
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Shape controlled iron oxide nanoparticles: inducing branching and controlling particle crystallinity†
Nur Hanisah
AbuTalib‡
a,
Alec P.
LaGrow‡
 *b,
Maximilian O.
Besenhard
c,
Oleksandr
Bondarchuk
b,
Andreas
Sergides
a,
Simone
Famiani
a,
Liliana P.
Ferreira
de,
M. Margarida
Cruz
e,
Asterios
Gavriilidis
c and
Nguyen Thi Kim
Thanh
*a
*b,
Maximilian O.
Besenhard
c,
Oleksandr
Bondarchuk
b,
Andreas
Sergides
a,
Simone
Famiani
a,
Liliana P.
Ferreira
de,
M. Margarida
Cruz
e,
Asterios
Gavriilidis
c and
Nguyen Thi Kim
Thanh
*a
aBiophysics Group, Department of Physics and Astronomy, University College London, Gower Street, London, UK and UCL Healthcare Biomagnetic and Nanomaterials Laboratories, 21 Albemarle Street, London W1S 4BS, UK. E-mail: ntk.thanh@ucl.ac.uk
bInternational Iberian Nanotechnology Laboratory, Braga, 4715-330, Portugal. E-mail: alec.lagrow@inl.int
cDepartment of Chemical Engineering, University College London, Torrington Place, London, WC1E 7JE, UK
dPhysics Department, University of Coimbra, 3004-516 Coimbra, Portugal
eBioISI-Biosystems and Integrative Sciences Institute, Faculdade de Ciências, Universidade de Lisboa, Campo Grande, 1749-016 Lisboa, Portugal
First published on 16th December 2020
Abstract
Anisotropic nanoparticles (NPs) have garnered a great deal of attention for their applications in catalysis, magnetism and biomedicine. However, synthetic strategies to grow such NPs are still limited as their growth mechanisms are poorly understood. This work presents the synthesis of iron oxide nanoparticles (IONPs) based on the decomposition of iron(III) acetylacetonate in organic solvents to form anisotropic IONPs that are branched or multiply branched. We fully explore their growth parameters to understand the effect of varying amounts of oleylamine (OAm), as well as a nitrogen purge on particle morphology. We show here the synthetic relationship between a wide range of sizes and shapes of IONPs that are both isotropic and anisotropic. Of all the parameters, the amount of oleylamine in the reaction is the key to tune the particle size while the effect of a nitrogen gas purge during synthesis was shown to be crucial for the formation of the branched and multiply branched NPs. Two multiply branched NP systems with only a small difference in the synthetic conditions were shown to have radically different magnetic properties, such as heating in an alternating magnetic field. This was attributed to the defects found in the structure of one and not in the other. By following their development during growth, crystal defects were observed in both systems during the early stages of the reaction. However, for the multiply branched structure that became single crystalline, the aggregation of the nuclei occurred earlier in the reaction, allowing more time for growth and crystallite rearrangement to occur. These results have wide ranging implications for controlling the properties of anisotropic nanomaterials with similar structures, including their magnetic behavior.
Introduction
Nanomaterials are now being used in a wide range of applications, and it is becoming increasingly obvious that control over the morphology of a nanomaterial is imperative for many applications.1,2 A class of nanomaterials which have seen increased interest is branched or clustered NPs.1 They have shown increased activity in catalysis due to the higher proportion of high index facets exposed.1,3–5 Another example is in biomedical applications where multi-core magnetic NPs, often called nanoflowers, have been shown to be ideal candidates for magnetic hyperthermia.6–10 For magnetic hyperthermia, the key characteristics making these shapes ideal is thought to be their magnetic anisotropy and core–core interactions, with the best properties coming from having multiple aligned crystalline domains.6Several mechanisms have been reported in the formation of anisotropic NPs, these include controlled aggregation, etching and branching. Etching typically occurs either during the growth or via a post-preparative step to remove material selectively from faceted surfaces and rearrange the particle into a multiply branched structure.5,11 In aggregative growth mechanisms the final particles can be made up of aligned6 or unaligned12 crystal domains. The alignment of the crystal domains to form a single crystal occurs via aggregation in solution called oriented attachment where the facets of growing particles align in solution.13–16 Anisotropic structures can also grow in solution where branches grow from a central core, this can lead to the formation of particles ranging from short rods up to multiply branched particles.1 The branches are grown off a central core, either from the introduction of a seed particle17 or via self-seeding.4,17–20 These branches are typically formed in twinned particles from symmetry breaking events,19–21 or via polymorphism where arms of an hcp polymorph are grown from seeds of an fcc polymorph.21,22
For IONPs, there are several synthetic strategies to form anisotropic morphologies, in particular nanoflowers have been formed via water based co-precipitation pathways,23 and the thermal decomposition synthesis in polyol solvents.24,25 For thermal decomposition synthesis of branched NPs in other (i.e., non-polar) organic solvents, there is one study on tetrapods.26 However, control over synthesis of anisotropic, branched or nanoflowers of iron oxide, including their total size and crystallinity, is still limited due to a lack of understanding over the growth mechanism at play to form these unique shapes.
Here we illustrate a high temperature synthesis expanding on the work of Sun et al.27 forming anisotropic NPs with varying levels of branching, as well as their isotropic counterparts by varying the amount of OAm, and purging nitrogen during synthesis. Of particular interest were branched, and multiply branched NPs which were characterized fully for their growth mechanisms and potential for magnetic hyperthermia. The two multiply branched systems explored were shown to have a similar morphology but very different magnetic properties which were attributed to their growth mechanism where one rearranged to form a single crystalline branched structure and the other did not.
Experimental
The IONPs were synthesized by the decomposition of iron(III) acetylacetonate [Fe(acac)3] in a mixture of OAm, oleic acid, 1,2-hexadecanediol (HDD), benzaldehyde and 1-octadecene at 280 °C with purging nitrogen (Fig. 1A). The mixture of benzaldehyde and 1-octadecene is used as a solvent, instead of benzyl ether to increase the reproducibility of the synthesis as reported by Qiao et al.28 From our experiments, the use of the benzaldehyde/ 1-octadecene solvent mixture gave the same results as what was seen with benzyl ether alone, while also showing that the benzylether/ benzaldehyde was a key component in forming anisotropic nanostructures (Fig. S1†). The precursor solution was composed of 0.35 g Fe(acac)3, made up with between 0 and 40 equivalents of OAm molecules to the metal precursor (eq. OAm). The other surfactants, oleic acid and HDD, equivalents to the metal precursor concentration were typically kept at 9 eq. and 4.5 eq. respectively, unless otherwise stated. The precursor solution was heated to ∼120 °C with a nitrogen purge and held there for 30 min to remove oxygen and water. Then the solution was rapidly heated to 280 °C at a rate of ∼9 °C min−1, with a vigorous purge of nitrogen (∼0.5 mL min−1). The sample was kept at 280 °C for 30 min before cooling to below 80 °C in ∼45 min. The reactions with the nitrogen purge are the standard configuration used (Fig. 1A), but experiments were also carried out under a blanket of nitrogen, in these cases the nitrogen purge was used at the start of the reaction to degas the system at ∼120 °C and then the system was put under a nitrogen blanket (Fig. 1B) before the reaction was heated from ∼120 °C to 280 °C.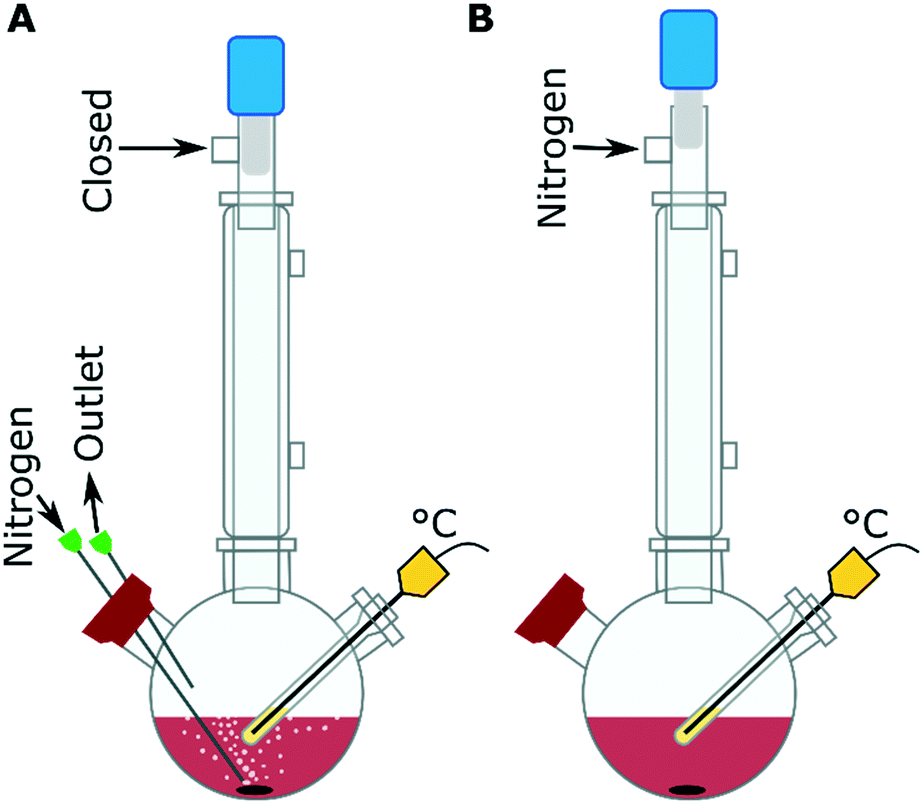 | ||
| Fig. 1 Schematic of the reaction set up showing a three neck flask with a stir bar in it, a condenser and a temperature probe in the right arm, with A) the nitrogen purge set up where a needle allowing the nitrogen in is run through the left arm into the solution and out through an outlet needle from the same arm. B) The nitrogen blanket set up, where the nitrogen is attached to the top of the flask allowing for a nitrogen blanket that can be replaced from the nitrogen manifold which is attached to the top of the condenser. | ||
Results and discussion
Synthesis of anisotropic and isotropic nanoparticles
Varying the OAm to precursor ratio from 0 to 40 eq. of OAm to the metal precursor showed a clear and significant change in shape anisotropy as seen in Fig. 2. The total size refers to the longest continuous diameter across the particles to allow comparison between the varied shapes formed, and the branch width was taken from the diameter of a branch. The measurement error given is the standard deviation of the particle size distribution. With a large excess of OAm (>40 eq.) almost spherical particles of 6 ± 1 nm are formed (Fig. 2A). For lower equivalencies, 25–19 eq. OAm branched particles (Fig. 2B and C, Table S1 and Fig. S2†) are formed with typically between 1 and 4 arms, with 25 eq. have a total size of 13 ± 3 nm and a branch width of 5.3 ± 0.7 nm (Fig. 2B) and 21 eq. being slightly larger with a size of 14 ± 3 nm and a branch width of 5.6 ± 0.8 nm (Fig. 2C). At 17 eq. multiply branched particles are formed having a total size of 21 ± 4 nm with branch width of 6.6 ± 0.9 nm (Fig. 2D). With decreasing amounts of OAm, the branched particles get thicker and the core larger. At 15 eq. OAm the multiply branched particles have a total size of 17 ± 3 nm and a branch width of 7 ± 1 nm (Fig. 2E). At 13 eq. OAm the branched structure becomes less obvious as the total size increases to 29 ± 4 nm with a branch width of 14 ± 3 nm (Fig. 2F). When the amount of OAm is decreased to 10 eq. the particles still have a degree of anisotropy and elongation and have a total size of 17 ± 3 nm with a ‘branch’ width of 12 ± 2 nm. Finally, faceted octahedral particles were formed at 5 eq. OAm (Fig. 2H). At 0 eq. OAm, NPs were not formed. Fe2p XPS spectra show main peaks at ∼711 and ∼725 eV corresponding to Fe2p3/2 and Fe2p1/2 respectively, and a small peak at ∼719 eV which is the shake-up satellite (Fig. S3 and S4, Tables S2 and S3†). The peak positions of the Fe2p3/2 and in particular the satellite peak are characteristic of Fe3+.29 An Fe2+ contribution is often observed when the satellite is less obviously present, and can also be inferred from the ratio of the iron to oxygen bound to a metal (Omet). The XPS O1s spectra showing a complex structure were fitted with up to three Lorentzian–Gaussian curves (Fig. S3†). The O1s component (Omet) centred at ∼530 eV is assigned to the oxygen–metal bonds. The molar ratio of Fe/Omet ranges from 0.5 (5 eq. OAm) to 0.72 (17 eq. OAm) as shown in Table S2,† Fe3O4 and Fe2O3 have ratios of 0.75 and 0.66 respectively, whereas the lower ratio for 5 eq. OAm is attributed to unreacted Fe(acac)3 which is difficult to remove via cleaning steps. | ||
| Fig. 2 TEM images of IONPs made with a constant nitrogen purge with A) 40 eq., B) 25 eq., C) 21 eq., D) 17 eq., E) 15 eq., F) 13 eq., G) 10 eq., and H) 5 eq. OAm to the metal precursor [Fe(acac)3]. | ||
The effect of the nitrogen purge was investigated. Instead of having a constant purge through the system, the reaction was held under a blanket of nitrogen after purging the system with nitrogen at 120 °C. Applying the nitrogen blanket system to the OAm series, the shape of the NPs are more isotropic than that with the nitrogen purge, forming spheres and cubes (Fig. 3). The NPs formed with 23 eq. OAm are 7 ± 1 nm and faceted (Fig. 3A and Table S4†). With 17 eq. OAm the particles increased to 8 ± 2 nm (Fig. 3B). With 15 eq. OAm the NPs were 10 ± 3 nm in size with ∼50% of faceted cubic particles formed (Fig. 3C). The particle size increases as the equivalents of OAm is decreased, until it reaches a maximum of 24 ± 5 nm at 13 eq. OAm (Fig. 3D). At 10 eq. the particle size decreases to 14 ± 2 nm and are highly faceted (Fig. 3E). At 5 eq. OAm the particles are 6.6 ± 0.8 nm in size (Fig. 3F). XPS of the particles are similar to that of the particles synthesized with the nitrogen purge, with the peaks in the Fe2p spectra being typical for Fe3+ and Fe![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :Omet ratios ranging from 0.7 for 23 eq. to 0.5 for 5 eq. OAm (Fig. S5 and S6, Tables S5 and S6†).
:Omet ratios ranging from 0.7 for 23 eq. to 0.5 for 5 eq. OAm (Fig. S5 and S6, Tables S5 and S6†).
 | ||
| Fig. 3 TEM images of IONPs made under a positive nitrogen pressure with A) 23 eq., B) 17 eq., C) 15 eq., D) 13 eq., E) 10 eq., and F) 5 eq. OAm to the metal precursor [Fe(acac)3]. | ||
Further experiments showed that to form the branched particles the nitrogen purge must be bubbling through the solution. With nitrogen flowing over the solution (∼0.5 mL min−1) the particles are anisotropic in shape, with bipod formation and some that are branched (Fig. S7A†). An increased flow rate of the nitrogen to 0.6 mL min−1 through the solution was tested by increasing the diameter of the needles used for the entry and exit of the nitrogen into the solution, this did not make an appreciable difference to the NPs size or shape compared to the particles synthesized under the typical flowing nitrogen configuration (Fig. S7B†).
To further characterize the system with the nitrogen purge, the effect of other reaction conditions were investigated. Increasing the concentration of the Fe(acac)3 by up to 50% more, while keeping all other parameters the same (Fig. S8†) showed a similar trend as seen when decreasing the OAm (Fig. 2), going from branched to multiply branched particles. Increasing the concentration of the iron precursor and the surfactants together, by up to 50% more, showed no change in the shape of the particles formed (Fig. S9†). Decreasing the amount of oleic acid from 9 eq. down to 0 eq. led to smaller more spherical particles (Fig. S10†). Finally, the effect of the diol surfactant was also investigated by altering its amount in the reaction mixture. Increasing the amount of HDD to 7 eq. formed small and slightly elongated NPs (Fig. S11†). While decreasing the amount of HDD, increased the particle size and maintained the shape for 23 eq. OAm and 15 eq. OAm, until controlled particles were not observed for 15 eq. OAm (Fig. S12 and Table S7†). At 23 eq. OAm and 0 eq. HDD, 33 nm octapods of FeO/FexOy were formed (Fig. S12 and Fig. S13†), similar to reports of octapods formed in the literature.30 For 5 eq. OAm smaller particles were formed with lower amounts of HDD until no particles were formed at all.
The effect of the surfactants in this system can be considered in two parts, the first is as reducing agents facilitating the decomposition of Fe(acac)3. OAm is well documented to work as a reducing agent,31 HDD has also been reported to be a reducing agent32 and benzaldehyde (replacing the early benzyl ether) has been reported to facilitate the reduction of Fe3+ to Fe2+.28 Previous reports have shown that thermal decomposition occurs via the reduction of Fe3+ to Fe2+ through a radical reaction between the Fe3+ center and the carboxylate ligand33 (in this case it would be either the acetylacetonate ligand or the oleic acid). Other possible reductants could come from the H2, CO and carbon formed during the breakdown of the precursor.34 Also in the reaction, decomposition of the precursor can be facilitated by radicals that occur at high temperatures during break down of the precursors.34 Under these reaction conditions, the large variation in the amount of OAm would cause a change in the reduction rate observed of the Fe(acac)3. This would also be true for the HDD, where in reactions with low amounts of OAm (5 eq.) and without HDD particles were observed only in negligible amounts indicating that only limited reduction/decomposition of the precursor was occurring.
The other effect of the role of surfactants in this synthesis is discussed with regards to the categories of surfactants described as nucleating agents and ripening agents.35,36 To summarize, the nucleating agent promotes the generation of nuclei through strong attachments on the surface of the NP thus stabilizing the nuclei. This lowers the barrier to nucleation and affects the survival probability of the nuclei. The ripening agent, on the other hand, stabilizes the monomers in solution, thus limiting the number of nuclei formed and leads to the formation of larger nuclei. Different surfactant behaviour shows variation over the control of the rate of nucleation of nanocrystals and the solubility of the surfactant monomer complex.
We hypothesize that the effect of OAm in the system with a nitrogen blanket atmosphere is to promote the decomposition of Fe(acac)3 for nanoparticle formation. Below 13 eq., it is hypothesized that there is not enough OAm to facilitate the full decomposition of the Fe(acac)3, versus the oleic acid in solution that is stabilizing the precursor, thus reducing the size of the particles that are formed with lower equivalents of OAm. As higher equivalents of OAm is added to the reaction mixture, decomposition of the precursor occurs to a greater extent. This allows for the NPs to grow in size until a maximum of 24 ± 5 nm at 13 eq. OAm; with further addition of OAm, a general decrease in size is observed. After 13 eq. OAm, the higher equivalents of OAm increases the rate of nuclei formation and reduces the critical size for nuclei to form, thus making smaller particles with increased amounts of OAm. The same general trend is seen for the total size of the particles synthesized with the nitrogen purge.
In this reaction, the purge of nitrogen was shown to give rise to the anisotropy. Nitrogen flow have been shown in the literature to have an effect on NP formation.25,37 One possible reason is the extra stirring due to the nitrogen bubbles, Hemery et al. showed that vigorous stirring formed nanoflowers, whereas the solution with only convective stirring formed spherical particles.25 However, in the case of this synthesis, all systems were vigorously stirred via a magnetic stir bar. Another possibility is the bubbles provoke nucleation or aggregation. This seems to be unlikely to be the explanation as experiments with flowing nitrogen over the solution (Fig. S7†) also lead to the formation of anisotropic NPs, just to a lesser degree. Finally, another effect of the purging nitrogen would be to allow volatile compounds generated during the decomposition of Fe(acac)3 to be removed. Belaïd et al. studied the thermal decomposition of Fe(acac)3 using thermogravimetric analysis-infrared (TGA-IR) spectroscopy, showing that decomposition began at 200 °C. At this temperature the loss of acetylacetonate ligands (2,4-pentadione) was observed,38 which unbound has a boiling point of 140 °C. Also, the formation of other products such as CO, CO2, H2, H2O, ketones and hydrocarbons has been reported,38,39 which all have a boiling point below 260 °C. When purging nitrogen though the solution volatile components are removed faster compared to the set-up without the nitrogen purge. This difference in components that bind to the forming nuclei in solution is likely to affect the synthesis. For example, acetylacetonate ligands and other ligand fragments have been suggested to stabilize nuclei during the early stages of a reaction.40,41 Of particular consideration is the CO formed during the break down of the precursor, CO binds strongly to Fe and is commonly used to form shape controlled NPs with their low index facets exposed (cubes and octahedra).42 CO generated from the breakdown of acetylacetonate ligands have been previously reported to act as a reducing agent in solution of reactions at 300 °C.43 In this context, the residual CO in the solution could be important for forming the isotropic nanostructures formed without the nitrogen purge, stabilizing the nuclei and the low index facets formed. Hence, the nitrogen gas purge in this synthesis is hypothesized to promote the rapid aggregation of the nuclei due to the loss of ligand fragments and reactive gases during the initial stages of particle formation.
Comparison of the branched nanoparticle types
The three branched nanoparticles systems were further studied for their magnetic and crystallographic properties, and were studied for their heating properties in an alternating magnetic field in water after a ligand exchange step. This exchange making the particles dispersible in water is essential for their use in biomedical applications. The branched nanoparticles formed with 25 eq. OAm which were considered to be the most pure in terms of not having multiply branched particles (more than 4 branches present in the reaction solution), was compared to the multiply branched systems formed with 15 and 17 eq. OAm, the particles formed with less OAm were not studied as they were either too large to make a stable dispersion (13 eq. OAm) or were no longer obviously anisotropic in shape. The NPs were ligand exchanged for dispersion in water with dopamine-functionalized poly(isobutylene-alt-maleic anhydride) as shown previously.44 The corresponding solutions were then characterized in regard of their heating performance for magnetic hyperthermia. The multiply branched NPs made with 15 eq. OAm were seen to have the best heating performance with an intrinsic loss property (ILP) of 1.55 nH m2 kgFe−1 and a specific absorption rate (SAR) of 457 W g−1 at 488 kHz and 24.5 kA m−1. The branched NPs (25 eq. OAm), and other multiply branched NPs (17 eq. OAm) had much lower ILP values of 0.13 and 0.11 nH m2 kgFe−1 respectively, with the corresponding SAR values of 37 and 50 W g−1 (Fig. 4 and Table S8†). | ||
| Fig. 4 Hyperthermia results for the branched (25 eq. OAm), and the two multiply branched NPs (17 eq. OAm and 15 eq. OAm) made with a constant nitrogen purge. The ILP values were all calculated at 488 kHz and 24.5 kA m−1. The average size and size distribution (error bars) of the long axis (total size) and the branch width of the particles is used to compare with the ILP. | ||
The branched NPs (25 eq. OAm) as well as the two multiply branched NPs (17 eq. OAm and 15 eq. OAm) were fully characterized with aberration corrected high resolution transmission electron microscopy (HRTEM). The branched NPs (25 eq. OAm) were single crystalline across the entire particle, shown by the fast Fourier transforms (FFTs) from the whole particle, the left arm (red box) and the right arm (blue box), all have the reflections for the [110] zone axis present in the same locations (Fig. 5A). For the multiply branched particle formed with 17 eq. OAm, the predominant zone axis the particle is being viewed down is the [100], as shown from the area taken in the blue box at the bottom of the particle (Fig. 5B). However the top area that is indicated by the red box, shows the [100] axis reflections as well as additional reflections indicated by the red circles in the FFT. The FFT of the whole particle shows some of these additional reflections still. For the multiply branched particle formed with 15 eq. OAm, the particle is shown to be single crystalline, with all of the FFT's showing the reflections for the [110] zone axis (Fig. 5C). For 15 eq. OAm it is also noticeable that the central core of the particle is much larger than for 17 eq. OAm. Comparing these results to the XRD of the three systems with the Scherrer equation gave crystallite sizes of 12 ± 1, 8.1 ± 0.8 and 8.8 ± 0.7 nm for 25 eq. OAm, 17 eq. OAm and 15 eq. OAm respectively (Fig. S14†). This also indicates that in the case of the 15 eq. OAm there is a slight increase in crystallite ordering increasing the particle size observed, corroborating the HRTEM results.
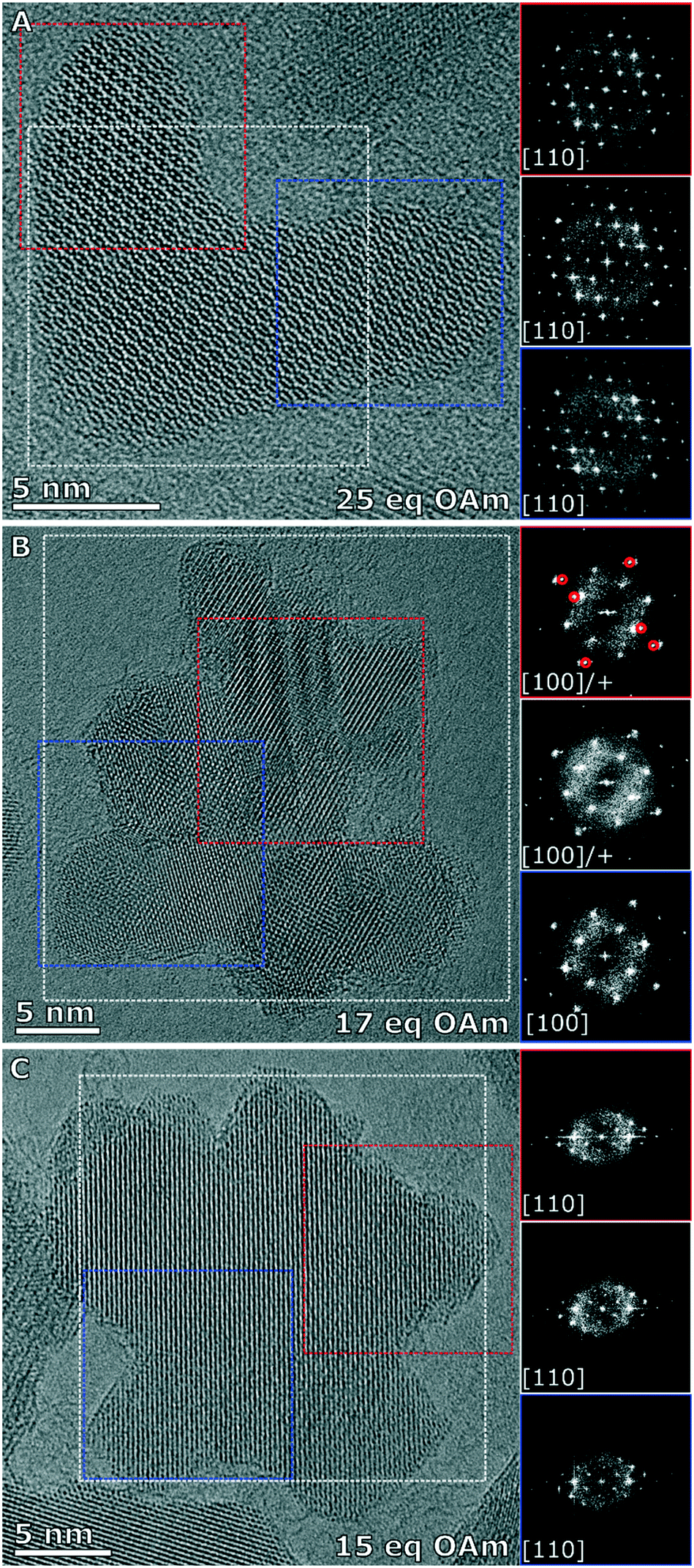 | ||
| Fig. 5 High resolution images of the IONPs synthesized with a nitrogen purge and A) 25 eq. OAm (branched), B) 17 eq. OAm (multiply branched) and C) 15 eq. OAm (multiply branched). Each particle has the FFT of the full particle in the white box, and the FFT's of two different regions of the particle indicated by the red and blue boxes respectively. | ||
It is interesting to note the large difference in values between the two multiply branched NPs, where the particles formed at 17 eq. OAm have a larger total size of the particles, 21 ± 4 nm vs. 17 ± 3 nm, and a slightly smaller branch width, 6.6 ± 0.9 nm vs. 7 ± 1 nm, yet drastically different heating properties. Studies performed with a superconducting quantum interference device (SQUID) showed a saturation magnetization of 73.7 Am2 kg−1 at 300 K for 17 eq. OAm which was lower than the 85.1 Am2 kg−1 for 15 eq. OAm (Fig. S15A†). Both systems had a negligible coercivity of less than ∼2.0 kA m−1 at 300 K, indicating superparamagnetic behavior. The blocking temperature (TB) was investigated for the particles via zero-field cooled/field cooled (ZFC-FC) measurements at 1.6 kA m−1 Oe−1. The TB was recorded at 159 K for the 17 eq. OAm, whereas for 15 eq. OAm the two curves overlap only at 300 K, showing that it's blocking temperature is higher and more varied amongst the population of NPs (Fig. S15B†).
There was no clear correlation when comparing the total particle size of the particles with their heating performance. The key effects explaining the data is the number of correlated domains forming a single crystalline structure, the short branches and the larger core seen in 15 eq. OAm. Whereas for 17 eq. OAm the core and branch sizes are roughly equivalent in size, and not all the crystalline domains are aligned. Different heating characteristics of these particles are in line with the work of Palchoudhury et al. and Lartigue et al., showing that the internal crystal ordering of the nanoflowers is essential for improved heating performance.8,45 This system unlike the reports with nanoflowers are smaller and more controlled with specific domains around a central core, whereas nanoflowers are made up of aligned but mis-oriented grains, which are made by in a high temperature polyol reaction and are typically larger and optimized to have higher heating response.6,25 However, it is interesting to note that having a single crystalline branched structure is increasing the ILP of the nanoparticles formed via this method, above what is seen for other materials synthesized via a derivative of the Sun method, in particular for spheres of the same size range.46 This is particularly of note as for the high temperature reaction methods carried out for iron oxide, the particles formed via the Sun method were shown to have the best magnetic properties as they are formed in a single crystalline manner without defects present in the structure.47
Growth studies of the anisotropic nanoparticle type
To understand the growth mechanism, 0.2 mL aliquots were taken over the course of the synthesis (during the temperature ramp of the synthesis and the aging time at 280 °C) starting at 260 °C for the branched particles (25 eq. OAm) and multiply branched particles (17 eq. OAm and 15 eq. OAm). Small nuclei were observed at 260 °C in all cases, and proceeded to grow over time till the particles reached their final size and shape after 30 min (Fig. 6A). At 260 °C in all cases some larger chunks of material are observed, and this is clear in the TEM image of the particles formed with 17 eq. OAm. The average particle size and size distribution was plotted with time for the three particle shapes with time 0 starting at 280 °C (Fig. 6B and C). The branched particles (25 eq. OAm) grow steadily from small anisotropic nuclei at 260 °C to larger branched structures after 30 min at 280 °C. This growth shows a small increase in the total size, which reaches a maximum after 5 min, while the branch width which continues to increase with time (Fig. 6). For the multiply branched particles formed at 17 eq. OAm, the nuclei formed are initially branched, and the number of branches appears to increase at 280 °C, while at 5 min the particles become fully aggregated and reach almost their full size. The total size of the multiply branched particles formed at 15 eq. OAm increased the most rapidly, reaching almost their final size at 0 min, indicating that aggregation occurs during the ramp up process (Fig. 6B). However, the branch width at 15 eq. OAm was observed to increase steadily after 0 min (Fig. 6C), indicating that growth was occurring on top of the aggregated particle. The branch width for the multiply branched particles at 17 eq. OAm is seen to increase with time, but to a smaller degree than for 15 eq. OAm.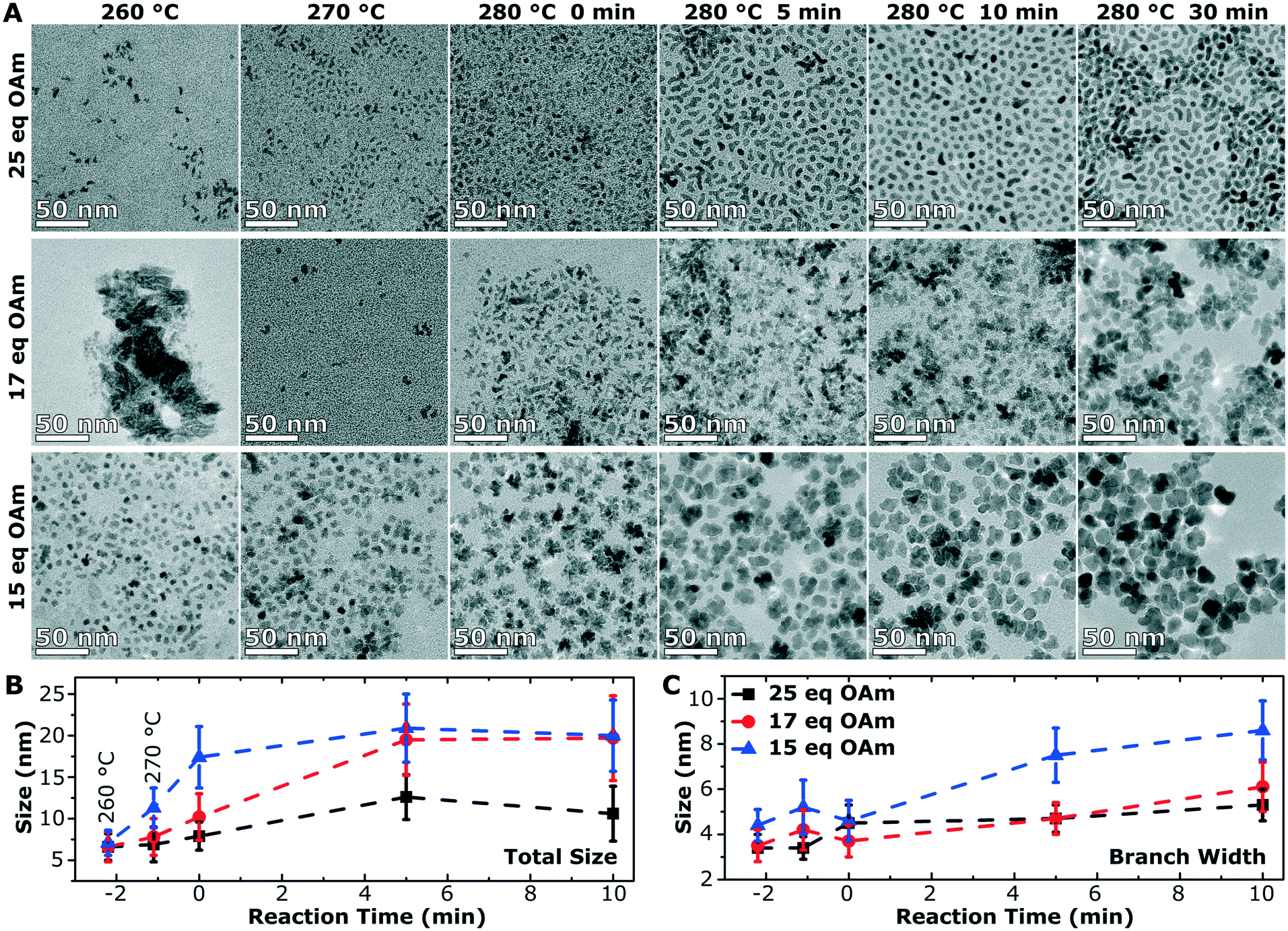 | ||
| Fig. 6 Growth studies of the branched and multiply branched IONPs synthesized with a constant nitrogen purge. A) TEM images of the aliquots taken from the synthesis of the branched nanoparticles (25 eq. OAm) and multiply branched NPs (17 eq. OAm and 15 eq. OAm) during the temperature ramp of the synthesis and during the aging time at 280 °C. Graphs of the particle sizes and their standard deviations (indicated by error bars) over time for the particles B) longest axis and C) the branch width, with time 0 starting at 280 °C. At negative reaction times the particle sizes for lower temperatures are shown, i.e., before the synthesis mixture reached 280 °C. | ||
HRTEM was performed for the particles obtained at various stages during growth to study how the shape and crystal structure evolve. At 260 °C, nuclei were observed at 25 eq. OAm, 17 eq. OAm and 15 eq. OAm. In all cases the nuclei were slightly anisotropic and polycrystalline with one or more defects observed in the structure (Fig. 7A–C, red arrows). For 25 eq. OAm, polycrystalline branched NPs are observed at 260 °C, with one particle showing a mis-alignment between one anisotropic particle and the second branch (Fig. 7A, red arrow). The branched particle at 0 min appears to be single crystalline and the one at 5 min still shows several defects present, however it is viewed down a single zone axis (Fig. 7D and G). For 17 eq. OAm the nuclei at 260 °C show a small anisotropic particle that has multiple defects present (Fig. 7B, red arrows). At 280 °C, 0 min and 5 min, the particles show an increased number of branches but each of the structures still have multiple defects present (Fig. 7E and H, red arrows). The FFT of the particle at 280 °C 0 min shows a small mis-orientation in the FFT (Fig. 7E, white arrow) between the main particle and the end of one of the arms. At 5 min looking at a larger particle with three distinct arms it can be seen that there are several additional reflections present in the FFT (Fig. 7H, FFT red circles) and each of the arms appear to have a defect present near where is attaches to the main particle (Fig. 7H, red arrows). For 15 eq. OAm at 260 °C a particle is seen with defects present where a second smaller particle appears to be attaching to the side of the particle, the rest of the particle is viewed down one zone axis with streaking in the FFT indicating defects present in the particle (Fig. 7C). At 280 °C 0 min the particles have already adopted a multiple branched morphology, which is all viewed down a single zone axis (Fig. 7F). After 5 min a larger multiply branched particle is observed, however the FFT shows additional reflections indicating the presence of mis-aligned domains. As shown above, after 30 min of reaction only the multiply branched particles formed with 17 eq. OAm have a large degree of defects in the structure. This indicates that for 25 eq. OAm and 15 eq. OAm, the particles are not formed as single crystal, but their crystal domains undergo a degree of rearrangement and ordering during the 30 min reaction time at 280 °C. For the multiply branched particles formed at 17 eq. OAm this rearrangement does not completely occur. XPS shows that for all synthesis systems, after exceeding 260 °C, the material consisted of Fe2+/3+ and O2− in a metal oxide lattice with a Fe:O ratio between 0.72 and 0.76 (Fig. S16–S18 and Tables S9–S12). This is consistent with the nucleated particles being magnetite/maghemite, and without any obvious change in the chemical structure after nucleation.
 | ||
| Fig. 7 HRTEM images of particles formed with 25 eq. OAm (A, D and G), 17 eq. OAm (B, E and H) and 15 eq. OAm (C, F and I) taken from different stages in the reaction at 260 °C, 280 °C at 0 min and at 5 min respectively. The red arrows point defects in the structure, white arrows indicating streaking or slight misalignments in the FFTs, and the red circles in the FFTs correspond to additional reflections coming from mis-oriented grains. | ||
Finally, the octahedra formed by reducing the amount of OAm to 5 eq. were studied. The initial particles were observed only after 5 min at 280 °C with a size of 13 ± 3 nm, which continues to grow to their final size of 16 ± 2 nm (Fig. S19†). The initial particles formed have a degree of anisotropy that disappears as the reaction progresses, ending up with faceted octahedra.
Two significant effects of the OAm are shown. First, increasing the OAm causes the decomposition of the precursor to occur at lower temperatures and shorter times, by favoring the nucleation of the IONPs. At low OAm concentrations (5 eq.), nucleation occurs at a much later time due to an increased critical size required before the nuclei are stable in solution. The initial NPs formed are polydisperse and aggregated, however as the concentration of monomers in solution (and iron still in the precursor) is high, growth on top of the initial nuclei allows the initial random shapes formed with 5 eq. OAm to continue to grow until only isotropic octahedra are present.
Increasing the amount of OAm lowers the nucleation temperature and forms smaller anisotropic nuclei, these anisotropic nuclei aggregate earlier in the reaction (Fig. 8). Once the aggregated nuclei are formed, the NPs continue to grow with the remaining monomers in solution. Comparing the two systems with multiply branched NPs, the 15 eq. OAm synthesis had the aggregation occurring earlier in the reaction, and thus more growth would occur on top of the aggregated nuclei. The monomers in solution would then attach to “fill up the holes” within the aggregates and allow for the minimization of the surface energy during growth, causing the formation of larger cores with shorter branches. This growth on top of the aggregated nuclei would also favor crystallization and lattice orientation (Fig. 8). For the multiply branched NPs formed at 17 eq. OAm, the final particle is polycrystalline, this can be explained through the decreased number of monomers available for growth after the aggregated multiply branched NPs are formed. The particle would not be supplied with enough monomers during growth for the rearrangement of the crystal domains. At high concentrations, 25 eq. OAm, the initial nuclei formed are anisotropic and aggregate into branched particles during the early stages of reaction, but do not continue to aggregate to form the multiply branched particles. The OAm could stabilize the nuclei decreasing the amount of aggregation that occurs. After aggregation occurs, rearrangement also occurs which is further facilitated by the continued growth of the particles (Fig. 8).
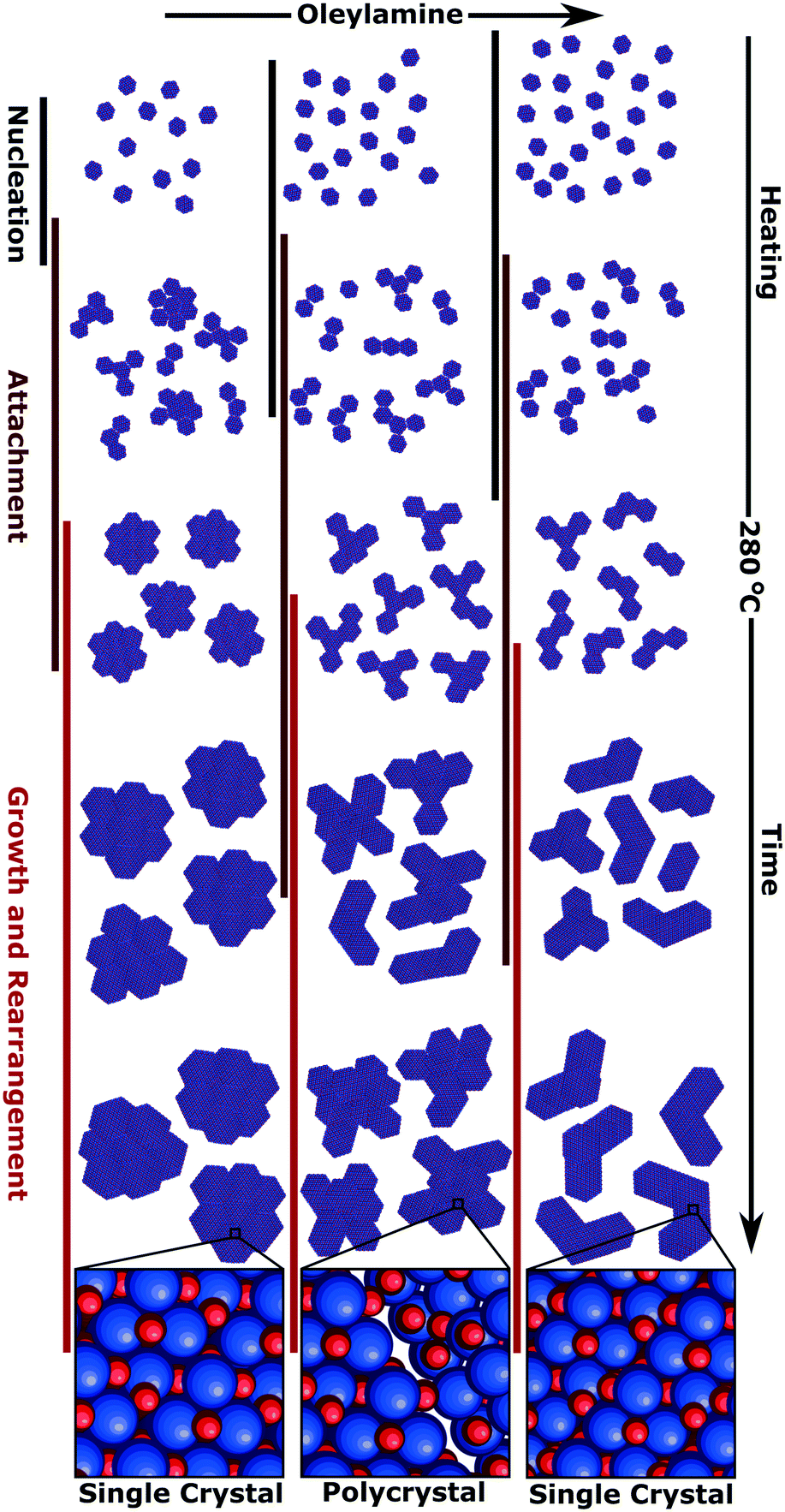 | ||
| Fig. 8 Schematic of the growth mechanism of the branched nanoparticles formed with 15, 17 and 25 eq. OAm with the nitrogen purge, detailing the overlapping stages of nucleation, attachment, growth and rearrangement. | ||
From the growth studies, the NPs are shown to aggregate into branched particles early and then grow from the initial branched nuclei. Our growth mechanism departs from studies of iron oxide nanoflowers in the literature, that show that the particulates are formed and then align to produce the nanoflowers.48,49 In nanoflowers the crystal domains successfully rearrange as the reaction progresses, forming aligned domains with a small degree of misalignment present between domains.10 The growth mechanism we observed is more similar to the work of Cozzoli et al. who showed small tetrapods of iron oxide formed shortly after nucleation at high temperatures in organic solvents and then grew only in branch length.26 Unlike the work of Cozzoli et al. we report here an initial nucleation of anisotropic iron oxide that goes through an aggregation step and then rearrange to form single crystalline particles. Once the initial nucleation and aggregation occurs, then the particles continue to grow in all directions. This leads to the rearrangement and orientation of crystalline domains.
Conclusions
This work studies the effect of synthetic conditions of IONPs on the particle morphology and size. Due to the wide range of the conditions studied, anisotropic IONPs (branched particles, and multiply branched particles) as well as isotropic particles were observed. The isotropic particles commonly observed, seem to be only a particular case of the broader synthetic landscape. It must be highlighted, that the presence of the purge of nitrogen through the solution was essential to form the anisotropic IONPs. In the formation of the anisotropic NPs, the OAm concentration was the key parameter having a large effect on the NP morphology. For the isotropic particles the OAm controlled the particle size and polydispersity while for the anisotropic particles it also controlled the degree of branching observed. Of particular interest was the two multiply branched systems studied that showed that small changes in the equivalents of OAm (17 eq. vs. 15 eq. OAm) in the synthesis was shown to significantly affect the NPs' heating properties. These changes were linked to one key difference in the growth mechanisms, the time that aggregation took place which then dictated the degree of growth and crystallite rearrangement that occurred on top of the aggregated nuclei.The insights from the growth mechanism indicate the importance of the nucleation rate and growth period to allow crystallite rearrangement in the creation of a single crystalline particle. Even small differences in growth parameters resulted in particles with similar morphologies that had radically different magnetic properties. This indicates that a deep understanding of a NPs growth mechanism and appropriate synthetic measures must be taken to allow for crystallite rearrangement to occur. This can also help to explain and thus limit problems with reproducibility in a nanomaterials properties and structure, which are common within the synthesis of anisotropic nanostructures. It also illustrates the necessity of understanding the growth mechanism to make sure that time and feed source of precursors/monomers is sufficient to allow for the rearrangement of the crystallites to occur, so that an extended single crystalline domain of iron oxide materials can be formed for efficient magnetic hyperthermia.
Finally, we have expanded the high temperature reaction system, that has been used to form spheres, cubes, octahedra and tetrapods, to branched nanoparticles and multiply branched nanoparticles. Using a single procedure, thus excluding system dependent effects, to fabricate a wider range of nanoparticle shapes and sizes will allow further study on the effect of synthetic parameters on the materials properties. The insights from this study will also allow for the other nanoparticle growth systems such as the polyol method to be further expanded to be able to form other shape controlled nanoparticles.
Methods
Chemicals
Iron(III) acetylacetonate (≥99%), HDD (90%), oleic acid (90%), OAm (70%), benzaldehyde (≥99%), 1-octadecene (90%), poly(isobutylene-alt-maleic anhydride) (Mw ≈ 6000), dopamine hydrochloride, triethylamine, dimethylformamide (anhydrous), and tetrahydrofuran (THF) were purchased from Sigma-Aldrich. Toluene (HPLC grade) was purchased from Fisher Chemical. All chemicals were used as perceived.Nanoparticle synthesis
In a typical synthesis 0.35 g (0.04 mol L−1) of iron(III) acetylacetonate [Fe(acac)3] and 1.3 g (4.5 molar equivalents to Fe) of HDD was dissolved in 15 mL of 1-octadecene, 7.5 mL of OAm, 2.85 mL (9 molar equivalents to Fe) of oleic acid and 0.5 mL of benzaldehyde. The reaction mixture was heated to 120 °C with purging nitrogen and dewatered and degassed for 10 min. The solution was then heated to 280 °C and the reaction was run with purging nitrogen for 30 min. Once the reaction was finished the solution was cooled to room temperature. Once cool, the solution was removed from the flask and mixed with the same proportion of acetone and then the particles were separated with a magnet. A second and third wash was carried out by re-dispersing the particles in a minimum amount of toluene and then adding three times as much ethanol and separated with a magnet.Transmission electron microscopy (TEM)
Transmission electron microscopy was carried out on a JEOL JEM 1200-EX operating at 120 kV with a tungsten filament. The high-resolution TEM (HRTEM) was carried out on an FEI Model Titan G2 60-300 and FEI Model Titan Themis 60-300 equipped with a Cs image corrector operating at 200 kV. TEM samples were made by depositing the nanoparticle solutions onto 200 mesh carbon coated copper grids (EM Resolutions Ltd.).X-ray diffraction (XRD)
Measurements were carried out on an X-ray diffractometer from PanAnalytical, using a CoKα radiation source with a wavelength of 1.789 Å. Samples were prepared by pressing dried powders onto a zero-background silicon wafer.X-ray photoelectron spectroscopy (XPS)
Measurements were carried out on an ESCALAB 250Xi with a monochromatic Al Kα X-ray source. The XPS spectra were acquired with a hemispherical analyzer with pass energies 20 eV and 200 eV for high resolution and survey spectra, respectively. A dual beam charge neutralization technique with 0.5 eV Ar ions and 0.5 eV electrons was used for eliminated surface charging during the XPS measurements. XPS spectra were peak-fitted using Avantage (Thermo Fisher Scientific) data processing software. For peak fitting smart-type background subtraction was used. Quantification has been done using sensitivity factors provided by Avantage library.Magnetic measurements
Isothermal magnetic hysteresis curves up to 5.5 T and field cooled/zero field cooled measurements in increasing temperature, between 10 K and 300 K under 1.6 kA m−1, were carried out using a Quantum Design superconducting quantum interference device (SQUID-MPMS).Ligand exchange
The synthesis of the dopamine-functionalized poly(isobutylene-alt-maleic anhydride) and subsequent ligand exchange with it, was carried out as in the work of Thanh et al.44 In this work the ligand exchange was carried out by dissolving ∼50 mg of the dopamine-functionalized poly(isobutylene-alt-maleic anhydride) in 10 mL of THF and 0.5 mL of ethanol. Then ∼10 mg of NPs that had been precipitated and dried were dispersed in 2 mL of THF. The NP dispersion was mixed with the solution containing the polymer and then it was agitated via a rotating shaker for 48 h. The NPs were then precipitated via centrifugation (5 min at 4000 rpm) using excess hexane. The supernatant was discarded and the precipitate was re-dispersed in 6 mL of THF and 1 mL of ethanol. The nanoparticles were again precipitated out with centrifugation and excess hexane. The precipitate was then dried under vacuum before 10 mL of di-sodium tetraborate buffer solution at pH = 9.2 was added to it and it was agitated via the rotating shaker for up to 3 d. The resulting dispersion was filtered through a 0.45 μm disposable syringe filter with VWR polyethersulfone membrane. The excess ligands were removed through a centrifugal filter (Sartorius, Mw cut-off 50 kDa) and washing three times with Milli-Q water (>18.0 MΩ cm−1).Magnetic hyperthermia
The particles heating abilities in an AC magnetic field were tested using a G2 driver D5 series calorimetric analyzer from nB nanoScale Biomagnetics which was equipped with the CAL1 coil set featuring a sealed glass Dewar flask (pressure 0.01–0.1 Pa) providing good thermal insulation (pseudo-adiabatic system). The analyzer facilitated measurements for variable frequencies and field strength while recording the temperature with GaAs-based fiber optic probe immersed in a vial containing ∼1 mL of the aqueous IONP solution, i.e., after the ligand exchange step.The particles' specific absorption rate (SAR), i.e., the power dissipation of the magnetic material, was obtained based by
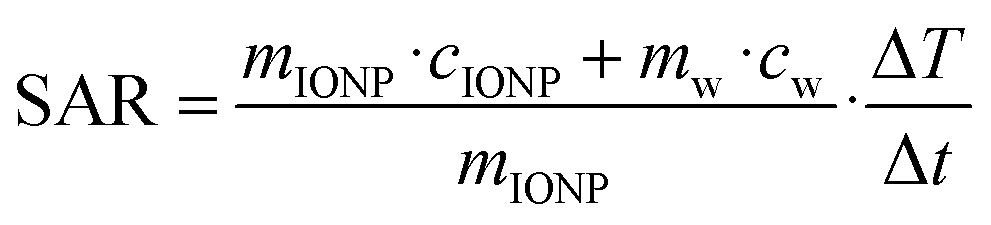
where ΔT/dt was determined using the initial slope method via a linear fit through the first 20 s of the heating profile T(t) after applying the AC magnetic field, cw is the specific heat capacity of water, and msample/mIONP is the sample to IONP mass ratio.
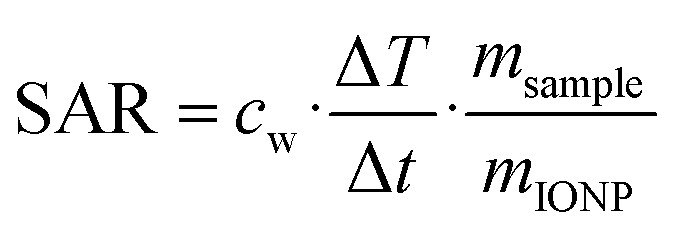
Since SAR values depend on the used field strength and frequencies the intrinsic loss power50 was determined as
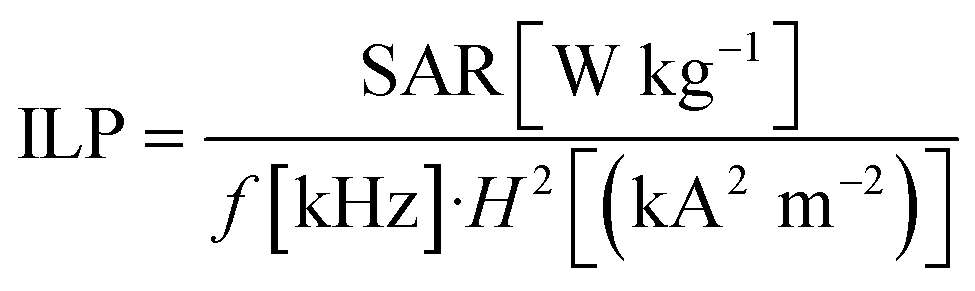
Iron quantification
To determine the concentration of iron in the NP sample for hyperthermia analysis, an iron quantification assay method is used. This was conducted through the formation of an iron complex with 1,10-phenanthroline forming a deep red colour which was quantified using UV/vis spectroscopy. A serial dilution was used to find the concentration of the unknown iron sample. Using a volumetric flask, a stock solution of 0.2 mg mL−1 of [Fe] was made up using Milli-Q water. This solution was used to form a calibration curve for the UV/vis measurements by creating a dilution series.In a glass vial, 500 μg mL−1 concentration sample was added with 500 μg mL−1 of 4 M HCL and the solutions were heated for 1 h in a 70 °C heating bath to ensure that surfactants attached on the iron were completely digested. In a glass vial, 200 μg mL−1 of the sample was added with 450 μL of sodium acetate (125 mg mL−1), 50 μg mL−1 of hydroxylamine hydrochloride (10 mg mL−1) and 300 μmL of 1,10-phenanthroline monohydrate (10 mg mL−1). The sample is left for 30 min to ensure that the complexing process is complete. Each sample was then analysed using a SpectraMax M2e UV/vis spectrometer. The absorbance was measured at 510 nm and compared to the calibration curve to find the concentration of iron in the sample.
Author contributions
NHAT and APL carried out the syntheses, TEM, and writing of the article. MOB carried out the magnetic hyperthermia measurements. OB carried out and analyzed the XPS. NHAT and SF carried out the iron quantification and ligand exchange. APL and AS carried out the aberration corrected HRTEM. LPF and MMC carried out and analyzed the SQUID data. NTKT and APL conceived of the study and supervised NHAT. NHAT, APL, MB, NTKT and AG carried out the analysis of the data as a whole for the discussion of the synthesis, magnetic hyperthermia properties and growth mechanism.Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
APL, MOB, AG, NTKT would like to thank EPSRC for funding (grant EP/M015157/1). AS thanks the EPSRC CDT for the Advanced Characterization of Materials (grant EP/L015277/1) for his studentship. NTKT thanks AOARD (FA2386-17-1-4042 award). LPF and MMC thank Portuguese funding through FCT-Fundação para a Ciência e a Tecnologia, I. P., in the scope of projects UID/MULTI/04046/2019 (BioISI) and PTDC/NAN-MAT/28785/2017. This work was carried out in part through the use of the INL Advanced Electron Microscopy, Imaging and Spectroscopy Facility.Notes and references
- E. Ye, M. D. Regulacio, S. Y. Zhang, X. J. Loh and M. Y. Han, Chem. Soc. Rev., 2015, 44, 6001 RSC.
- Y. Xia, Y. Xiong, B. Lim and S. E. Skrabalak, Angew. Chem., Int. Ed., 2009, 48, 60 CrossRef CAS.
- J. Watt, S. Cheong and R. D. Tilley, Nano Today, 2013, 8, 198 CrossRef CAS.
- J. Watt, S. Cheong, M. F. Toney, B. Ingham, J. Cookson, P. T. Bishop and R. D. Tilley, ACS Nano, 2010, 4, 396 CrossRef CAS.
- D. Gao, S. Yang, L. Xi, M. Risch, L. Song, Y. Lv, C. Li, C. Li and G. Chen, Chem. Mater., 2020, 32, 1581 CrossRef CAS.
- P. Hugounenq, M. Levy, D. Alloyeau, L. Lartigue, E. Dubois, V. Cabuil, C. Ricolleau, S. Roux, C. Wilhelm, F. Gazeau and R. Bazzi, J. Phys. Chem. C, 2012, 116, 15702 CrossRef CAS.
- P. Bender, J. Fock, C. Frandsen, M. F. Hansen, C. Balceris, F. Ludwig, O. Posth, E. Wetterskog, L. K. Bogart, P. Southern, W. Szczerba, L. Zeng, K. Witte, C. Grüttner, F. Westphal, D. Honecker, D. González-Alonso, L. Fernández Barquín and C. Johansson, J. Phys. Chem. C, 2018, 122, 3068 CrossRef CAS.
- L. Lartigue, P. Hugounenq, D. Alloyeau, S. P. Clarke, M. Lévy, J.-C. Bacri, R. Bazzi, D. F. Brougham, C. Wilhelm and F. Gazeau, ACS Nano, 2012, 6, 10935 CrossRef CAS.
- C. Blanco-Andujar, D. Ortega, P. Southern, Q. A. Pankhurst and N. T. K. Thanh, Nanoscale, 2015, 7, 1768 RSC.
- C. Blanco-Andujar, D. Ortega, P. Southern, S. A. Nesbitt, N. T. K. Thanh and Q. A. Pankhurst, Nanomedicine, 2015, 11, 121 CrossRef.
- S. Cheong, J. Watt, B. Ingham, M. F. Toney and R. D. Tilley, J. Am. Chem. Soc., 2009, 131, 14590 CrossRef CAS.
- N. M. Alyami, A. P. Lagrow, D. H. Anjum, C. Guan, X. Miao, L. Sinatra, D.-J. Yuan, O. F. Mohammed, K.-W. Huang and O. M. Bakr, Cryst. Growth Des., 2018, 18, 1509 CrossRef CAS.
- J. Lee, J. Yang, S. G. Kwon and T. Hyeon, Nat. Rev. Mater., 2016, 1, 16034 CrossRef CAS.
- N. T. K. Thanh, N. Maclean and S. Mahiddine, Chem. Rev., 2014, 114, 7610 CrossRef CAS.
- D. Li, M. H. Nielsen, J. R. I. Lee, C. Frandsen, J. F. Banfield and J. J. De Yoreo, Science, 2012, 336, 1014–1018 CrossRef CAS.
- K. S. Cho, D. V. Talapin, W. Gaschler and C. B. Murray, J. Am. Chem. Soc., 2005, 127, 7140 CrossRef CAS.
- L. Graham, G. Collins, J. D. Holmes and R. D. Tilley, Nanoscale, 2016, 8, 2867 RSC.
- A. P. Lagrow, L. Sinatra, A. Elshewy, K.-W. Huang, K. Katsiev, A. R. Kirmani, A. Amassian, D. H. Anjum and O. M. Bakr, J. Phys. Chem. C, 2014, 118, 19374 CrossRef CAS.
- S. Maksimuk, X. Teng and H. Yang, J. Phys. Chem. C, 2007, 111, 14312 CrossRef CAS.
- J. Watt, N. Young, S. Haigh, A. Kirkland and R. D. Tilley, Adv. Mater., 2009, 21, 2288 CrossRef CAS.
- L. Manna, E. C. Scher and A. P. Alivisatos, J. Am. Chem. Soc., 2000, 122, 12700 CrossRef CAS.
- A. P. Lagrow, S. Cheong, J. Watt, B. Ingham, M. F. Toney, D. A. Jefferson and R. D. Tilley, Adv. Mater., 2013, 25, 1552 CrossRef CAS.
- F. Shubitidze, K. Kekalo, R. Stigliano and I. Baker, J. Appl. Phys., 2015, 117, 94302 CrossRef.
- D. Caruntu, G. Caruntu, Y. Chen, C. J. O'Connor, G. Goloverda and V. L. Kolesnichenko, Chem. Mater., 2004, 16, 5527–5534 CrossRef CAS.
- G. Hemery, A. C. Keyes, E. Garaio, I. Rodrigo, J. A. Garcia, F. Plazaola, E. Garanger and O. Sandre, Inorg. Chem., 2017, 56, 8232 CrossRef CAS.
- P. D. Cozzoli, E. Snoeck, M. A. Garcia, C. Giannini, A. Guagliardi, A. Cervellino, F. Gozzo, A. Hernando, K. Achterhold, N. Ciobanu, F. G. Parak, R. Cingolani and L. Manna, Nano Lett., 2006, 6, 1966 CrossRef CAS.
- S. Sun and H. Zeng, J. Am. Chem. Soc., 2002, 124, 8204–8205 CrossRef CAS.
- L. Qiao, Z. Fu, J. Li, J. Ghosen, M. Zeng, J. Stebbins, P. N. Prasad and M. T. Swihart, ACS Nano, 2017, 11, 6370 CrossRef CAS.
- M. C. Biesingera, B. P. Payne, A. P. Grosvenor, L. W. M. Laua, A. R. Gerson and R. St. C. Smart, Appl. Surf. Sci., 2011, 257, 2717–2730 CrossRef.
- H. Khurshid, W. Li, S. Chandra, M.-H. Phan, G. C. Hadjipanayis, P. Mukherjee and H. Srikanth, Nanoscale, 2013, 5, 7942 RSC.
- S. Mourdikoudis and L. M. Liz-Marzán, Chem. Mater., 2013, 25, 1465 CrossRef CAS.
- C. Moya, M. del P. Morales, X. Batlle and A. Labarta, Phys. Chem. Chem. Phys., 2015, 17, 13143 RSC.
- N. Pérez, F. López-Calahorra, A. Labarta and X. Batlle, Phys. Chem. Chem. Phys., 2011, 13, 19485 RSC.
- S. G. Kwon, Y. Piao, J. Park, S. Angappane, Y. Jo, N.-M. Hwang, J.-G. Park and T. Hyeon, J. Am. Chem. Soc., 2007, 129, 12571 CrossRef CAS.
- C. R. Bullen and P. Mulvaney, Nano Lett., 2004, 4, 2303 CrossRef CAS.
- J. van Embden and P. Mulvaney, Langmuir, 2005, 21, 10226 CrossRef CAS.
- D. Ung, L. D. Tung, G. Caruntu, D. Delaportas, I. Alexandrou, I. A. Prior and N. T. K. Thanh, CrystEngComm, 2009, 11, 1309 RSC.
- S. Belaïd, D. Stanicki, L. Vander Elst, R. N. Muller and S. Laurent, Nanotechnology, 2018, 29, 165603 CrossRef.
- J. Von Hoene, R. G. Charles and W. M. Hickam, J. Phys. Chem., 1958, 62, 1098 CrossRef CAS.
- S. Özkar and R. G. Finke, Langmuir, 2002, 18, 7653 CrossRef.
- A. P. LaGrow, T. M. D. Besong, N. M. AlYami, K. Katsiev, D. H. Anjum, A. Abdelkader, P. M. F. J. Costa, V. M. Burlakov, A. Goriely and O. M. Bakr, Chem. Commun., 2017, 53, 2495 RSC.
- J. Wu, A. Gross and H. Yang, Nano Lett., 2011, 11, 798 CrossRef CAS.
- S.-W. Kim, J. Park, Y. Jang, Y. Chung, S. Hwang and T. Hyeon, Nano Lett., 2003, 3, 1289 CrossRef CAS.
- S. Famiani, A. P. LaGrow, M. O. Besenhard, S. Maenosono and N. T. K. Thanh, Chem. Mater., 2018, 30, 8897 CrossRef CAS.
- S. Palchoudhury, Y. Xu, A. Rushdi, R. A. Holler and Y. Bao, Chem. Commun., 2012, 48, 10499 RSC.
- Z. Nemati, J. Alonso, I. Rodrigo, R. Das, E. Garaio, J. Á. García, I. Orue, M.-H. Phan and H. Srikanth, J. Phys. Chem. C, 2018, 122, 2367 CrossRef CAS.
- Z. Nedelkoski, D. Kepaptsoglou, L. Lari, T. Wen, R. A. Booth, S. D. Oberdick, P. L. Galindo, Q. M. Ramasse, R. F. L. Evans, S. Majetich and V. K. Lazarov, Sci. Rep., 2017, 7, 45997 CrossRef CAS.
- B. C. Park, J. Cho, M. S. Kim, M. J. Ko, L. Pan, J. Y. Na and Y. K. Kim, Nat. Commun., 2020, 11, 298 CrossRef CAS.
- C. Cheng, F. Xu and H. Gu, New J. Chem., 2011, 35, 1072–1079 RSC.
- R. R. Wildeboer, P. Southern and Q. A. J. Pankhurst, J. Phys. D: Appl. Phys., 2014, 47, 495003 CrossRef.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0ce01291b |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2021 |