
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA supramolecular double-helix based on complementary phosphate–guanidinium pairing†
Maike
Thiele
a,
Frescilia
Octa-Smolin
a,
Simon
Thölke
a,
Christoph
Wölper
b,
Jürgen
Linders
c,
Christian
Mayer
c,
Gebhard
Haberhauer
 *a and
Jochen
Niemeyer
*a
*a and
Jochen
Niemeyer
*a
aFaculty of Chemistry, Organic Chemistry and Center for Nanointegration Duisburg-Essen (CENIDE), University of Duisburg-Essen, Universitätsstrasse 7, 45141 Essen, Germany. E-mail: jochen.niemeyer@uni-due.de
bFaculty of Chemistry, Inorganic Chemistry, University of Duisburg-Essen, Germany
cFaculty of Chemistry, Physical Chemistry, University of Duisburg-Essen, Germany
First published on 27th August 2021
Abstract
A double-helical supramolecular structure was formed by self-assembly of 1,1′-binaphthyl-based bisguanidines and bisphosphoric acids. Interestingly the homochiral (S,S) + (S,S)-pair forms a left-handed double-helix, while the heterochiral (S,S) + (R,R)-pair forms a non-helical dimer.
The DNA double-helix is one of the most common supramolecular structures in nature. Its left-handed helical structure is formed by cooperative non-covalent forces such as hydrogen bonds and π–π interactions. The biological functions of DNA are tightly connected to its helical super-structure, e.g. in DNA self-repair and DNA replication.1
Inspired by DNA, there have been numerous efforts to generate artificial double- or multi-stranded helical structures by supramolecular self-assembly.2 This was pioneered by the works of Lehn and others on metallosupramolecular double-helices, which feature ligand strands that wrap around an array of central metal ions.3 Anionic centers can also be exploited as templates, e.g. by wrapping oligo-guanidine strands around dianionic sulfates4 or by formation of spiroborates from oligocatechols in a dynamic covalent approach.5 Purely organic frameworks can also be employed, as shown for oligoamide-based foldamers6 or linked bisphosphoric acids,7 both of which self-assemble into helical superstructures.
However, there is limited precedence for the selective formation of artificial supramolecular double-helices by recognition of complementary binding motifs, as found in DNA based on the A–T and G–C base pairs. In this context, Yashima and coworkers reported a modular strategy towards double-helical structures based on complementary interstrand-recognition. Based on aminidium- and carboxylate-binding motifs, which interact via two hydrogen-bonds assisted by Coulomb interactions, supramolecular double-helices of different composition and length could be generated.8 It was even shown that a template strand can effect the formation of its complementary counterpart, mimicking the DNA replication process.9 In Yashima's systems, the amidine groups are functionalized with centrochiral substituents. Upon pairing with the (achiral) carboxylates, the chiral amidines can control the supramolecular helicity of the resulting double-helices.
For this work, we now targeted hydrogen-bonded supramolecular double-helices based on complementary guanidinium–phosphate binding. The guanidinium–phosphate ion-pair has a strong biological relevance10 and has also found application in supramolecular chemistry,11 albeit mostly in achiral form. We now envisaged the use of axially chiral 1,1′-binaphthyl-phosphoric acids and 1,1′-binaphthyl-guanidines (see Fig. 3). This resembles Yashima's amidinium–carboxylate pairing, with the added benefits that the lower acidity of the guanidinium-unit (pKa = 13.6 for Me2–guanidine vs. pKa = 12.4 for acetamidine10b) favors its protonation and the fact that both strands are chiral in our setup. Thus, the single strands can be combined in a homochiral or heterochiral fashion in order to investigate the following points: (a) Does any of the two combinations lead to the desired double-helical structure? (b) Does the strand-chirality influence the helix-sense, so that the double-helix is generated in a diastereoselective fashion?
We first tested the supramolecular interaction of our designated binding motifs in a crystallographic study. Thus, 1,1′-binaphthyl-phosphate (rac)-1 (used as the Na-salt) was cocrystallized with 1,1′-binaphthyl-based guanidine (rac)-2 (used as the HBr salt) to yield to corresponding heterodimeric complex (rac)-1 + (rac)-2 (see Fig. 1).
 | ||
| Fig. 1 Molecular structures of (rac)-1, (rac)-2 and complex 1 + 2 in the solid state (only (S)-isomers shown, nonpolar hydrogen atoms and solvent molecules omitted for clarity, thermal ellipsoids shown at the 60% probability level, H atoms (refined as riding atoms): light grey, C atoms: dark grey, N atoms: blue, O atoms: red, Na atoms: purple, P atoms: orange, Cl atoms: brown, hydrogen-bonds shown as red dashed lines). | ||
In the solid state, we find homochiral pairs of (S)-1 + (S)-2 and (R)-1 + (R)-2. The complexes feature the expected cyclic arrangement of two PO⋯HN hydrogen-bonds in the phosphate–guanidinium pair, leading to the placement of the two binaphthyl units on opposite sides, as desired (albeit in a slightly tilted geometry, dihedral angle between the two binaphthyl C1–C1′ axes: 23.0°).
Based on this successful proof of complementary binding, we then aimed for the formation of double-helical structures from covalently linked bisphosphates and bisguanidines. The synthesis of the 3,3-linked bisphosphate 9 has previously been developed in our group,7b,12 and both the (S,S)- and (R,R)-enantiomer were used for this study. As for the bisguanidine (S,S)-8, we had to establish a suitable synthetic route (see Fig. 2A). We started from 3,3′-diiodo-2,2′-di(bromomethyl)-1,1′-bi-naphthyl (S)-3, which was generated in seven steps from BINOL according to literature procedures.13 Reaction with tert-butylcarbamate in the presence of sodium hydride gave the seven-membered azepine derivative (S)-4. This was followed by mono-deiodination by halogen–lithium exchange and protonation in a carefully controlled reaction to give monoiodide (S)-5, which underwent twofold Sonogashira coupling with 1,4-diethynylbenzene to give the desired Boc-protected precursor (S,S)-6. The synthesis was finalized by Boc-deprotection with TFA to give the ammonium salt (S,S)-7 and installation of the guanidine groups by deprotonation and reaction with diisopropylcarbodiimide to give the bisguanidine (S,S)-8 (as the (TFA−)2-salt). All compounds were fully characterised by standard analytical techniques (see ESI† for details). Furthermore, the solid-state structures of the 4, 5 and 6 were determined by X-ray single crystal structure analyses (see Fig. 2B).
 | ||
| Fig. 2 (A) Synthesis of the bis-guanidine (S,S)-8. Reagents and conditions: (i) NaH, tert-butyl carbamate, 0 °C, THF/DMF, 77%; (ii) n-BuLi, 0 °C, toluene, then MeOH, 63%; (iii) 1,4-diethynylbenzene, CuI, Pd(PPh3)4, 80 °C, ACN:NEt3, 86%. (iv) TFA, RT, DCM, 76%; (v) NaOH, then n-BuLi, diisopropylcarbodiimide, RT, toluene, 33%. (B) Molecular structures of (S)-4, (R)-5 and (S,S)-6 in the solid state (hydrogen atoms and solvent molecules omitted for clarity, thermal ellipsoids shown at the 50% probability level, C atoms: dark grey, N atoms: blue, O atoms: red). For (S,S)-6, only one orientation of the disorder is displayed for clarity. Monoiodide 5 was analyzed as the (R)-isomer. | ||
For the synthesis of the hydrogen-bonded supramolecular double-helices, bisguanidine (S,S)-8 (used as the (TFA−)2-salt) was mixed in a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 stoichiometry with either bisphosphate (S,S)-9 or its enantiomer (R,R)-9 (used as the (N+Bu4)2-salts). We expected that the chirality of the building blocks would strongly influence their ability to self-assemble, so that possibly only the matched (S,S)-8 + (S,S)-9 pair would be able to form a supramolecular complex. Surprisingly, NMR analysis reveals formation of supramolecular complexes both for the (S,S)-8 + (S,S)-9 and for the (S,S)-8 + (R,R)-9 case (see Fig. 3 and Fig. S48–S64, incl. VT spectra, ESI†). The resonances of the guanidinium NH protons are shifted downfield (δ(NH) = 9.85/9.93 + 9.85 ppm for the complexes, cf. δ(NH) = 9.32 + 9.08 ppm for (S,S)-8), as are the signals for the NBu4 countercations (δ(N+Bu4, Me-signals) = 0.96/0.92 ppm for the complexes, cf. 0.83 ppm for (S,S)-9). Altogether this strongly suggests formation of guanidinium–phosphate interactions and liberation of NBu4+CF3COO− in a salt metathesis type process. Interestingly, the benzylic methylene signals are shifted downfield for the homochiral (S,S)-8 + (S,S)-9 pair, while upfield shifts can be observed for the heterochiral (S,S)-8 + (R,R)-9 pair, indicating a difference in complex geometry for both cases.
:1 stoichiometry with either bisphosphate (S,S)-9 or its enantiomer (R,R)-9 (used as the (N+Bu4)2-salts). We expected that the chirality of the building blocks would strongly influence their ability to self-assemble, so that possibly only the matched (S,S)-8 + (S,S)-9 pair would be able to form a supramolecular complex. Surprisingly, NMR analysis reveals formation of supramolecular complexes both for the (S,S)-8 + (S,S)-9 and for the (S,S)-8 + (R,R)-9 case (see Fig. 3 and Fig. S48–S64, incl. VT spectra, ESI†). The resonances of the guanidinium NH protons are shifted downfield (δ(NH) = 9.85/9.93 + 9.85 ppm for the complexes, cf. δ(NH) = 9.32 + 9.08 ppm for (S,S)-8), as are the signals for the NBu4 countercations (δ(N+Bu4, Me-signals) = 0.96/0.92 ppm for the complexes, cf. 0.83 ppm for (S,S)-9). Altogether this strongly suggests formation of guanidinium–phosphate interactions and liberation of NBu4+CF3COO− in a salt metathesis type process. Interestingly, the benzylic methylene signals are shifted downfield for the homochiral (S,S)-8 + (S,S)-9 pair, while upfield shifts can be observed for the heterochiral (S,S)-8 + (R,R)-9 pair, indicating a difference in complex geometry for both cases.
 | ||
| Fig. 3 (A) Double-helix formation from (S,S)-8 + (S,S)-/(R,R)-9. (B) 1H NMR spectra of (S,S)-9, (S,S)-8 and the 1:1 complexes (S,S)-8 + (S,S)-9 and (S,S)-8 + (R,R)-9 (400 MHz (components) 500 MHz (complexes, 1 mM for each component), CDCl3, 298 K). | ||
To further elucidate the nature of the supramolecular complexes, we performed NMR titrations and DOSY measurements (see Table 1). Titration of (S,S)-8 with (S,S)-/(R,R)-9 in CDCl3 gave sigmoidal binding isotherms (see Fig. S11 and S13, ESI†), indicating competitive displacement of the triflate counteranions upon addition of the phosphate guest. We determined the relative binding strengths14 and found that the homochiral and the heterochiral complex have an identical association constant within the margin of error (Krel = 0.90 ± 0.18). Similarly, DOSY measurements show an almost identical decrease in diffusion coefficients upon complex formation for both the homochiral and the heterochiral case (D = 6.00/6.05 × 10−10 m2 s−1 for (S,S)-8 + (S,S)-9/(S,S)-8 + (R,R)-9; cf. 6.79 × 10−10 m2 s−1 and 6.27 × 10−10 m2 s−1 for the components (S,S)-8 and (S,S)-9).
| Compound | (S,S)-8 | (S,S)-9 | (S,S)-8 + (S,S)-9 | (S,S)-8 + (R,R)-9 | |
|---|---|---|---|---|---|
| Association constants Krel | — | — | 1 | 0.90 ± 0.18 | |
| Diffusion coefficients | CHAr | 6.79 ± 0.32 | 6.27 ± 0.05 | 6.00 ± 0.14 | 6.05 ± 0.23 |
| D [10−10 m2 s−1] | N+Bu4 | — | 6.89 ± 0.09 | 8.77 ± 0.60 | 8.69 ± 0.35 |
However, the overall changes in diffusion coefficients are small, which we attribute to the presence of large counteranions (in case of the N+Bu4-salts of (S,S)-9/(R,R)-9) and the formation of compact supramolecular complexes that only possess a rather small size-difference in comparison to their components.7b However, we can also observe a drastic change in diffusion coefficient for the N+Bu4-cations, indicating their displacement upon formation of the guanidinium–phosphate complexes (see Table 1). Thus, all data strongly hints at the formation of supramolecular complexes of similar size and binding strength upon mixing (S,S)-8 with either (S,S)-9 or (R,R)-9.
To investigate the geometry of the supramolecular complexes, we calculated the structure of the monomers 8 and 9 as well as the complexes by means of quantum chemical methods. The geometrical parameters of the monomers and complexes were optimized using B3LYP together with the dispersion correction with Becke–Johnson damping (D3BJ). As basis set 6-31G(d) was applied. Chloroform was considered as solvent by using the SMD model.15 The thus obtained molecular structures are depicted in Fig. 4 (see Fig. S21–S25 for additional views, ESI†). A comparison of the supramolecular complexes (S,S)-8 + (S,S)-9 and (S,S)-8 + (R,R)-9 shows that two guanidinium–phosphate interactions are formed in both cases. For the homochiral complex this leads to a compact double-helical structure with both subunits adopting an S-type conformation with an anti-orientation of the 1,1′-binaphthyl units with respect to the central linker (Fig. 4A). Although this structure only constitutes a half-turn of a left-handed double-helix (similar to Yashima's pioneering double-helix8b), our structure can in principle be extended by substitution in the 3′-positions in order to generate longer double helices (see Fig. S32 (ESI†) for the calculated structure of an extended helix). A different picture is obtained for the heterochiral complex (S,S)-8 + (R,R)-9. Here, (S,S)-8 adopts a U-type conformation with a cis-orientation of the 1,1′-binaphthyl units with respect to the central linker (Fig. 4B). Thus, this structure represents a non-helical dimeric complex. However, both complexes do feature two strong ionic phosphate–ammonium interactions, which might explain their similar stability. Moreover, the overall size of both complexes is very similar, as can be seen from the calculated diffusion coefficients (D = 5.2/5.5 × 10−10 m2 s−1 for (S,S)-8 + (S,S)-9/(S,S)-8 + (R,R)-9, see ESI† for details), explaining the unexpected similarity of the DOSY results.
 | ||
| Fig. 4 Molecular structures of the dimeric complexes as calculated by means of B3LYP-D3BJ(SMD)/6-31G* C atoms: light grey for (S,S)-8, dark grey for (S,S)/(R,R)-9, N atoms: blue, O atoms: red, P atoms: orange, nonpolar hydrogen atoms and Me groups omitted for clarity. (A) Double-helical homochiral complex (S,S)-8 + (S,S)-9. (B) Non-helical heterochiral complex (S,S)-8 + (R,R)-9. The structures on the right show the C3′–C1′–C1–C3–C11–C12–C13–C13–C12–C11–C3–C1–C1′–C3′ trajectory highlighted for each subunit, for numbering see Fig. 3. | ||
Both monomers exhibit a bent diethynylbenzene unit in the complexes. Such a bend should also be noticeable in the CD spectra of the compounds and could be utilized to confirm the calculated structures. Therefore, ECD measurements of the monomers as well as the complexes (S,S)-8 + (S,S)-9 and (S,S)-8 + (R,R)-9 were performed in chloroform at 10 μM concentration. Furthermore, the CD spectra were simulated with time-dependent density functional theory (TD-DFT), using the functional cam-B3LYP, the basis set 6-31G* and the SMD model (chloroform as solvent).15 In the following, the lowest energy π–π* transition in the monomer units is considered. This electronic excitation is found at around 350 nm and is essentially caused by the diethynylbenzene unit. For both monomers (S,S)-8 and (S,S)-9, this π–π* transition exhibits a positive Cotton effect in the experimental CD-spectra, which is in good agreement with the simulated spectra (Fig. S19 and S30, ESI†). In the case of the supramolecular complexes (see Fig. 5 for the calculated and experimentally determined spectra), there are accordingly two bands in this area: the lower energy band (ca. 360 nm) can be assigned to the bisphosphate (Fig. S26 and S28, ESI†), the higher energy band at ca. 320 and 340 nm, respectively, to the bisguanidinium species (Fig. S27 and S29, ESI†). Both in the computed and in the measured spectra, the lower energy band shows a negative and the higher energy band a positive Cotton effect. In the calculated spectrum of the complex (S,S)-8 + (R,R)-9, however, the negative band is less pronounced than in the experiment. Qualitatively, the calculated CD spectra of both complexes agree with the measured ones in this area and thus confirm the structures found by the calculations.
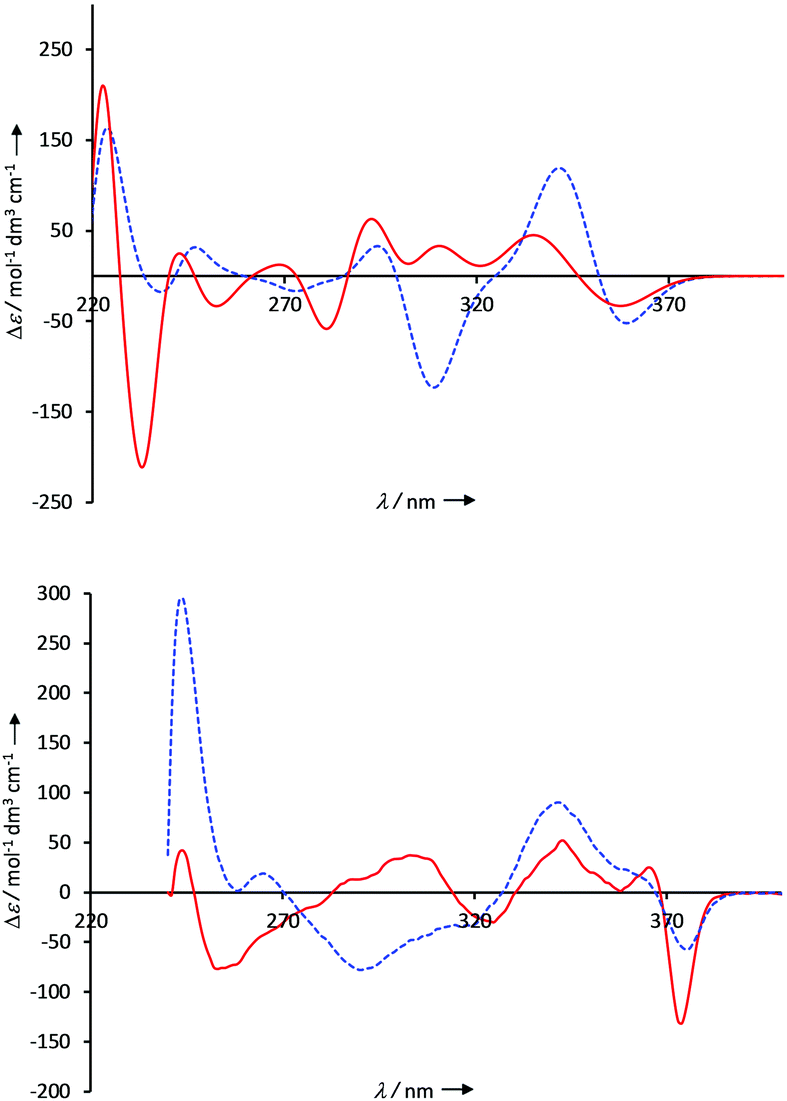 | ||
| Fig. 5 Above: CD spectra of the homochiral complex (S,S)-8 + (S,S)-9 (blue, dashed line) and the heterochiral complex (S,S)-8 + (R,R)-9 (red, solid line) in CHCl3 (c = 10−5 M). Below: TD-cam-B3LYP(SMD)/6-31G*-calculated CD spectra of the homochiral complex (S,S)-8 + (S,S)-9 (blue, dashed line) and the heterochiral complex (S,S)-8 + (R,R)-9 (red, solid line). | ||
In summary, we reported that guanidinium–phosphate pairing can be employed for the formation of a complementary double-helical structure. In case of the homochiral pairing (S,S)-8 + (S,S)-9 an intertwined double-helical structure with left-handed helicity is formed, while the heterochiral case leads to formation of a non-helical dimeric structure.
Funding from the DFG (NI1273/4-1, Heisenberg-Professorship to J. N.) is gratefully acknowledged.
Conflicts of interest
There are no conflicts to declare.Notes and references
- J. M. Berg, L. Stryer, J. L. Tymoczko and G. Gatto, Biochemistry, WH Freeman, New York, 2019 Search PubMed.
- (a) E. Yashima, N. Ousaka, D. Taura, K. Shimomura, T. Ikai and K. Maeda, Chem. Rev., 2016, 116, 13752–13990 CrossRef CAS PubMed; (b) E. Yashima, K. Maeda and Y. Furusho, Acc. Chem. Res., 2008, 41, 1166–1180 CrossRef CAS PubMed; (c) Y. Furusho and E. Yashima, Macromol. Rapid Commun., 2011, 32, 136–146 CrossRef CAS PubMed; (d) D. Haldar and C. Schmuck, Chem. Soc. Rev., 2009, 38, 363–371 RSC.
- (a) J. M. Lehn, A. Rigault, J. Siegel, J. Harrowfield, B. Chevrier and D. Moras, Proc. Natl. Acad. Sci. U. S. A., 1987, 84, 2565–2569 CrossRef CAS PubMed; (b) B. Kersting, M. Meyer, R. E. Powers and K. N. Raymond, J. Am. Chem. Soc., 1996, 118, 7221–7222 CrossRef CAS; (c) M. Albrecht and S. Kotila, Angew. Chem., Int. Ed., 1995, 34, 2134–2137 CrossRef CAS.
- J. Sánchez-Quesada, C. Seel, P. Prados, J. de Mendoza, I. Dalcol and E. Giralt, J. Am. Chem. Soc., 1996, 118, 277–278 CrossRef.
- Y. Furusho, K. Miwa, R. Asai and E. Yashima, Chem. – Eur. J., 2011, 17, 13954–13957 CrossRef CAS PubMed.
- V. Berl, I. Huc, R. G. Khoury and J.-M. Lehn, Chem. – Eur. J., 2001, 7, 2810–2820 CrossRef CAS.
- (a) H. Yamada, K. Maeda and E. Yashima, Chem. – Eur. J., 2009, 15, 6794–6798 CrossRef CAS PubMed; (b) F. Octa-Smolin, R. Mitra, M. Thiele, C. G. Daniliuc, L. Stegemann, C. Strassert and J. Niemeyer, Chem. – Eur. J., 2017, 23, 10058–10067 CrossRef CAS PubMed.
- (a) H. Yamada, Z.-Q. Wu, Y. Furusho and E. Yashima, J. Am. Chem. Soc., 2012, 134, 9506–9520 CrossRef CAS PubMed; (b) Y. Tanaka, H. Katagiri, Y. Furusho and E. Yashima, Angew. Chem., Int. Ed., 2005, 44, 3867–3870 CrossRef CAS PubMed; (c) H. Ito, Y. Furusho, T. Hasegawa and E. Yashima, J. Am. Chem. Soc., 2008, 130, 14008–14015 CrossRef CAS PubMed.
- (a) H. Yamada, Y. Furusho and E. Yashima, J. Am. Chem. Soc., 2012, 134, 7250–7253 CrossRef CAS PubMed; (b) H. Yamada, Y. Furusho, H. Ito and E. Yashima, Chem. Commun., 2010, 46, 3487–3489 RSC.
- (a) A. Pantos, I. Tsogas and C. A. Paleos, Biochim. Biophys. Acta, 2008, 1778, 811–823 CrossRef CAS PubMed; (b) K. A. Schug and W. Lindner, Chem. Rev., 2005, 105, 67–113 CrossRef CAS PubMed.
- (a) T. Schrader, G. Bitan and F.-G. Klärner, Chem. Commun., 2016, 52, 11318–11334 RSC; (b) R. J. T. Houk, S. L. Tobey and E. V. Anslyn, Top. Curr. Chem., 2005, 255, 199–229 CrossRef CAS.
- F. Octa-Smolin, M. Thiele, R. Yadav, A. Platzek, G. H. Clever and J. Niemeyer, Org. Lett., 2018, 20, 6153–6156 CrossRef CAS PubMed.
- (a) M. Gingras and F. Dubois, Tetrahedron Lett., 1999, 40, 1309–1312 CrossRef CAS; (b) H. Konishi, Chem. Pharm. Bull., 2018, 66, 1–19 CrossRef CAS PubMed; (c) M. Widhalm, M. Abraham, V. B. Arion, S. Saarsalu and U. Maeorg, Tetrahedron: Asymmetry, 2010, 21, 1971–1982 CrossRef CAS; (d) T. Ooi, Y. Uematsu and K. Maruoka, J. Org. Chem., 2003, 68, 4576–4578 CrossRef CAS PubMed.
- S. A. Kadam, K. Haav, L. Toom, T. Haljasorg and I. Leito, J. Org. Chem., 2014, 79, 2501–2513 CrossRef CAS PubMed.
- (a) A. D. Becke, Phys. Rev. A: At., Mol., Opt. Phys., 1988, 38, 3098–3100 CrossRef CAS PubMed; (b) C. Lee, W. Yang and R. G. Parr, Phys. Rev. B: Condens. Matter Mater. Phys., 1988, 37, 785–789 CrossRef CAS PubMed; (c) B. Miehlich, A. Savin, H. Stoll and H. Preuss, Chem. Phys. Lett., 1989, 157, 200–206 CrossRef CAS; (d) S. Grimme, S. Ehrlich and L. Goerigk, J. Comput. Chem., 2011, 32, 1456–1465 CrossRef CAS PubMed; (e) A. V. Marenich, C. J. Cramer and D. G. Truhlar, J. Phys. Chem. B, 2009, 113, 6378–6396 CrossRef CAS PubMed; (f) T. Yanai, D. P. Tew and N. C. Handy, Chem. Phys. Lett., 2004, 393, 51–57 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2078130–2078135. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d1cc04644f |
| This journal is © The Royal Society of Chemistry 2021 |