With the increase of irradiation time, with a xenon lamp, the fluorescence intensity of the emission decreased gradually and finally reached 79% and 70% of their original intensity for compounds 1 and 2, respectively.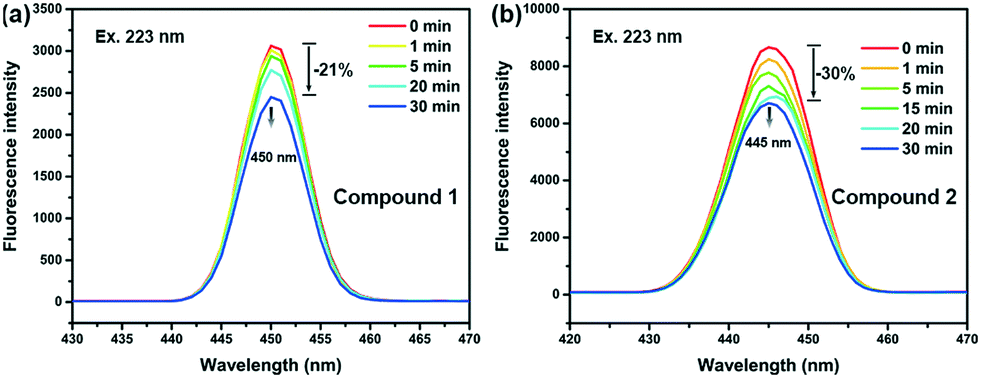 | ||
| Fig. 5 Time-dependent fluorescence spectra of 1 (a) and 2 (b) upon UV-visible light irradiation. | ||
Mechanism of photoresponsive behaviour
NDI derivatives are famous for their photochromic properties due to the strong ability of accepting and stabilizing electrons. However, the ligand m-DPNDI can hardly change its colour upon irradiation. The substitutional group of pyridines in m-DPNDI is not an ideal electron donor. The electron withdrawing effect of pyridyl N greatly reduced the ability of accepting electrons of the aromatic rings. Hence, almost no colour change can be detected when illuminating m-DPNDI ligands.13 The introduction of electron-rich carboxylate ligands will complete this electron transfer process and enhance the photochromic behaviour. The variation trend in the UV-vis absorption spectra was similar to some other NDI based photochromic compounds, which were derived by a photoinduced electron transfer (PET) process.24 Unlike other photochromic species with significant structural variation between two states upon irradiation such as azo-compounds, spiropyrane and diarylethene, photochromism in the PET mechanism occurs only when there is a difference in electron configurations via electron transfer from the donor's HOMO to the acceptor's LUMO, which formed charge separated (CS) states.25 In particular, the new absorption bands at around 610 nm are typical characteristic of NDI˙− radicals.26 Hence, the photoresponsive behaviour in colour and luminescence of both compounds was probably caused by PET.To further investigate the mechanism of the photoresponsive behaviour, we firstly excluded photolysis or rearrangement reactions of both compounds during irradiation according to the results of PXRD (Fig. S1†) and FT-IR (Fig. S2†) experiments. No obvious variation was found between the samples before and after irradiation. Then, EPR measurements were performed to affirm the existence of NDI˙− radicals. As shown in Fig. 6a, a weak signal at g = 2.0021 was found for the unirradiated samples of 1, which is very close to the value of free electrons (g = 2.0023). The original signal found in the EPR spectra indicates that there is a small number of radicals, which are properly generated from the stimulation of ambient light. After irradiation, the intensity of this peak greatly increased and shifted to g = 2.0005 with a width of 30 Gauss, suggesting that many radicals were generated during this process. Similar results were also found in the EPR spectra of 2 (Fig. 6b). As mentioned above in the crystal structure, abundant π⋯π interactions between the bdpc2− and m-DPNDI ligands exist in both compounds, which is an ideal electron transfer pathway for the PET reaction. Upon irradiation, electrons transfer from electron-rich units (bpdc2−) to electron-deficient units (m-DPNDI) and are stabilized by NDI rings forming long-lived charge separated states.27 Obviously, some excitons jumped to metastable CS states rather ground states directly in this process, while the latter one corresponds to the transition for luminescence. Hence, besides difference in colour between ground states and CS states, the fluorescence intensity is also affected.28,29
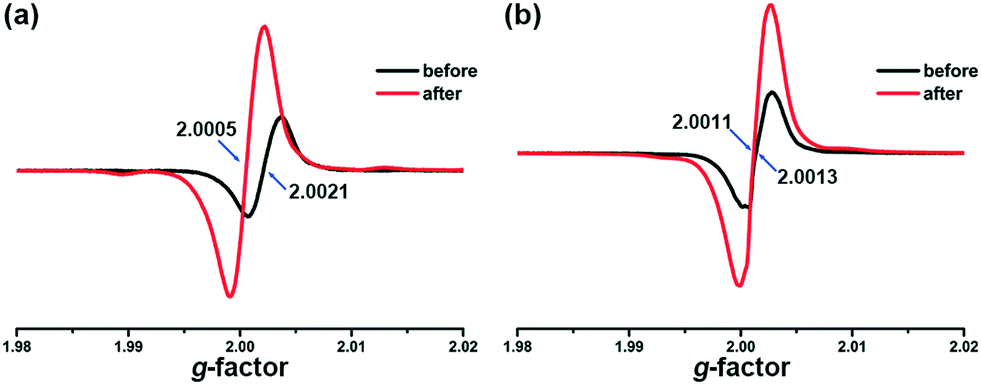 | ||
| Fig. 6 EPR spectra of 1 (a) and 2 (b) before and after irradiation. | ||
Conclusions
In summary, we successfully isolated two coordination polymers based on NDI ligands. The combination of N-donor ligands and carboxylic ligands constitutes a complete electron donor and acceptor pair. A photoinduced electron transfer process can occur in both compounds upon irradiation and was confirmed by PXRD, IR and EPR measurements. Abundant and strong π⋯π interactions between the aromatic rings of the donor and acceptors in the structure provide a suitable electron transfer pathway. The photoresponsive behaviours of both are triggered by the PET process. Upon irradiation, the colour of compounds 1 and 2 changed and the fluorescence intensity reduced. Besides, the existence of interpenetrating networks and abundant π⋯π interactions in 1 gives it excellent thermostability. Both compounds are promising candidates for the design in molecular switches, information storage and anti-counterfeiting.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We are grateful to Prof. Yang-Ping Liu and Master Kai-Yun Ji from Tianjin Medical University who supplied the EPR spectrometer and conducted EPR measurements. This work was supported by the National Natural Science Foundation of China (No. 21771111, 21601092, 21371104, 21471084 and 21801137) and the MOE Innovation Team (IRT13022) of China.Notes and references
- M. Irie, Chem. Rev., 2000, 100, 1685–1716 CrossRef CAS.
- J. Wang, S. L. Li and X. M. Zhang, ACS Appl. Mater. Interfaces, 2016, 8, 24862–24869 CrossRef CAS.
- C. Sun, G. Xu, X. M. Jiang, G. E. Wang, P. Y. Guo, M. S. Wang and G. C. Guo, J. Am. Chem. Soc., 2018, 140, 2805–2811 CrossRef CAS.
- H. Tian and S. Wang, Chem. Commun., 2007, 781–792, 10.1039/b610004j.
- H. Y. Li, H. Xu, S. Q. Zang and T. C. Mak, Chem. Commun., 2016, 52, 525–528 RSC.
- Y. J. Ma, J. X. Hu, S. D. Han, J. Pan, J. H. Li and G. M. Wang, J. Am. Chem. Soc., 2020, 142, 2682–2689 CrossRef CAS.
- O. Sato, Nat. Chem., 2016, 8, 644–656 CrossRef CAS.
- D. Aguila, Y. Prado, E. S. Koumousi, C. Mathoniere and R. Clerac, Chem. Soc. Rev., 2016, 45, 203–224 RSC.
- A. J. McConnell, C. S. Wood, P. P. Neelakandan and J. R. Nitschke, Chem. Rev., 2015, 115, 7729–7793 CrossRef CAS.
- H. Cheng, J. Yoon and H. Tian, Coord. Chem. Rev., 2018, 372, 66–84 CrossRef CAS.
- F. M. Raymo and M. Tomasulo, Chem. Soc. Rev., 2005, 34, 327–336 RSC.
- M. A. Kobaisi, S. V. Bhosale, K. Latham, A. M. Raynor and S. V. Bhosale, Chem. Rev., 2016, 116, 11685–11796 CrossRef CAS.
- S. Guha, F. S. Goodson, L. J. Corson and S. Saha, J. Am. Chem. Soc., 2012, 134, 13679–13691 CrossRef CAS.
- J. E. Rogers and L. A. Kelly, J. Am. Chem. Soc., 1999, 121, 3854–3861 CrossRef CAS.
- M. Ding, X. Cai and H. L. Jiang, Chem. Sci., 2019, 10, 10209–10230 RSC.
- A. J. Howarth, Y. Liu, P. Li, Z. Li, T. C. Wang, J. T. Hupp and O. K. Farha, Nat. Rev. Mater., 2016, 1, 1–15 Search PubMed.
- C. J. Zhang, Z. W. Chen, R. G. Lin, M. J. Zhang, P. X. Li, M. S. Wang and G. C. Guo, Inorg. Chem., 2014, 53, 847–851 CrossRef CAS.
- J. Z. Liao, H. L. Zhang, S. S. Wang, J. P. Yong, X. Y. Wu, R. Yu and C. Z. Lu, Inorg. Chem., 2015, 54, 4345–4350 CrossRef CAS.
- G. M. Sheldrick, Acta Crystallogr., Sect. C: Struct. Chem., 2015, 71, 3–8 Search PubMed.
- A. L. Spek, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2009, 65, 148–155 CrossRef CAS.
- G. Koshkakaryan, L. M. Klivansky, D. Cao, M. Snauko, S. J. Teat, J. O. Struppe and Y. Liu, J. Am. Chem. Soc., 2009, 131, 2078–2079 CrossRef CAS.
- C. Fu, G.-S. Zhang, H.-Y. Wang, L. Li, J.-W. Fu, Y.-N. Sun and H. Zhang, CrystEngComm, 2018, 20, 6821–6827 RSC.
- X. H. Jin, J. K. Sun, L. X. Cai and J. Zhang, Chem. Commun., 2011, 47, 2667–2669 RSC.
- Y. X. Xie, W. N. Zhao, G. C. Li, P. F. Liu and L. Han, Inorg. Chem., 2016, 55, 549–551 CrossRef CAS.
- S. Guha, F. S. Goodson, S. Roy, L. J. Corson, C. A. Gravenmier and S. Saha, J. Am. Chem. Soc., 2011, 133, 15256–15259 CrossRef CAS.
- M. Pan, X.-M. Lin, G.-B. Li and C.-Y. Su, Coord. Chem. Rev., 2011, 255, 1921–1936 CrossRef CAS.
- M. S. Wang, G. Xu, Z. J. Zhang and G. C. Guo, Chem. Commun., 2010, 46, 361–376 RSC.
- J. K. Sun and J. Zhang, Dalton Trans., 2015, 44, 19041–19055 RSC.
- C. Chen, X.-H. Jin, X.-J. Zhou, L.-X. Cai, Y.-J. Zhang and J. Zhang, J. Mater. Chem. C, 2015, 3, 4563–4569 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available: CCDC 2026863 and 2026864. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d0ce01317j |
| ‡ The peak around 44° in the variable temperature PXRD patterns is a background peak of the sample sink. Due to the air stream and thermal expansion of the samples during the test, the sample sink would be exposed to X-ray more or less in an uncontrollable way. This residual noise signal is difficult to remove in a reasonable way. |
| This journal is © The Royal Society of Chemistry 2021 |