
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Entering new chemical space with isolable complexes of single, zero-valent silicon and germanium atoms
Shenglai
Yao
 ,
Yun
Xiong
,
Artemis
Saddington
and
Matthias
Driess
*
,
Yun
Xiong
,
Artemis
Saddington
and
Matthias
Driess
*
Department of Chemistry, Metalorganics and Inorganic Materials, Technische Universität Berlin, Strasse des 17. Juni 135, Sekr. C2, D-10623 Berlin, Germany. E-mail: matthias.driess@tu-berlin.de
First published on 9th September 2021
Abstract
Monatomic zero-valent silicon and germanium complexes (silylones and germylones), stabilised by neutral donating ligands, emerged only recently as a new class of low-valent group 14 element compounds. Featuring four valence electrons in the form of two lone pairs at a single site, silylones and germylones represent a molecular resting state of single Si and Ge atoms, which are typically only observed at high temperature in the gas phase or in interstellar matter. These species are capable of transferring single Si and Ge atoms to unsaturated substrates and acting as building blocks for novel group 14 species. After introducing this type of compound and the examples known to date, this feature article highlights some chelating bis N-heterocyclic carbene (bis(NHC)) and bis N-heterocyclic silylene (bis(NHSi)) supported Si0 and Ge0 complexes, for which a range of unprecedented reactivity has been discovered. The characteristic behaviour of these silylones and germylones discussed here consists of (i) coordination to Lewis acids, (ii) oxidation with elemental chalcogens, (iii) bond activation of common organic substrates and inert small molecules; and (iv) homocoupling of the Si0 and Ge0 centres. This wealth of reactivity has opened the door to a series of Si and Ge compounds, which would be otherwise difficult to realise.
1. Introduction
Our understanding of molecular complexes of single, zero-valent group 14 elements (ylidones, Chart 1) began following the calculations of Frenking and co-workers. They examined the donor–acceptor interactions in carbodiphosphorane Ph3P→:C:←PPh3 with quantum chemical analysis in 2006.1 The bonding situation between the carbon centre and both phosphorus atoms in C(PPh3)2, prepared already in 1961 by Ramirez,2,3 was previously described as containing electron-sharing bonds (double Wittig ylide). However, a theoretical analysis of the frontier orbitals of C(PPh3)2 demonstrated the presence of a p-type lone pair orbital corresponding to the HOMO (Highest Occupied Molecular Orbital) and an s-type lone pair orbital as the HOMO−1, both located at the central carbon atom.1 As a result, C(PPh3)2 was reinterpreted as a single carbon atom complex due to the donor–acceptor interactions between two electron-donating phosphines and a “bare” carbon(0) atom, which retains its four valence electrons as two lone pairs. The term “carbones” was subsequently suggested by Frenking for such monatomic C0 complexes coordinated by Lewis donors.4,5 In addition, the terms silylone, germylone, stannylone, and plumbylone were suggested to label such complexes of the corresponding heavier group 14 elements in the zero oxidation state.6 Carbones, featuring two lone pairs at the carbon centre, differ significantly electronically from carbenes with a singlet carbon centre in the +2 oxidation state. Such carbenes possess only one lone pair of electrons and one formally vacant orbital at the carbon centre, with the remaining two electrons involved in two electron-sharing bonds.7 In 2007, Tonner and Frenking predicted that carbodicarbenes NHC→:C:←NHC (NHC = N-heterocyclic carbene)8 should be stable enough to be synthesised.9 They were in fact correct with the group of Bertrand reporting the synthesis of the first isolable carbodicarbene in 2008, which has two resonance forms as shown in Chart 1 (bottom). This carbodicarbene is able to coordinate to RhCl(CO)2via the carbon(0) centre, demonstrating the predominance of the carbone resonance form A, with active lone pairs at the carbon centre, over the bent allene B.10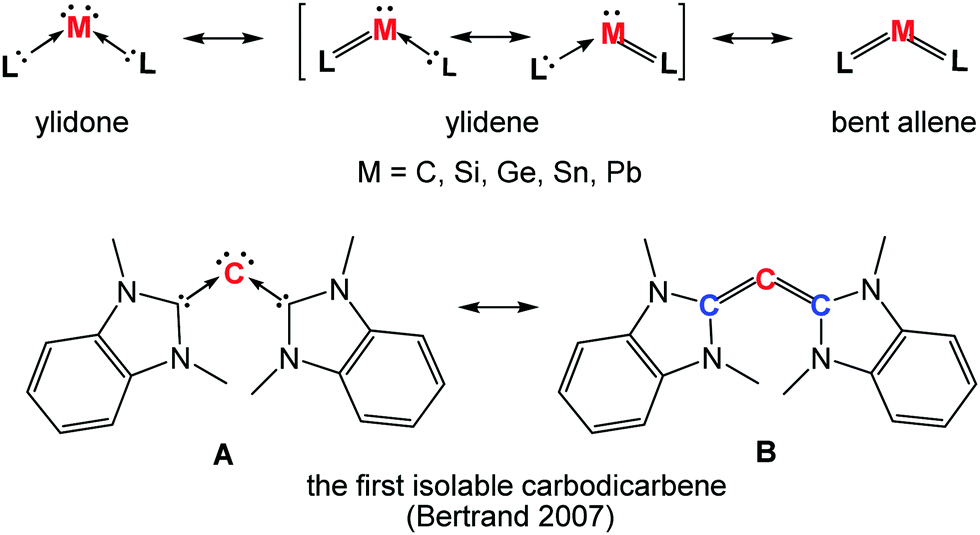 | ||
| Chart 1 The general form of ylidone and its ylidene and bent allene forms, and the resonance structures A and B of the first isolable carbodicarbene. | ||
Moving down group 14 to silicon and germanium, in 2003 heavy ylidones began to be explored following the investigations of Kira and co-workers, who reported the first trisilaallene C (M′ = Si, Chart 2).11–14 Similar to the above-mentioned case of carbodiphosphorane, a theoretical analysis of the bonding situation in related model compounds suggested that the Si![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) Si bonding in C should also be reconsidered as donor–acceptor interactions between the central silicon atom and two silylene ligands.15 Accordingly, the trisilaallene C (M′ = Si) was then described as a silylone and the 1,3-disilagermaallene D (M′ = Si, Chart 2)16 as a germylone. In 2013, experimental realisation of the silylone E (also known as a siladicarbene, Chart 2) supported by two cyclic alkyl amino carbenes (cAACs)17 was achieved by Roesky, Stalke, and Frenking.18 In the same year, our group reported the first cyclic silylone 1 (Chart 2) with the central silicon atom coordinated by a neutral chelating bis(NHC) ligand,19 followed by the analogous germylone 2 (Chart 2, also known as germadicarbene) with the same bis(NHC) ligand.20 Meanwhile, the isolation of cAAC-supported acyclic germylone F (Chart 2) was achieved by Roesky, Zhu, Stalke, and Andrada,21 and subsequently the germanium(0) complexes G,22H,23 and I24 ligated by imino containing systems were obtained by Nikonov and Kinjo, respectively.
Si bonding in C should also be reconsidered as donor–acceptor interactions between the central silicon atom and two silylene ligands.15 Accordingly, the trisilaallene C (M′ = Si) was then described as a silylone and the 1,3-disilagermaallene D (M′ = Si, Chart 2)16 as a germylone. In 2013, experimental realisation of the silylone E (also known as a siladicarbene, Chart 2) supported by two cyclic alkyl amino carbenes (cAACs)17 was achieved by Roesky, Stalke, and Frenking.18 In the same year, our group reported the first cyclic silylone 1 (Chart 2) with the central silicon atom coordinated by a neutral chelating bis(NHC) ligand,19 followed by the analogous germylone 2 (Chart 2, also known as germadicarbene) with the same bis(NHC) ligand.20 Meanwhile, the isolation of cAAC-supported acyclic germylone F (Chart 2) was achieved by Roesky, Zhu, Stalke, and Andrada,21 and subsequently the germanium(0) complexes G,22H,23 and I24 ligated by imino containing systems were obtained by Nikonov and Kinjo, respectively.
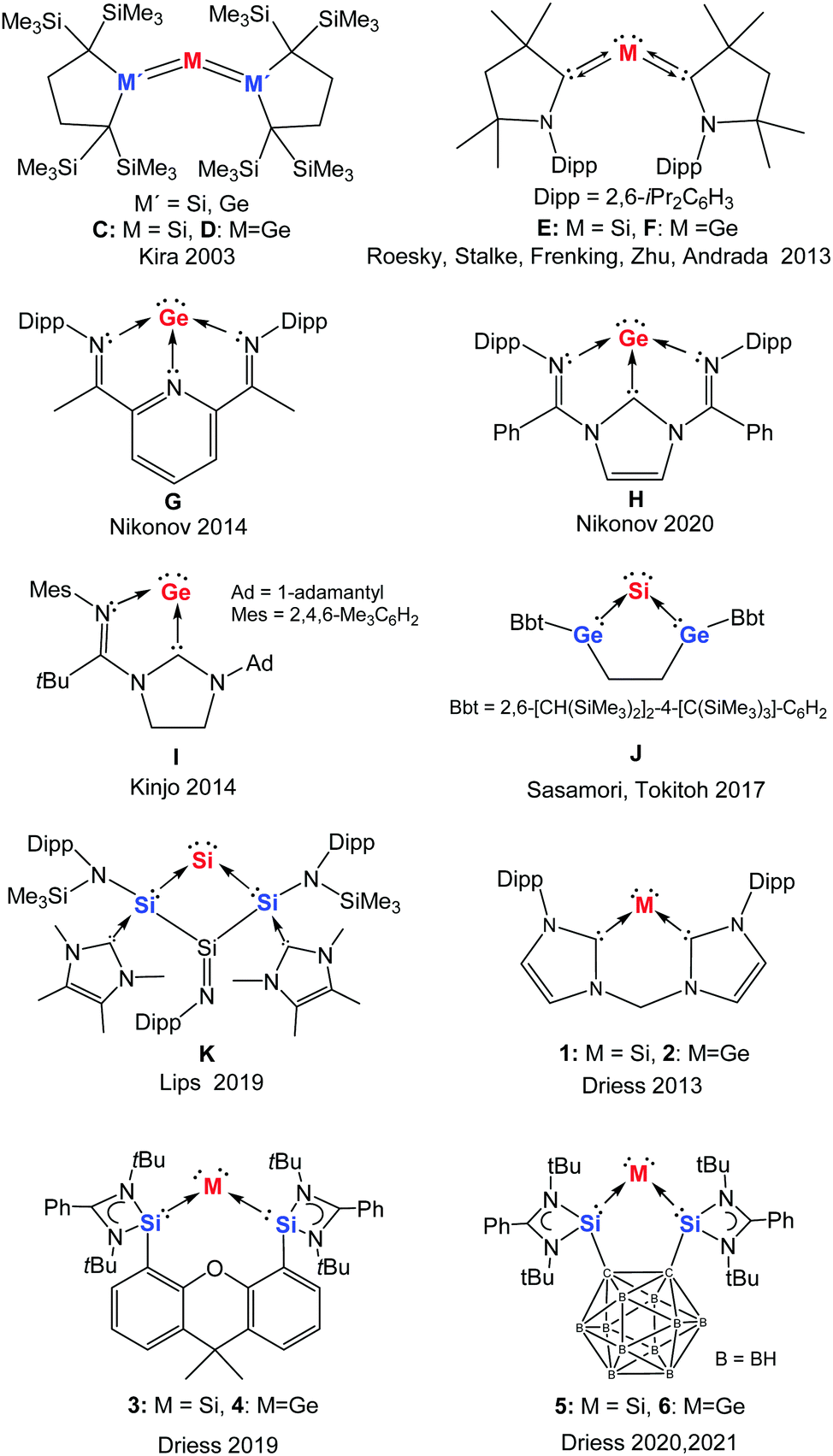 | ||
| Chart 2 Known monatomic silicon(0) and germanium(0) complexes C–K and 1–6. | ||
A deeper understanding of the bonding situation in heavy group 14 zero-valent complexes was probed by Turek et al. in 2017. By combining valence bond (VB) theory and maximum probability domain (MPD) approaches, they concluded that the bonding between the central group 14 atom and the donating ligands should be described as a resonating combination of “ylidone” and “ylidene” structures with a minor contribution of the “bent allene” structure (Chart 1 top).7 The stabilisation of silicon(0) and germanium(0) complexes with various silylene and germylene ligands was also investigated by Phukan and Gadre using density functional theory (DFT) and molecular electrostatic approaches. They concluded that such Si0 and Ge0 complexes contain very strong donor–acceptor bonds and are thermodynamically stable.25 In 2017 Sasamori and Tokitoh isolated the bent 1,3-digerma-2-silaallene J which could be better described as a germylene-coordinated Si0 compound with pronounced silylone character, rather than as a ![[double bond splayed left]](https://www.rsc.org/images/entities/char_e009.gif) GeSiGe
GeSiGe![[double bond splayed right]](https://www.rsc.org/images/entities/char_e00a.gif) heterocumulene, due to the cyclic skeleton and the σ-donating properties of the germylene moieties.26 In addition, the group of Lips synthesised the bis-silylene stabilised silylone K.27 Very recently, utilising the strongly donating bis(NHSi) (N-heterocyclic silylene) ligands we developed two new silylones (328 and 529) and two new germylones (430 and 631).
heterocumulene, due to the cyclic skeleton and the σ-donating properties of the germylene moieties.26 In addition, the group of Lips synthesised the bis-silylene stabilised silylone K.27 Very recently, utilising the strongly donating bis(NHSi) (N-heterocyclic silylene) ligands we developed two new silylones (328 and 529) and two new germylones (430 and 631).
Akin to carbones, the central silicon and germanium atoms of silylones and germylones are in the zero oxidation state and retain their four valence electrons as two lone pairs. They represent a molecular resting state of single Si and Ge atoms, which are typically only observed at high temperature in the gas phase or in interstellar matter.32–34 This type of chemical species was proposed to represent a soluble “allotrope” of the respective elements.35 Owing to the peculiar bonding situation and high electron-richness of the central atoms, these complexes exhibit diverse reactivity with access to novel compounds. Cyclic ylidones developed in our group using bis(NHC) and bis(NHSi) ligands have allowed us to more widely explore the reactivity of genuine silylones and germylones, as well as enabling some comparison with already existing acyclic analogues. This feature article is focused on the chemistry of existing isolable monatomic silicon(0) and germanium(0) complexes, highlighting their synthesis, structure, and reactivity, with a particular focus on the cyclic bis(NHC) and bis(NHSi) supported silylones and germylones. Previous results of isolable zero-valent Si, Ge, and Sn were reviewed by Sasamori36 and our group.37 The dative bonding in zero-valent group 14 complexes has also been reviewed by Frenking and co-workers recently.38–40
2. Synthesis of monatomic silicon(0) and germanium(0) complexes
2.1 Silylones and germylones C–K
The first isolable silicon compound with silylone character to be reported (mentioned above) is the trisilaallene C (Chart 2, M′ = Si) obtained by Kira and co-workers in 2003.11 It was synthesised from silicon tetrachloride and a cyclic dialkylsilylene41 in a two-step reaction, including reduction with potassium graphite. The molecular structure of C established by X-ray diffraction analysis (XRD) at −150 °C revealed a significantly bent Si–Si–Si angle [136.49(6)°] and two relatively short Si–Si bonds [2.177(1)/2.188(1) Å]. With a similar synthetic strategy, Kira and co-workers developed the first examples of 1,3-digermasilaallene C (Chart 2, M′ = Ge),42 1,3-disilagermaallene D (M′ = Si),16 and trigermaallene D (M′ = Ge).42 It is of note that the related tristannaallene (tBu3Si)4Sn3 was actually already reported by Wiberg and co-workers a few years earlier in 1999.43The acyclic silylone (cAAC)2Si E (Chart 2) was isolated by the groups of Roesky, Stalke, and Frenking in 2013.18,44 The precursor of this silylone is a stable carbene-centred biradical (cAAC)2SiCl245 resulting from the reaction of SiCl2(DNHC)46 [DNHC = 1,3-bis(Dipp)-imidazol-2-ylidene, Dipp = 2,6-diisopropylphenyl] and the free cAAC17 ligand. Reduction of the Si(IV) species (cAAC)2SiCl2 with potassium graphite in THF furnished the silylone E in good yield. It was later revealed that this silylone can additionally be obtained from either the reaction of cAAC with H2SiI2 or the reduction of the Si(I) radical (cAAC)2SiI with LiN(iPr)2.47 Although the silylone E features a silicon atom in the formal oxidation state zero, it exhibits remarkable stability. The amorphous powder of E is stable for an hour while its crystals are stable for about a day in air, according to the observed colour change. The germanium homologue (cAAC)2Ge F (Chart 2) was isolated by Roesky, Zhu, Stalke, and Andrada through a one-pot synthesis with GeCl2(dioxane), cAAC, and potassium graphite in a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2:2.1 molar ratio.21 According to DFT calculations of (cAAC)2Si and (cAAC)2Ge, the predominant contribution to the HOMO−1 is an s-type lone pair orbital and to the HOMO is a p-type orbital with largest extension at the central atoms, indicating their ylidone character.18,21 However, as a result of the highly π-accepting nature of the cAAC ligand, the ylidone character of E and F is considerably reduced, which accounts for the remarkable stability of silylone E.
:2:2.1 molar ratio.21 According to DFT calculations of (cAAC)2Si and (cAAC)2Ge, the predominant contribution to the HOMO−1 is an s-type lone pair orbital and to the HOMO is a p-type orbital with largest extension at the central atoms, indicating their ylidone character.18,21 However, as a result of the highly π-accepting nature of the cAAC ligand, the ylidone character of E and F is considerably reduced, which accounts for the remarkable stability of silylone E.
Utlising a tridentate bis(imino)pyridine ligand, which has also been employed for stabilising iron(0) complexes,48 Nikonov and co-workers were able to isolate the germanium(0) complex G (Chart 2).22 This three-coordinate germanium complex G was generated in moderate yield by the reduction of the bis(imino)pyridine [GeCl]+ adduct resulting from autoionisation of germanium dichloride. Owing to the non-innocent character of the bis(imino)pyridine platform, this formal zero-valent germanium species possesses some multiple-bond character between the Ge atom and imine-N atoms as a result of delocalisation of one Ge lone pair into the π*(CN) orbitals. A related three-coordinate tin(0) complex, resulting from the transamination reaction of a bis(imino)pyridine ligand with Sn[N(SiMe3)2]2 in diethyl ether, was described by Fischer and Flock.49
Very recently, the group of Nikonov succeeded in the synthesis of the germanium(0) complex H, in which the germanium atom is stabilised by a tridentate diimino-NHC (dimNHC) ligand.23 In contrast to the bis(imino)pyridine-supported Ge0G, complex H exhibits some interesting reactivity towards organic substrates with the involvement of the NHC-carbon(II) atom [C(II) oxidised to C(IV), vide infra].
Starting from a related bidentate (mono)imino-NHC ligand system, Kinjo and co-workers prepared a chlorogermyliumylidene via autoionisation of germanium dichloride as a suitable precursor for the germanium(0) complex I (Chart 2).24 Reduction of this precursor with potassium graphite afforded the cyclic germanium complex I. DFT calculations revealed that one of the lone pairs of the central Ge atom significantly delocalises over the five-membered C2N2Ge ring. Thus compound I may be viewed as both a germanium(0) species and a mesoionic germylene.
Sasamori and Tokitoh recently achieved the highly bent 1,3-digerma-2-silaallene J (mentioned previously, Chart 2).26 The (tetra)chlorinated precursor of J (1,1,2,5-tetrachloro-2,5-digerma-1-sila-cyclopentane) was synthesised by heating the germanium-based ligand 1,2-Bbt2-1,2-digermacyclobutene50 (Bbt = 2,6-[CH(SiMe3)2]2-4-[C(SiMe3)3]-C6H2) in neat SiCl4. The subsequent reduction of this precursor with four molar equiv. of potassium graphite in benzene afforded J in moderate yield. Significantly enhanced silylone character has been assumed compared to that of acyclic trimetallallene such as C and D, based on the relatively smaller Ge–Si–Ge angle [80.08(4)°].
Finally, Lips and co-workers described recently a carbene-induced elimination of the tertiary amine NDipp(SiMe3)2, to afford the bicyclic amido- and carbene-substituted silicon ring compound K (Chart 2) with a SiN double bond and a two-coordinate Si atom.27 Spectroscopic investigations and DFT calculations of this bifunctional compound revealed significant silylone properties for the two-coordinate Si atom.
2.2 Bis(NHC)-supported silylone 1 and germylone 2
Since the beginning of last decade, we have been interested in developing silicon(0) and germanium(0) complexes with strongly sigma-donating and chelating ligands. Such compounds would allow us to realise a highly bent angle on the central Si0 and Ge0 atoms with enhanced reactivitiy, compared with non-chelating ligands and ligands featuring poorer sigma donors e.g. imines. Our first attempt was in 2012 with the isolation of the first chlorosilyliumylidene [:SiCl]+ complex 7 with bis(tributylphosphazenyl)naphthalene as the chelating ligand (Scheme 1).51 Compound 7 seemed to be a promising precursor for the hypothetic complex 9, however, its reduction with different reducing reagents such as potassium graphite and sodium naphthalenide led merely to unidentified mixtures of products. DFT calculations of 7 revealed that the lowest unoccupied molecular orbital (LUMO) was mainly localised on the naphthalene moiety of the supporting ligand, implying that the naphthalene backbone would be reduced over the silicon(II) centre. The same is true for our first attempt to prepare germanium(0) complex 10, starting from the corresponding bis(tributylphosphazenyl)naphthalene-ligated chlorogermyliumylidene [:GeCl]+8 with its LUMO localised also on the supporting ligand (Scheme 1).52 | ||
| Scheme 1 Reduction of chlorosilyliumylidene 7 and chlorogermyliumylidene 8 supported by bis(tributylphosphazenyl)naphthalene. | ||
Considering the fact that NHCs have been employed for stabilising the diatomic silicon(0)53 and germanium(0)54 complexes in the form of (DNHC):MM:(DNHC) (M = Si, Ge), we envisioned that a chelating bis(NHC) might be capable of supporting a monatomic silicon(0) or germanium(0) species. Starting with this bidentate bis(NHC),55 a ligand exchange reaction with SiCl2(DNHC)46 in THF afforded the expected chlorosilyliumylidene [:SiCl]+ complex 11 (Scheme 2).19 As intended, both the HOMO and LUMO of 11 are mainly localised on the Si centre. A similar synthetic approach resulted in isolation of the analogous complex, bis(NHC)-ligated chlorogermyliumylidene 12 (Scheme 2).20 The HOMO and LUMO of this germanium(II) compound are also mainly located on the Ge(II) atom. With 11 and 12 as long-sought precursors for the corresponding zero-valent complexes in hand, we conducted their dechlorination with two molar equiv. of sodium naphthalenide. Indeed, the reduction of 11 in THF at −60 °C furnished the desired silylone 1 which was isolated as a dark red powder (Scheme 2).19
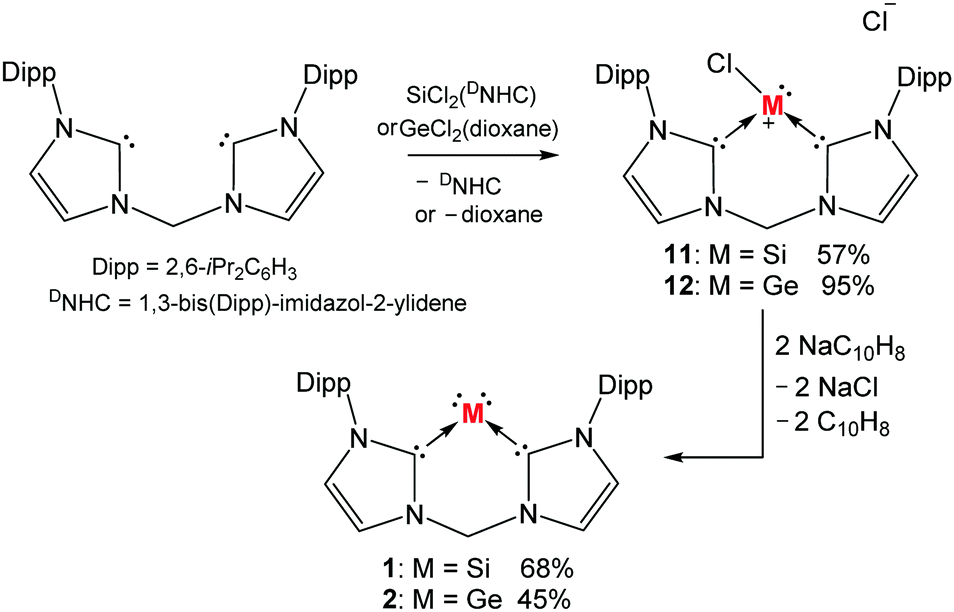 | ||
| Scheme 2 Synthesis of the bis(NHC)-supported silylone 1 and germylone 2via chlorosilyliumylidene 11 and chlorogermyliumylidene 12, respectively, starting from the bis(NHC) ligand. | ||
The molecular structure of silylone 1 established by XRD (Fig. 1) reveals two Si–C distances (av. 1.869 Å), both slightly longer than those observed for the acyclic silylone (cAAC)2Si [1.841(2) Å] reported by Roesky, Stalke, and Frenking,18 in which the cAACs act as much stronger π electron acceptors than NHCs. Consequently, the Si0 centre of 1 is more electron-rich than that in (cAAC)2Si as shown by the drastic upfield shift of the 29Si NMR resonance [1: δ = −80.1 ppm vs. δ = 66.7 ppm for (cAAC)2Si]. Moreover, silylone 1 shows larger first and second proton affinities [PA(1) = 283.4, PA(2) = 168.3 kcal mol−1] in comparison to those of (cAAC)2Si (268.8, 155.3 kcal mol−1) as suggested by DFT calculations based on molecular models.19 In line with these calculations, two lone pairs of electrons are confirmed at the silicon centre of silylone 1. The HOMO involves a silicon π-orbital with small degree of Si–C π bonding and the HOMO−1 corresponds to a silicon σ-lone pair orbital. Notably, silylone 1 shows a deep-red colour in toluene and an absorption maximum at λ = 547 nm (ε = 7.5 × 103) was observed in the UV-vis spectrum.
 | ||
| Fig. 1 Molecular structures of the bis(NHC)-supported silylone 1 (M = Si) and germylone 2 (M = Ge) (hydrogen atoms are omitted for clarity). | ||
As expected, the reduction of the bis(NHC)-supported chlorogermyliumylidene [:GeCl]+12 with two molar equiv. of sodium naphthalenide in THF at −30 °C furnishes the germylone 2 as a dark red powder (Scheme 2).20 The molecular structure established by XRD is isostructural with that of the silicon analogue 1 (Fig. 1). The two Ge–C distances of 1.967(2) and 1.962(2) Å in 2 are slightly longer than those in the acyclic (cAAC)2Ge complexes [1.9386(16)–1.954(2) Å] described by Roesky, Zhu, Stalke, and Andrada,21 indicating again the stronger electron accepting effect of cAACs vs. NHCs as supporting ligands. As suggested by the DFT calculations, the HOMO of 2 corresponds to a π-type orbital located at the Ge centre with appreciable Ge–C π bonding character, while the HOMO−1 involves a σ lone pair orbital at the Ge centre.20 The proton affinities of compound 2 [PA(1) = 279.6, PA(2) = 175.0 kcal mol−1] confirm the presence of two lone pairs at the germanium(0) centre.
2.3 Bis(NHSi)-ligated silylones and germylones 3–6
Silylenes are divalent silicon species with two valence electrons as a lone pair and a formally empty orbital at the silicon centre.56–59 Three-coordinate amidinato silylenes, PhC(NtBu)2SiR (R = organic substituent), derived from [PhC(NtBu)2SiCl],60 can serve as strong donating ligands in coordination chemistry due to the intramolecular electron donation of an imino moiety to the silylene centre. For instance, a digermanium(0) complex stabilised by the tricoordinate silylene [PhC (NtBu)2]Si(N(SiMe3)2) was reported by So and co-workers.61 Based on this type of NHSis, we recently developed several chelating bis(NHSi) systems and successfully applied them in transition metal coordination and catalysis.62–65 As the latter NHSis are generally stronger Lewis donors with respect to NHCs,66 we realised that such chelating NHSi ligands could also be applied for synthesising more reactive silylones and germylones. Starting with the pincer-type tridentate bis(NHSi)pyridine 13,67 we synthesised the germanium(II) complex 14 as a precursor for a bis(NHSi) supported germylone (Scheme 3). While attempts for bis(NHSi)pyridine-Ge0 complex 15via reductive dechlorination of 14 failed, the reaction with K2Fe(CO)4 resulted in the isolation of 16 as an iron(0) complex of the desired bis(NHSi)pyridene-Ge0 species, whereby the Fe(CO)4 moiety acts as a Lewis acid to stabilise one of the germylone lone pairs. The molecular structure of 16 shows a three-coordinate germanium atom bearing an Fe(CO)4 moiety in addition to two NHSis, adopting a trigonal-pyramidal geometry with a lone pair of electrons occupying the apex. Interestingly, insertion of GeCl2 into the Ge→Fe donor–acceptor bond of 16 occurred when 16 was reacted with GeCl2(dioxane), furnishing complex 17 (Scheme 3). | ||
| Scheme 3 Attempt to synthesise the bis(NHSi) supported germylone 15via chlorogermyliumylidene 14, starting from bis(NHSi) 13, and the formation of 16 and 17, respectively. | ||
Considering that the failure to achieve the bis(NHSi)-supported germylone 15 may be owing to the flexibility of the bis(NHSi)pyridine ligand, we selected the rigid bis(NHSi) 1868 with a xanthene scaffold in order to synthesise a bis(NHSi)-supported silylone and germylone (Scheme 4). Dropwise addition of one molar equiv. of SiCl2(DNHC)46 in diethyl ether into a solution of 18 at room temperature leads to the formation of chlorosilyliumylidene chloride 19 as a yellow precipitate.28 It is of note that further reaction of 19 with one additional molar equiv. of SiCl2(DNHC) can occur via the insertion of :SiCl2 into the Si–Cl bond of 19, affording the chlorosilane complex 20.
 | ||
| Scheme 4 Reaction of the bis(NHSi)xanthene 18 with SiCl2(DNHC) to afford the silicon(II) chloride complexes 19 and 20 as well as the synthesis of bis(NHSi)-supported silylone 3 from 19. | ||
Starting from 19, reduction with two molar equiv. of potassium graphite in THF at ambient temperature affords a dark purple solution, from which the desired bis(NHSi)-supported silylone 3 is isolated as dark purple crystals (Scheme 4).28 Compound 3 is considerably sensitive towards air and moisture. Its 29Si NMR spectrum exhibits a resonance at δ −187.5 ppm in C6D6 assignable to the silicon(0) atom. This signal is significantly upfield shifted with respect to that of the bis(NHC)-supported silylone 1 (δ −80.1 ppm in C6D6), indicating the stronger σ-donating nature of the NHSi–Si(II) atoms.
In the crystal structure, two independent molecules of 3 with nearly identical geometric parameters are present in the asymmetric unit (Fig. 2).28 The Si–Si bond distances of 3 ranging from 2.2451(7) to 2.2586(7) Å are substantially longer than the analogous Si–Si distances observed in trisilaallene C (Chart 2, M′ = Si) [2.177(1) and 2.188(1) Å] with two cyclic alkyl silylenes,11 implying the weaker π-accepting properties of an NHSi compared to that in a cyclic alkyl silylene. The Si–Si–Si bond angles [104.38(3)° and 103.87(3)°] in the two independent molecules of 3 are considerably narrower than that in trisilaallene C (Chart 2 M′ = Si) [136.49(6)°].11 The electronic structure of silylone 3 has been investigated by DFT calculations and Natural Bond Orbital (NBO) analysis, which revealed the presence of two perpendicular lone-pairs of electrons on the central silicon(0) atom, i.e., an sp0.41-type lone-pair and a delocalised p lone-pair.28 Despite the longer Si–Si distances mentioned above, the Wiberg Bond Index (WBI) of the Si–Si bonds in 3 is 1.40, indicating there is still some double bond character to the Si–Si bonds.
 | ||
| Fig. 2 Molecular structures of the xanthene-based bis(NHSi)-supported silylone 3 (M = Si) and germylone 4 (M = Ge) (hydrogen atoms are omitted for clarity). | ||
Akin to the silicon analogue 19, the chlorogermyliumylidene chloride 21 supported by (bis-NHSi)xanthene can be prepared by treatment of the bis(NHSi) ligand 18 with one molar equiv. of GeCl2(dioxane) in diethyl ether at room temperature (Scheme 5).30 In contrast to the silicon analogue, no further reaction of 21 with GeCl2(dioxane) was observed, which makes the synthesis of 21 easier. Dechlorination of 21 with two molar equiv. of potassium graphite in THF at room temperature furnishes the desired bis(NHSi)-supported germylone 4 as dark blue crystals (Scheme 5). In the molecular structure of 4, established by XRD (Fig. 2), the Ge–Si distances of 2.3147(9) and 2.3190(9) Å are considerably longer with respect to those in 2-germadisilaallene D [Chart 2, M′ = Si, 2.2366(7) and 2.2373(7) Å] again due to the weaker π-accepting ability of the NHSis compared to the cyclic alkyl silylenes. Moreover, the Si–Ge–Si angle [102.87(3)°] of 4 is much more acute than that of D [132.38(2)°]. The calculated WBI of the Si–Ge bonds in 4 is 1.34, indicating a partial double bond character.30 Similar to the silicon analogue 3, the computational analysis suggests that germylone 4 features a Ge0 atom with two perpendicular lone pairs, with some electron delocalisation into both silicon(II) formally vacant orbitals.
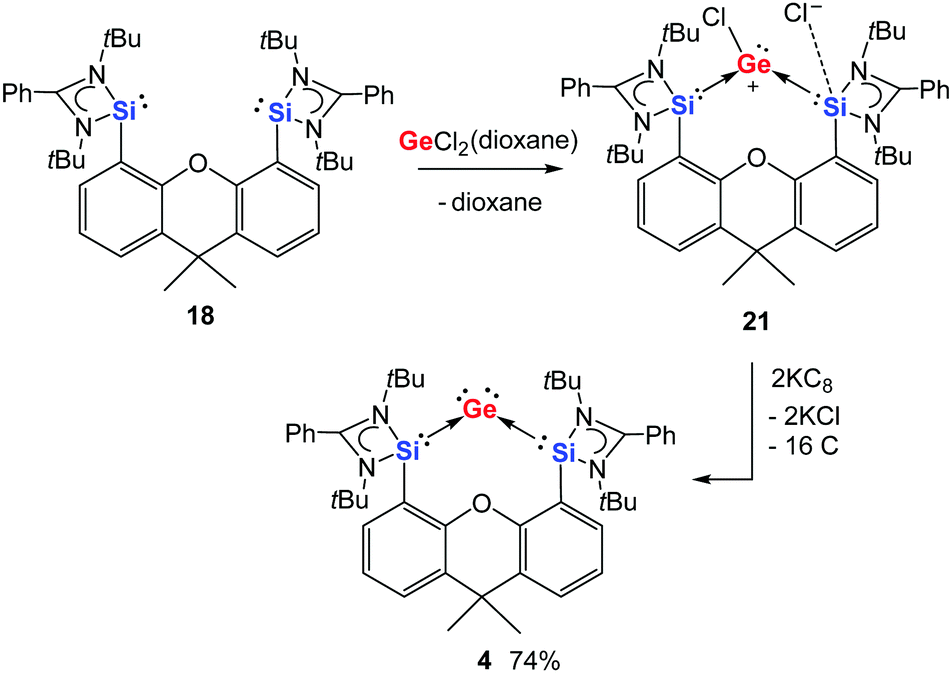 | ||
| Scheme 5 Synthesis of bis(NHSi)xanthene-supported germylone 4 from 18via the chlorogermyliumylidene 21. | ||
To realise a silylone and a germylone with an even more rigid bis(NHSi) ligand, we recently paid our attention to the bis(NHSi)-o-carborane 2269 (Scheme 6) with a Si⋯Si distance of ca. 3.3 Å documented by our group in 2016. Notably, o-carboranes can easily undergo cluster core opening via two-electron reduction with alkaline metals to form nido-carborane dianions.70,71 Accordingly, the reaction of 22 with two molar equiv. of potassium graphite leads expectedly to the desired dipotassium 1,2-bis(silylenyl)-nido-carborane salt 23. Compound 23 undergoes a metathesis reaction with SiCl2(DNHC)46 or better SiI2(DNHC)72 in THF to afford the bis(NHSi)-supported silylone 5 as a red powder.29
 | ||
| Scheme 6 Synthesis of the (bis-NHSi)carborane supported silicon(0) (5) and germanium (6) complexes from bis(NHSi) ligand 22via the bis(NHSi) dianion 23. | ||
In the 29Si{1H} NMR spectrum of 5, the signal of the central Si0 atom was observed at δ −263.8 ppm, which is even more strongly shielded by 76 ppm compared to that of NHSi-supported silylone 3 (δ −187.5 ppm).29 The molecular structure of 5 has a C2v symmetry with a planar five-membered C2Si3 ring with two Si–Si distances of [2.2272(6) and 2.2225(6) Å] (Fig. 3), which are comparable to those observed for 3 [2.2451(7)–2.2586(7) Å]. Remarkably, silylone 5 exhibits a very narrow Si–Si–Si angle [82.75(2)°] relative to that in 3 (104°).28 According to quantum chemical calculations, the HOMO and the HOMO−1 of 5 correspond to the silylone π- and σ-symmetry lone pairs, respectively, resembling the situations of silylones 119 and 3.28
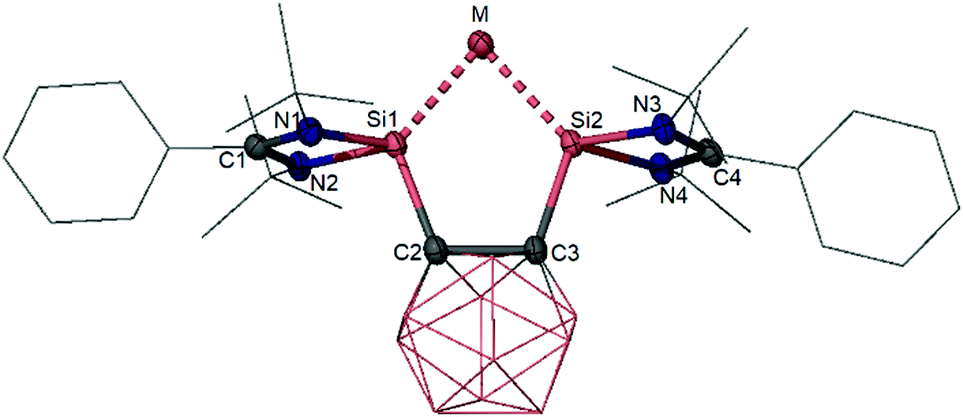 | ||
| Fig. 3 Molecular structures of the o-caborane-based bis(NHSi) supported silylone 5 (M = Si) and germylone 6 (M = Ge) (hydrogen atoms are omitted for clarity). | ||
Treatment of the nido-carborane dianions 23 with one molar equiv. of GeCl2(dioxane) at room temperature in THF resulted in formation of the carborane-based germylone 6 which is isolated as a brown-red powder (Scheme 6).31 Germylone 6 is isostructural with its silicon analogue 5 (Fig. 3) with Ge–Si distances [2.2896(5) and 2.2846(5) Å] close to those observed for the xanthene-based germylone 4 [2.3147(9) and 2.23190(9) Å].30 However, the Si–Ge–Si angle of 80.59(2)° in 6 is much more acute with respect to that in the xanthene-based germylone 4 [102.87(3)°]30 and even smaller than that of bis(NHC)-supported germylone 2 [86.6(1)°].20 As a genuine germylone, the HOMO and the HOMO−1 of 6 correspond to the germylone lone pairs with π- and σ-symmetry respectively.31
3. Reactivity of monatomic silicon(0) and germanium(0) complexes
3.1 Acting as a Lewis base
Since silylones and germylones feature two lone pairs of electrons at the central silicon and germanium atoms, they are expected to act as Lewis bases to form stable donor–acceptor adducts with appropriate Lewis acids. This was first proven by the reaction of silylone 1 with one molar equiv. of GaCl3 in THF, affording the silylone–GaCl3 complex 24 as a yellow solid (Scheme 7).73 The molecular structure of 24 exhibits a three-coordinate silicon atom adopting a pseudotetrahedral coordination geometry with a lone pair of electrons at the vertex position (Fig. 4). This suggests that one of the lone pairs at the silicon centre is donated to the GaCl3 moiety.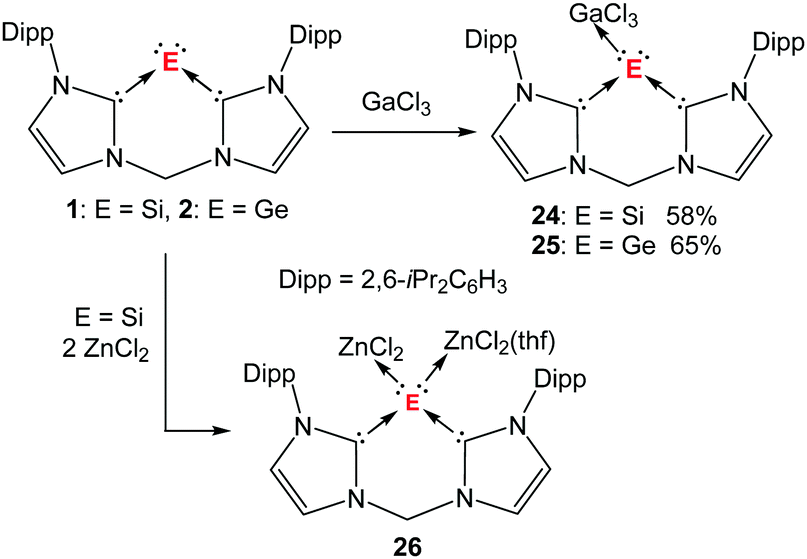 | ||
| Scheme 7 Formation of GaCl3 adducts of bis(NHC)-supported silylone 24 and germylone 25 from 1 and 2, respectively, and the two-fold ZnCl2 adduct 26 from 1. | ||
 | ||
| Fig. 4 Molecular structure of the silylone–GaCl3 adduct 24 (hydrogen atoms are omitted for clarity). | ||
By analogy, coordination of 2 with GaCl3 yields the germanium complex 25 as a yellow solid (Scheme 7).74 Compound 25 is isostructural to its silicon analogue 24 and possesses also a lone pair on the germanium centre. It is worth mentioning that coordination of the germanium compound I (Chart 2) with M(CO)5 (M = Cr, Mo, W) leads also to 1:1 adducts.75 As shown by Kinjo and co-workers, however, the germanium centres therein possess a trigonal planar geometry without any indication for a lone pair on the germanium centre.
The bis(NHC)-supported silylone 1 and germylone 2 react also readily with other group 13 Lewis acids such as AlBr3 and BCl3, but the desired products could not be isolated, presumably due to the formation of both mono and bis-Lewis-acid adducts. To our delight, a bis-Lewis-acid adduct 26 with silylone acting as a 2-fold donor toward ZnCl2 has been obtained from silylone 1 and characterised structurally (Scheme 7).37 In the molecular structure of 26 the central silicon atom, coordinated by a chelating bis(NHC) and two ZnCl2 molecules, adopts a pseudotetrahedral coordination environment (Fig. 5). The coordination environment of the two Zn atoms in 26 is different: one in a trigonal planar geometry, the other additionally coordinated by a THF molecule.
 | ||
| Fig. 5 Molecular structure of the silylone–ZnCl2 adduct 26 (hydrogen atoms are omitted for clarity). | ||
Although the xanthene-based bis(NHSi)-supported silylone 3 and germylone 4 possess some double bond character between the silylene–Si atoms and the central silicon or germanium atom, they show coordination ability toward one and even two Lewis acids. This has been demonstrated by the Lewis adduct formation with AlBr3. The reaction of germylone 4 with one molar equiv. of AlBr3 leads smoothly to the mono-AlBr3 adduct 27 which can be isolated as an orange solid (Scheme 8).30 In the molecular structure of 27, the germanium atom adopts a pseudotetrahedral coordination geometry, implying the presence of a lone pair of electrons at the apex.
 | ||
| Scheme 8 Formation of AlBr3 adducts 27 and 28 with xanthene-based bis(NHSi) germylone 4. | ||
Further coordination of AlBr3 with the mono-AlBr3 adduct 27 to form the bis-AlBr3 adduct 28 is dependent on the solvent used (Scheme 8).30 Due to the stronger coordination ability of the donor solvents than that of 27, the desired bis-AlBr3 adduct 28 could not be formed in ethereal solvent such as Et2O and THF even in the presence of the 20-fold molar excess of AlBr3. In benzene solutions, however, the adduct 28 can be obtained in the form of a white precipitate and characterised with XRD.
Interestingly, the o-carborane-bis(NHSi)-supported germylone 6 can react with GeCl2-dioxane, yielding exclusively complex 29 as a yellow solid regardless of the ratio of starting materials (Scheme 9).31 The crystal structure of 29 reveals a four-coordinate germanium centre attached to two germylone–Ge atoms and two chlorine atoms. The central germanium centre adopts a seesaw coordination geometry with both chloride atoms located at the axial positions, suggesting that one of the three equatorial positions is occupied by a lone pair (Fig. 6). Compound 29 can be viewed as a Ge02GeII adduct, in which each Ge0 atom still features a stereochemically active lone pair, as indicated by its pyramidal coordination geometry. Notably, this complex can serve as a precursor for the novel bis(NHSi)-supported neutral Ge2 complex 30 (Scheme 9), which is obtained as a minor isolable product along with germylone 6 when reduced using two molar equiv. of potassium naphthalenide.
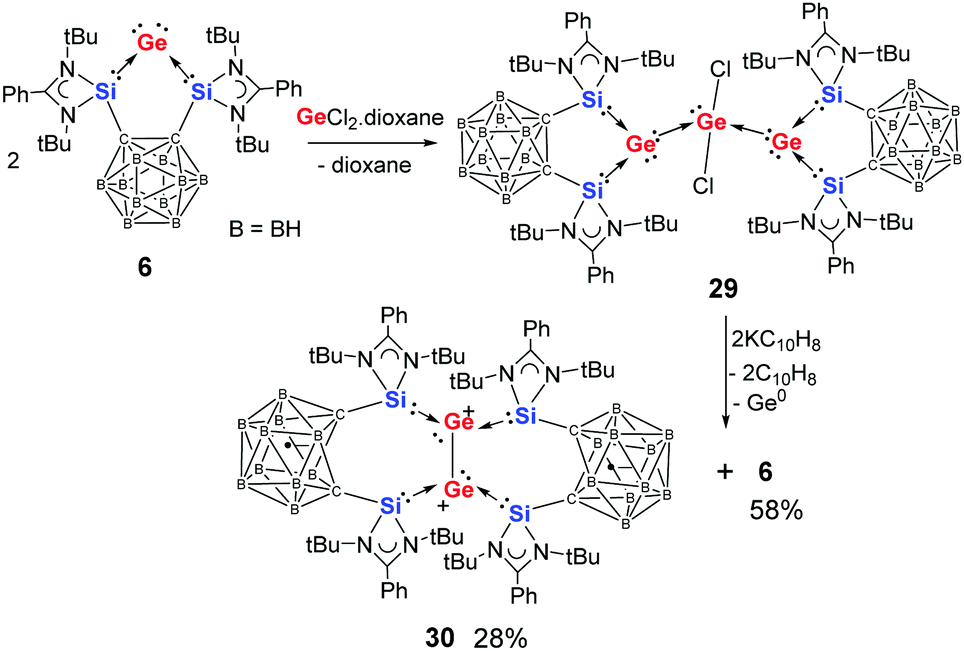 | ||
| Scheme 9 Formation of the two-fold germylone–GeCl2 adduct 29 and its reduction affording germylone 6 and the diradical dimer 30. | ||
 | ||
| Fig. 6 Molecular structure of the GeCl2–germylone adduct 29 (hydrogen atoms are omitted for clarity). | ||
It is of note, due to the electron-rich nature of silylones and germylones, some of them can also act as reducing agents towards germanium(II) and silicon(II) dichloride complexes to generate elemental germanium and silicon, respectively, as shown in Scheme 10. For instance, the reaction of silylone 1 or germylone 2 with SiCl2(DNHC) leads to the known disilicon(0) complex (DNHC)SiSi(DNHC),53 along with elemental silicon. In both cases, 1 and 2 convert back to their respective precursors 11 and 12 during the process. Similarly, utilising the germylone K (Chart 2), Nikonov and co-workers explored very recently the reaction with GeCl2(dioxane), giving rise to the corresponding [GeCl]+ complex with [GeCl3]− as counterion.23
 | ||
| Scheme 10 Reaction of silylone 1 and germylone 2 with GeCl2(dioxane) and SiCl2(DNHC)complexes, respectively. | ||
3.2 Oxidation with elemental chalcogens
Since silylones bear a silicon(0) centre, they may be transformed with elemental chalcogens to monomeric silicon(II) and silicon(IV) chalcogenide complexes. In fact, facile reaction between silylone 1 with elemental sulphur leads to the bis(NHC)-supported SiS2 complex 31 as a colourless powder (Scheme 11).73 Moreover, compound 31 can coordinate with GaCl3 to form a push–pull complex 32 as colourless crystals suitable for XRD characterisation. Alternatively, adduct 32 can be prepared through the reaction of the GaCl3 complex 24 with elemental sulphur. The molecular structure of 32 reveals a strongly bent SSiS unit with GaCl3 coordinating to one of the sulphide atoms and the new silicon(IV) centre remains ligated to the bis(NHC).
 | ||
| Scheme 11 Reaction of silylone 1 and its GaCl3 complex 24 with elemental sulphur, selenium, and TeP(nBu)3, respectively. | ||
In contrast to the reaction of 24 with sulphur, which leads directly to a silicon(IV) species, the reaction of 24 with red selenium (Se8) is controllable and solvent dependent, affording the two silicon selenide complexes 33 and 34 (Scheme 11).76 In acetonitrile, 24 reacts with red selenium at room temperature for 3 h to afford the silicon(II) selenide 33 as an isolable product, while the reaction of 24 with red selenium in THF furnishes directly the silicon(IV) selenide 34. Conversion of the silicon monoselenide 33 to the diselenide 34 with red selenium is observed in THF. As expected, compound 34 is isostructural with its sulphur analogue 32, baring a strongly bent SiSe2 moiety. The molecular structure of 33 features an :SiSe moiety with a terminal Se and a tetrahedrally coordinated silicon centre, ligated by the bis(NHC) and the gallium atom of GaCl3. The silicon–selenium distance (ca. 2.14 Å) of 33 suggests appreciable Si–Se double bond character. It should be mentioned that complexes of heavier analogues of CO are rare. Known examples include SnO,77 PbO,77 and PbSe78 coordinated by supporting ligands at both ends of the heavy CO analogues. Complex 33 represents the first example of a heavy homologue of CO complexes only supported at the group 14 (silicon) site.
In sharp contrast to the reaction of 24 with elemental sulphur and selenium, 24 does not react with elemental tellurium. Utilising TeP(nBu)3 as a more reactive tellurium source, however, 24 can be transformed to the isolable bis(NHC)SiTe235 as orange crystals (Scheme 11).76 The molecular structure of 35 established by XRD exhibits a strongly bent TeSiTe moiety (Te–Si–Te angle: 128.4°) with the silicon centre additionally coordinated by the bis(NHC) ligand (Fig. 7). The two Si–Te distances are slightly different [2.389(4) and 2.436(2) Å]. Compounds 32, 34, and 35 represent the first series of monomeric SiX2 (X = S, Se, Te) complexes as heavy homologues of CO2. Very recently, starting from the silicon(0) complex K, Lips obtained another series of SiX2 complexes K-1 (Chart 3) with one of the chalcogen atoms interacting with the silicon atom of the SiN(Dipp) moiety of the ligand as well.79 It is noted that dimeric SiX2 (X = S,80 Se,81 Te82) stabilised by NHC or cAAC have also been structurally characterised. A donor and acceptor supported SiO2 complex has also been described recently.83
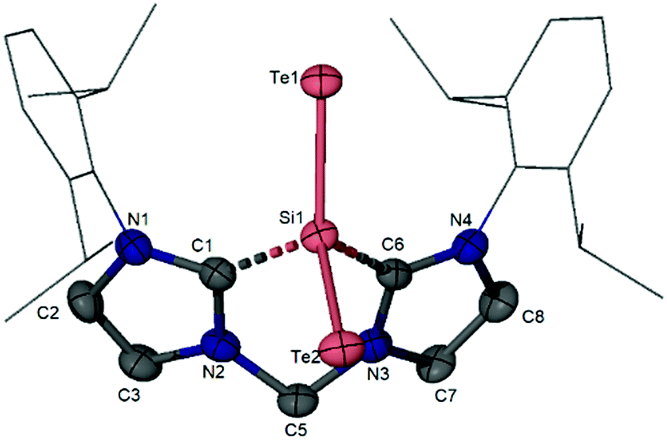 | ||
| Fig. 7 Molecular structure of the bis(NHC)-supported SiTe2 complex 35 (hydrogen atoms are omitted for clarity). | ||
 | ||
| Chart 3 Oxidation products K-1, E-1, I-1,2, H-1,2,3,4,5 resulting from silylones K and E as well as germylones I and H, respectively. | ||
Akin to 24, the germanium analogue 25 reacts readily with elemental sulphur to afford the GeS2 complex 36 as a colourless solid (Scheme 12).74 While attempts to get the germanium(II) monosulphide complex from 25 failed, the complexes of germanium(II) monoselenide 37 and monotelluride 38 result from the reaction of 25 with red selenium and elemental tellurium, respectively. Moreover, the germanium(II) complex 37 can be converted to the germanium(IV) species 39 with red selenium in THF. Remarkably, treatment of 37 and 39 with elemental sulphur leads to the GeS2 complex 36 under elimination of elemental selenium and tellurium, respectively.
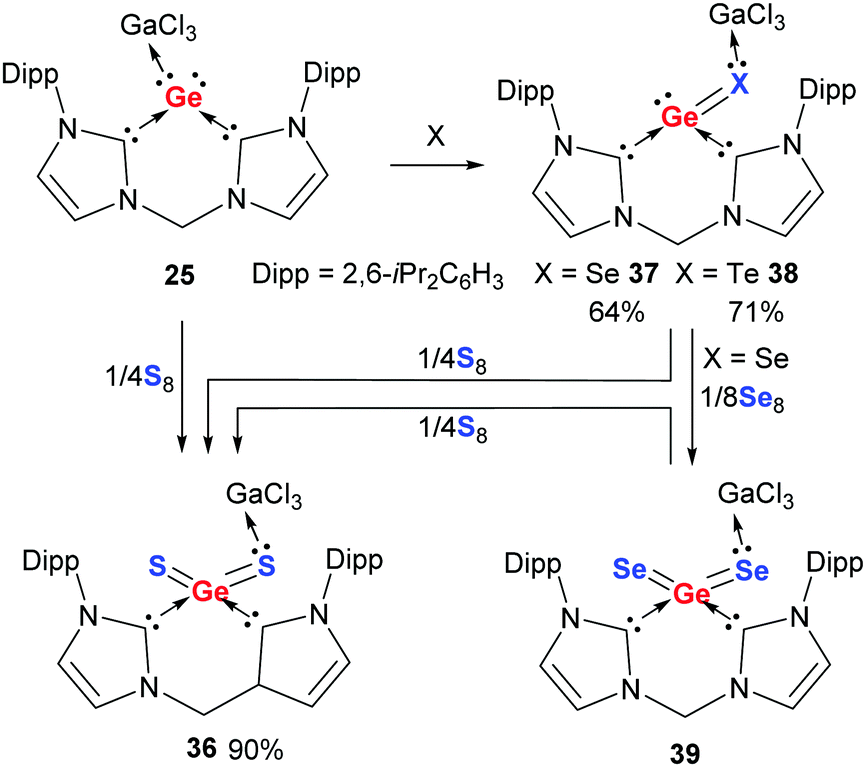 | ||
| Scheme 12 Reaction of germylone–GaCl3 adduct 25 with elemental Sulphur, selenium, and tellurium to afford 36–39, respectively. | ||
The molecular structures of the germanium(IV) dichalcogenides 36 and 39 resemble those of the silicon analogues 32 and 34. In contrast, the molecular structures of germanium(II) complexes 37 and 38 show completely different structures compared with that of the silicon monoselenide complex 33. Complexes 37 and 38 possess a lone pair at the germanium centre with the GaCl3 moiety coordinated to the Se and Te atoms, while 33 features a SiSe bond and has the GaCl3 attached to the silicon centre. This marks a significant difference between divalent silicon and germanium centres in terms of coordination to a Lewis acid: the lone pair of silicon(II) is much more active than that of germanium(II).
3.3 Activation of CO2 and N2O
Although silicon(0) and germanium(0) complexes are extremely sensitive toward dioxygen, leading to SiO2 or GeO2 with the liberation of their respective ligands,44 the bis(NHC)- and bis(NHSi)-supported silylones allowed us to control the extent of reaction, from which we were able to isolate several novel silicon compounds by using CO2 and N2O as oxygenation agents. For instance, the reaction of silylone 1 with CO2 at low temperature affords the silicon(IV) dicarbonate complex (bis-NHC)Si(CO3)240 (Scheme 13).84 Indeed, the latter product represents the first isolable molecular silicon dicarbonate complex. According to DFT calculations, (bis-NHC)SiO 41 and (bis-NHC)SiO242 are formed as reactive intermediates during the reaction. The formation of 42 proceeds exergonically by only −88 kJ mol−1 and 41 is even formed endergonically (12 kJ mol−1). Due to the extremely polar Si–O bonds in 42, the reaction with two additional molecules of CO2 to afford 40 occurs with a free-energy gain of −81 kJ mol−1. The calculated reaction free energies for the reaction of 1 with dioxygen are −236 and −583 kJ mol−1 for the mono- and di-oxygenation respectively, rationalising the uncontrollable reaction with O2 mentioned above. It should be mentioned that the reaction of germylone 2 with CO2 led merely to the Lewis adduct (bis-NHC)(CO2)2 along with a precipitate of GeO2.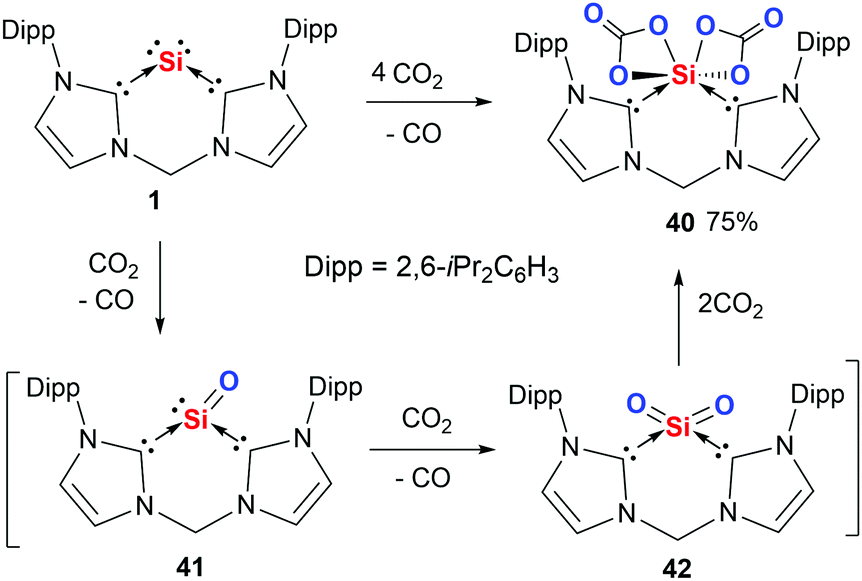 | ||
| Scheme 13 Reaction of silylone 1 with CO2 to furnish 40via the SiO and SiO2 intermediates 41 and 42. | ||
While the reaction of bis(NHC)-supported silylone 1 with N2O remains uncontrollable, slow exposure of a diethyl ether solution of the bis(NHSi)-stablised silylone 3 to two molar equiv. of N2O leads to the gradual crystallisation of the novel silicon oxide complex 43 as yellow crystals (Scheme 14).28 In the presence of excess N2O, silylone 3 is also partially oxidised to 43 by only two equiv. of N2O. Presumably, further oxidation by N2O is prevented by the poor solubility of the product 43. In contrast, when a diethyl ether solution of 3 is mixed with N2O gas in a molar ratio of 1:1, compound 44 precipitates gradually as colourless crystals. Interestingly, the Ph group of the amidinato group is reduced/undergoes a cycloadditon. Notably, exposing 44 to excess N2O only causes its decomposition and no further oxidation to 43 is observed, excluding compound 44 as an intermediate in the formation of 43.
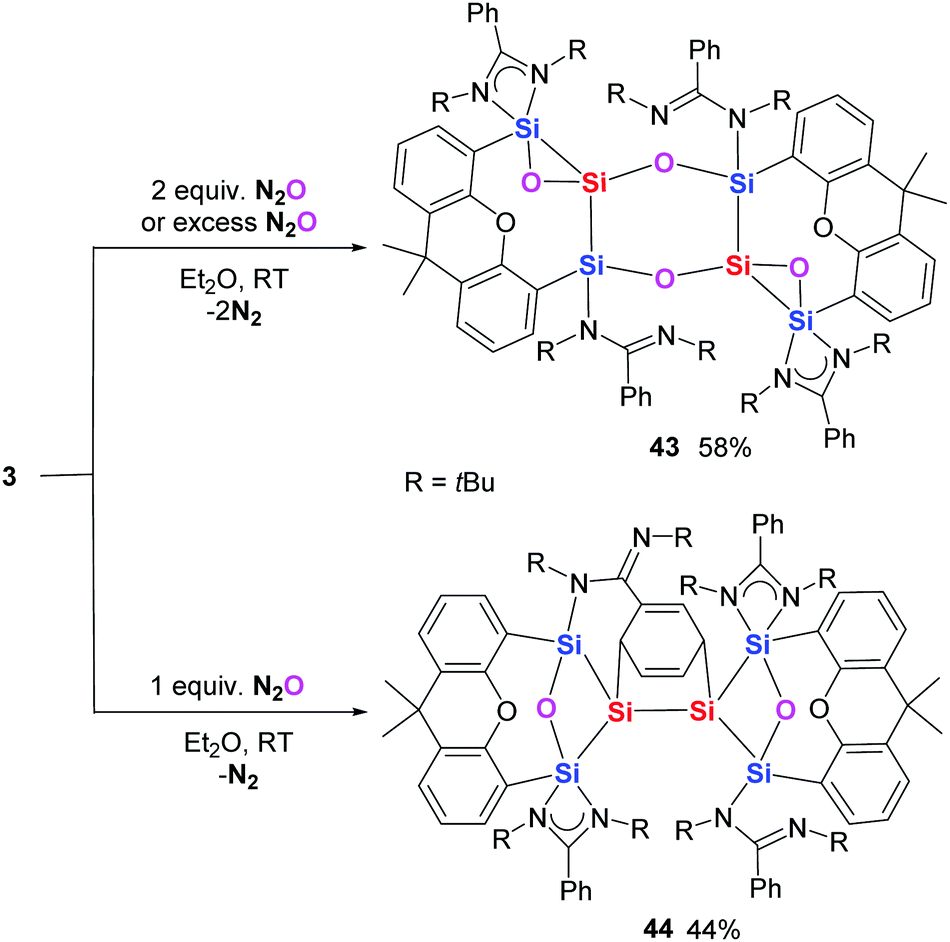 | ||
| Scheme 14 Reaction of silylone 3 with N2O under different conditions to yield 43 and 44. | ||
3.4 Activation of σ bonds in organic substrates
With two high energy lone pairs, silylones and germylones are expected to readily activate a range of chemical bonds from suitable organic substrates, including E–H (E = B, C, N) and C–X (X = halogen) bonds. In these addition reactions, the group 14(0) centre can be oxidised to the +2 or +4 oxidation state, whilst the ligand acts in a cooperative manner. With the reported ylidones featuring a variety of supporting ligands, the acceptor orbitals are specific to each complex and substrate, affecting the outcome of reaction significantly. Although (cAAC)2Si E is relatively stable, Roesky and co-workers described an intramolecular insertion of the silicon(0) centre into one of the Dipp C–H bonds of the supporting cAAC ligands, yielding the cyclic cAAC-stabilised silylene E-1 (Chart 3).85 This transformation was induced by treating the silylone E with elemental potassium and believed to proceed via a cAAC-carbon-centred radical anion, rather than the result of an intrinsic reactivity of the Si0 site in the silylone. In a nucleophilic substitution type reaction, germylone I can be controllably methylated with one molar equiv. of methyl trifluoromethanesulfonate (MeOTf) to furnish the germyliumylidene ion I-1 (Chart 3) and with two equiv. of MeOTf to afford the Ge(IV) species I-2, shown by Kinjo and co-workers.75 In the case of the (dimNHC)Ge H developed in 2020 by Nikonov,23 the carbene donor of the tridentate ligand is involved in H/C–X (X = halogen) activation. In reactions with HCl, MeI, PhI and C5F5N, a Ge–X bond and a C–H or C–C bond form at the carbene-C to give the new species H-1-4 (Chart 3) respectively.23 Additionally, the germylone H also undergoes a cycloaddition reaction with the CO moieties in tetrachloro-o-benzoquinone to afford an NHC-supported Ge(IV) bis(catecholate) H-5 (Chart 3).
Using the bis(NHSi)-supported germylone 4, B–H activation can be achieved. Treating compound 4 with 1 molar equiv. of 9-borabicyclo[3.3.1]nonane (9-BBN) in toluene at room temperature yields the unexpected silylene-stabilised boryl(silyl)germylene 45 as dark red crystals (Scheme 15).30 It became evident from the structure of 45 that the boron-bound hydrogen atom of 9-BBN is added onto the carbon atom of the amidinate ring. At the Ge0 site we see oxidation of Ge0 to GeII with a new Ge–B and Ge–Si bond. The NHSi ligand acts as the hydride acceptor in this case leading to the formation of an electron-sharing Ge–Si bond and saturation of the amidinato ring.
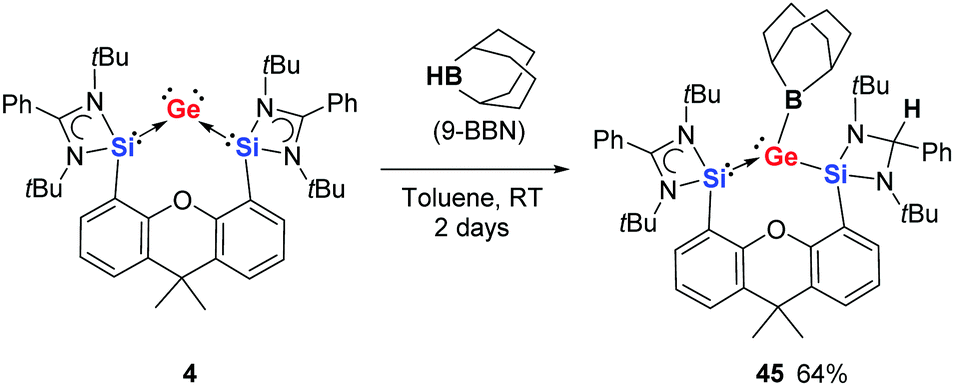 | ||
| Scheme 15 Reaction of germylone 4 with 9-BBN to give 45. | ||
Inspired by the recent reports using low-valent group 14 species for activating NH3,86–88 we investigated the reactivity of silylone 3 toward NH3. Exposure of 3 to 1 bar of ammonia at room temperature leads to the isolation of 1,3-diaminotrisilane 46 as colourless crystals (Scheme 16).28 Here we see two N–H bonds from two ammonia molecules add across the Si0–SiII bond to form two new Si–H bonds and two new Si–N bonds. All the silicon centres are fully oxidised to the +4 oxidation state. Notably, activation of NH3 by germylone 4 is also possible according to in situ NMR, affording a germanium analogue of 46 that is unstable and gradually converted to an unidentified mixture.
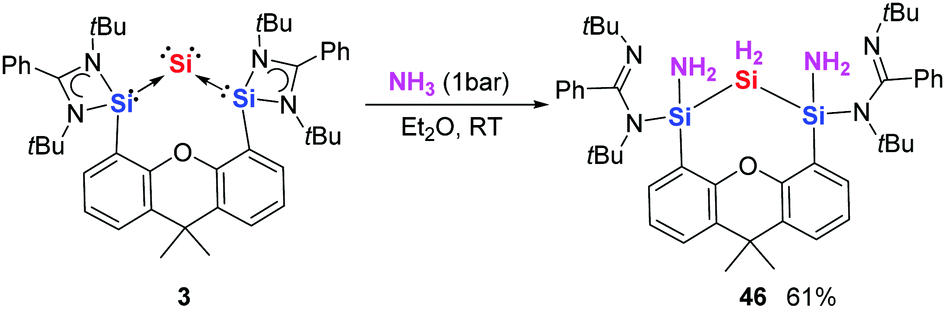 | ||
| Scheme 16 Activation of NH3 with silylone 3 to generate 46. | ||
3.5 Forming FLPs for small molecule activation
Low valent silicon compounds such as acyclic silylenes and silicon–silicon multiple bond-containing species have recently demonstrated their ability for activating H2 and ethylene.89–95 This progress prompted us to examine the reactivity of silylones toward these gases. Unfortunately, monitoring of the 1H NMR spectra of silylone 3 in H2 or C2H4 atmosphere reveals that the silylone is inert toward either of these gases, due to the lack of an appropriate acceptor orbital at the Si0 centre or on the ligand. Frustrated Lewis Pairs (FLPs) take advantage of the potential for cooperation between unquenched bulky Lewis bases and Lewis acids to activate poorly reactive small molecules and have recently attracted much attention.96,97 Considering the electron-rich character of the bulky silylone 3 and germylone 4, we expected that it either could behave as a suitable Lewis base partner in conjunction with a bulky Lewis acid for the activation of H2. Accordingly, a mixture of 3 and BPh3 was exposed to 1 bar of hydrogen gas, which results in heterolytic H2 cleavage and the formation of compound 47 as a yellow salt (Scheme 17).28 Formation of the Si–H and B–H moieties have been unambiguously confirmed by NMR spectroscopy and XRD (Fig. 8). The cation of 47 represents the first [:SiH]+ complex stabilised by a bis-silylene. | ||
| Scheme 17 Activation of H2 and C2H4 with silylone 3 and germylone 4, respectively, in the presence of BPh3 to generate 47–49. | ||
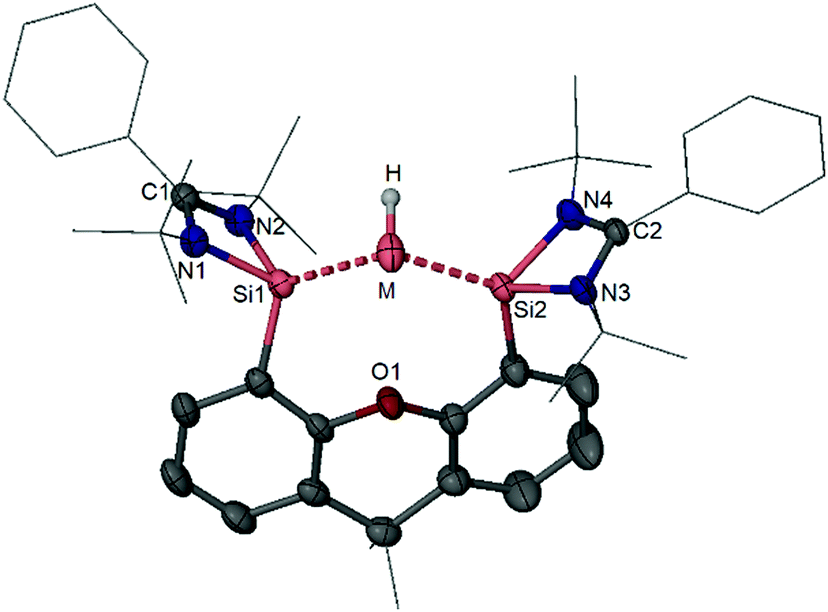 | ||
| Fig. 8 Molecular structures of the cations of 47 (M = Si) and 48 (M = Ge) [hydrogen atoms (except for that at M atom) are omitted for clarity]. | ||
Similarly, the bis(NHSi)-supported germylone 4 can also serve as a bulky base with BPh3 to perform FLP activity for hydrogen activation, affording compound 48 as an isolable product (Scheme 17 and Fig. 8).30 We have also shown that silylone 3 and BPh3 can activate ethene via a similar FLP mechanism leading to the novel zwitterionic complex 49.28 For the first time, we have shown that ylidones can also be applied as bulky bases in FLP chemistry, following the previous success of silylenes and germylenes in this area already.98–101
3.6 Si–Si and Ge–Ge coupling reactions
Generally, disilicon and digermanium complexes result from the reduction of monatomic silicon or germanium halides with alkaline metals through Si–Si and Ge–Ge coupling, respectively.53,54,102,103 However, the homocoupling of isolable monatomic Si0 and Ge0 was still unknown. Remarkably, treatment of germylone 4 with Ni(cod)2 (cod = 1,5-cyclooctadiene) in a molar ratio of 2:1 in diethyl ether allows the isolation of the novel Ge2Ni complex 50 as dark-brown crystals (Scheme 18).30 In the crystal structure, 50 features a three-membered Ge2Ni ring with the Ni centre adopting a square planar geometry (Fig. 9). The Ge–Ge distance [2.470(1) Å] and the Ni–Ge distances [2.425(2) and 2.443(2) Å] suggest that 50 can be described as a bis(NHSi)-stabilised Ge2Ni complex bearing a Ge(I)–Ge(I)–Ni(II) metallacycle. This metallacycle must be favoured over a [η2-(digermene)]Ni(0) π-complex.104 It is generated after Ge→Ni coordination through the reductive coupling of the two germylone–Ge0 centres and insertion of the Ni atom to two of the Si→Ge dative bonds.
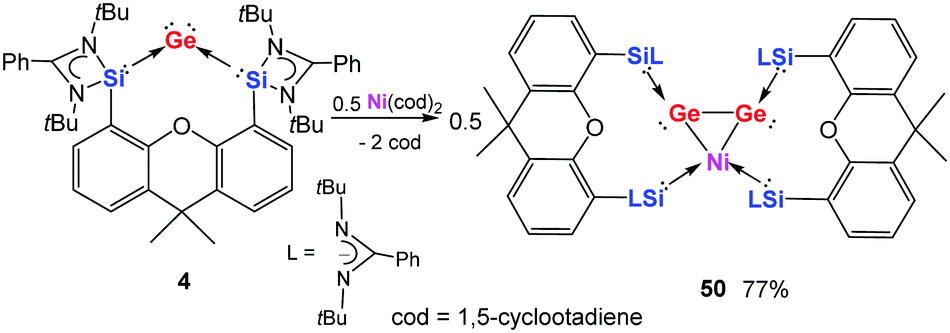 | ||
| Scheme 18 Formation of the NiGe2 complex 50via reaction of 4 with Ni(cod)2. | ||
 | ||
| Fig. 9 Molecular structure of 50 (hydrogen atoms are omitted for clarity). | ||
Without the presence of transition metals, the Si–Si coupling of silylone 5 occurs when 5 is reduced with one molar equiv. of potassium naphthalenide, furnishing the intriguing bis(NHSi)-supported disilicon complex 51 as a red crystalline solid (Scheme 19).29 Overall, the one-electron reduction leads to the net movement of two electrons into the carborane cage with the loss of one electron at Si0 to give SiI. According to DFT calculations, the latter reduction process can be encoded with four sub-steps: (i) one-electron reduction of one amidinato ligand of 5 to form 5a (Scheme 19); (ii) electron transfer to the ortho-carborane cluster in 5a to afford 5b with a radical carborane cluster; (iii) intramolecular one-electron oxidation of the Si0 atom in 5b and rearrangement of the carborane cluster core to form the intermediate 5c with a radical silicon centre and a dianionic nido-carborane core; and (iv) Si–Si homocoupling of 5c to yield the final product 51.
 | ||
| Scheme 19 One-electron reduction of silylone 5 and germylone 6 to generate the bis(NHSi)-supported disillicon(I) complex 51 and digermanium(I) 52via the proposed radical species 5a–c. | ||
In the crystal structure of 51, the two coupled silicon atoms are linked by a normal Si–Si single bond and each adopts a trigonal-pyramidal geometry with a lone pair of electrons.29 Therefore, the coupling product 51 represents a bis(NHSi) supported [SiI–SiI]2+ dication, significantly different from the NHC-stabilised neutral [Si0Si0] complex described by Robinson.53 Starting from germylone 6, the digermanium analogue 52 can also be achieved by following the same synthetic protocol mentioned above for 51 (Scheme 19).31
Although the latter homocoupling of the silylone and germylone results from reduction with potassium naphthalenide, the silicon and germanium centres are actually oxidised from zero to +1. This prompted us to conduct the one-electron oxidation of germylone 6 with [Cp2Fe][B(C6H3{CF3}2)4] in a molar ratio of 1:1 (Scheme 20).31 To our delight, the oxidation led to the isolable [GeI–GeI] coupling compound 53 as an orange solid, with no reduction of the carborane. Compound 53 crystallised as a separate ion pair with a Ge2-containing dication and two borate counteranions. Similarly to the dianion of 52, the dication of 53 features two germanium atoms in a trigonal-pyramidal coordination environment (Fig. 10). The dication in 53 and the dianion in 52 both can be considered as a [GeI–GeI]2+ complex supported by the bis(NHSi) ligand. In sharp contrast to the dianionic nido-carborane core of 52, compound 53 possesses a neutral closo-carborane core.
 | ||
| Scheme 20 One-electron oxidation of germylone 6 to give the bis(NHSi)-supported digermanium(I) complex 53. | ||
 | ||
| Fig. 10 Molecular structure of the cation in 53 (hydrogen atoms are omitted for clarity). | ||
4. Conclusions and outlook
Judicious use of strongly donating ligands has enabled access to a new chemical space with isolable complexes of single, zero-valent silicon and germanium atoms. Several intriguing stable silylones and germylones have thus far been synthesised and structurally characterised by a few research groups in recent years. In this feature article, we have highlighted the chelating bis(NHC)- and bis(NHSi)-supported silylones and germylones. Due to their striking electronic and steric situations, the bis(NHC)- and bis(NHSi)-stabilised Si0 and Ge0 compounds allowed us to explore the intrinsic reactivities of such fascinating zero-valent species.As expected for a genuine silylone and a germylone, the silicon(0) and germanium(0) species can form 1:1 and even 1:2 Lewis adducts with the two lone pairs of electrons at the Si0 or Ge0 centres. With the central silicon and germanium atoms in the zero oxidation state, they can serve as intriguing building blocks with elemental chalcogens for synthesis of isolable heavy CO and CO2 homologues including complexes of SiSe, SiS2, and SiTe2. Moreover, these silylones and germylones can readily mediate the activation of small molecules such as CO2, N2O, and NH3 in a controlled manner and other bonds such as E–H (E = B, C) and H/C–X (X = halogen), etc. They can also act as bulky Lewis bases to form FLPs with BPh3 for H2 and C2H4 activation. Finally, homocoupling of the central atoms of silylones and germylones has also been observed under reducing or oxidising conditions. The impressive reactivity of these bis(NHC)- and bis(NHSi)-supported silylones and germylones results from both the strongly donating nature and chelating structural effect of the bis(NHC) and bis(NHSi) ligands, which make the central silicon or germanium atoms highly electron rich as well as providing necessary kinetic protection during each transformation.
Despite the current progress described in this feature article, the chemistry of monatomic zero-valent silicon and germanium complexes remains in its infancy. Discovery of their remarkable electronic structure, with two extremely active lone pairs at a single Si or Ge site, has begun to be translated to wide-ranging and unprecedented reactivity. With the increasing availability of new strongly donating ligands, more examples with differing reactivity are likely to be developed shortly. The coordination chemistry of silylones and germylones, in particular, is promising due to the electron-richness of the group 14(0) atom and its capacity to coordinate to 1 or 2 metal sites simultaneously. It seems possible that ylidones could act as verstatile ligands in the manner of cAACs or NHCs for stabilizing novel low valent main group and transition metal species. With these ylidones acting as soluble allotropes of silicon and germanium, they could also serve as reagents for unusual transformations – for instance as silicon or germanium atom transfer agents. Moreover, the strategy of synthesising silylones and germylones may also extend to other single, zero-valent main-group elements such as tin, lead, and even with group 2, 12, 13, and 15 elements. Beyond potential future applications, the unique bonding situation in these ylidones – where a central zero-valent Si or Ge atom is bound to neutral donating ligands – expands the field of main group chemistry, by demonstrating a new way in which very unusual oxidation states can be trapped and utilised.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank the Deutsche Forschungsgemeinschaft (Germany's Excellence Strategy–EXC 2008–390540038–UniSys-Cat and DR 226/21-1) for financial support.Notes and references
- R. Tonner, F. Öxler, B. Neumüller, W. Petz and G. Frenking, Angew. Chem., Int. Ed., 2006, 45, 8038–8042 CrossRef CAS.
- F. Ramirez, N. B. Desai, B. Hansen and N. McKelvie, J. Am. Chem. Soc., 1961, 83, 3539–3540 CrossRef CAS.
- G. E. Hardy, J. I. Zink, W. C. Kaska and J. C. Baldwin, J. Am. Chem. Soc., 1978, 100, 8001–8002 CrossRef CAS.
- G. Frenking and R. Tonner, Pure Appl. Chem., 2009, 81, 597–614 CAS.
- R. Tonner, G. Heydenrych and G. Frenking, ChemPhysChem, 2008, 9, 1474–1481 CrossRef CAS.
- N. Takagi, T. Shimizu and G. Frenking, Chem. – Eur. J., 2009, 15, 8593–8604 CrossRef CAS PubMed.
- J. Turek, B. Braïda and F. De Proft, Chem. – Eur. J., 2017, 23, 14604–14613 CrossRef CAS PubMed.
- A. J. Arduengo III, R. L. Harlow and M. Kline, J. Am. Chem. Soc., 1991, 113, 361–363 CrossRef.
- R. Tonner and G. Frenking, Angew. Chem., Int. Ed., 2007, 46, 8695–8698 CrossRef CAS PubMed.
- C. A. Dyker, V. Lavallo, B. Donnadieu and G. Bertrand, Angew. Chem., Int. Ed., 2008, 47, 3206–3209 CrossRef CAS PubMed.
- S. Ishida, T. Iwamoto, C. Kabuto and M. Kira, Nature, 2003, 421, 725–727 CrossRef CAS PubMed.
- M. Kira, T. Iwamoto, S. Ishida, H. Masuda, T. Abe and C. Kabuto, J. Am. Chem. Soc., 2009, 131, 17135–17144 CrossRef CAS PubMed.
- T. Veszprémi, K. Petrov and C. T. Nguyen, Organometallics, 2006, 25, 1480–1484 CrossRef.
- M. Kosa, M. Karni and Y. Apeloig, J. Chem. Theory Comput., 2006, 2, 956–964 CrossRef CAS PubMed.
- N. Takagi, T. Shimizu and G. Frenking, Chem. – Eur. J., 2009, 15, 3448–3456 CrossRef CAS PubMed.
- T. Iwamoto, T. Abe, C. Kabuto and M. Kira, Chem. Commun., 2005, 5190–5192 RSC.
- V. Lavallo, Y. Canac, C. Präsang, B. Donnadieu and G. Bertrand, Angew. Chem., Int. Ed., 2005, 44, 5705–5709 CrossRef CAS PubMed.
- K. C. Mondal, H. W. Roesky, M. C. Schwarzer, G. Frenking, B. Niepötter, H. Wolf, R. Herbst-Irmer and D. Stalke, Angew. Chem., Int. Ed., 2013, 52, 2963–2967 CrossRef CAS PubMed.
- Y. Xiong, S. Yao, S. Inoue, J. D. Epping and M. Driess, Angew. Chem., Int. Ed., 2013, 52, 7147–7150 CrossRef CAS PubMed.
- Y. Xiong, S. Yao, G. Tan, S. Inoue and M. Driess, J. Am. Chem. Soc., 2013, 135, 5004–5007 CrossRef CAS PubMed.
- Y. Li, K. C. Mondal, H. W. Roesky, H. Zhu, P. Stollberg, R. Herbst-Irmer, D. Stalke and D. M. Andrada, J. Am. Chem. Soc., 2013, 135, 12422–12428 CrossRef CAS PubMed.
- T. Chu, L. Belding, A. Van Der Est, T. Dudding, I. Korobkov and G. I. Nikonov, Angew. Chem., Int. Ed., 2014, 53, 2711–2715 CrossRef CAS PubMed.
- M. T. Nguyen, D. Gusev, A. Dmitrienko, B. M. Gabidullin, D. Spasyuk, M. Pilkington and G. I. Nikonov, J. Am. Chem. Soc., 2020, 142, 5852–5861 CrossRef CAS PubMed.
- B. Su, R. Ganguly, Y. Li and R. Kinjo, Angew. Chem., Int. Ed., 2014, 53, 13106–13109 CrossRef CAS PubMed.
- S. Sarmah, A. K. Guha, A. K. Phukan, A. Kumar and S. R. Gadre, Dalton Trans., 2013, 42, 13200–13209 RSC.
- T. Sugahara, T. Sasamori and N. Tokitoh, Angew. Chem., Int. Ed., 2017, 56, 9920–9923 CrossRef CAS PubMed.
- J. Keuter, A. Hepp, C. Mück-Lichtenfeld and F. Lips, Angew. Chem., Int. Ed., 2019, 58, 4395–4399 CrossRef CAS PubMed.
- Y. Wang, M. Karni, S. Yao, A. Kaushansky, Y. Apeloig and M. Driess, J. Am. Chem. Soc., 2019, 141, 12916–12927 CrossRef CAS PubMed.
- S. Yao, A. Kostenko, Y. Xiong, A. Ruzicka and M. Driess, J. Am. Chem. Soc., 2020, 142, 12608–12612 CrossRef CAS PubMed.
- Y. Wang, M. Karni, S. Yao, Y. Apeloig and M. Driess, J. Am. Chem. Soc., 2019, 141, 1655–1664 CrossRef CAS PubMed.
- S. Yao, A. Kostenko, Y. Xiong, C. Lorent, A. Ruzicka and M. Driess, Angew. Chem., Int. Ed., 2021, 60, 14864–14868 CrossRef CAS PubMed.
- M. Jadan, A. R. Chelyadins and V. Y. Yavid, Am. J. Appl. Sci., 2009, 6, 1242–1245 CrossRef CAS.
- F. J. Lovas, Astrophys. J., 1974, 193, 265–272 CrossRef CAS.
- J. L. Turner and A. Dalgarno, Astrophys. J., 1977, 213, 386–389 CrossRef CAS.
- C. A. Dyker and G. Bertrand, Science, 2008, 321, 1050–1051 CrossRef CAS PubMed.
- P. K. Majhi and T. Sasamori, Chem. – Eur. J., 2018, 24, 9441–9455 CrossRef CAS PubMed.
- S. Yao, Y. Xiong and M. Driess, Acc. Chem. Res., 2017, 50, 2026–2037 CrossRef CAS PubMed.
- G. Frenking, R. Tonner, S. Klein, N. Takagi, T. Shimizu, A. Krapp, K. K. Pandey and P. Parameswaran, Chem. Soc. Rev., 2014, 43, 5106–5139 RSC.
- G. Frenking, M. Hermann, D. M. Andrada and N. Holzmann, Chem. Soc. Rev., 2016, 45, 1129–1144 RSC.
- L. Zhao, M. Hermann, N. Holzmann and G. Frenking, Coord. Chem. Rev., 2017, 344, 163–204 CrossRef CAS.
- M. Kira, S. Ishida, T. Iwamoto and C. Kabuto, J. Am. Chem. Soc., 1999, 121, 9722–9723 CrossRef CAS.
- T. Iwamoto, H. Masuda, C. Kabuto and M. Kira, Organometallics, 2005, 24, 197–199 CrossRef CAS.
- N. Wiberg, H. W. Lerner, S. K. Vasisht, S. Wagner, K. Karaghiosoff, H. Nöth and W. Ponikwar, Eur. J. Inorg. Chem., 1999, 1211–1218 CrossRef CAS.
- K. C. Mondal, P. P. Samuel, M. Tretiakov, A. P. Singh, H. W. Roesky, A. C. Stückl, B. Niepötter, E. Carl, H. Wolf, R. Herbst-Irmer and D. Stalke, Inorg. Chem., 2013, 52, 4736–4743 CrossRef CAS PubMed.
- K. C. Mondal, H. W. Roesky, M. C. Schwarzer, G. Frenking, I. Tkach, H. Wolf, D. Kratzert, R. Herbst-Irmer, B. Niepötter and D. Stalke, Angew. Chem., Int. Ed., 2013, 52, 1801–1805 CrossRef CAS PubMed.
- R. S. Ghadwal, H. W. Roesky, S. Merkel, J. Henn and D. Stalke, Angew. Chem., Int. Ed., 2009, 48, 5683–5686 CrossRef CAS PubMed.
- Y. Li, Y. C. Chan, B. X. Leong, Y. Li, E. Richards, I. Purushothaman, S. De, P. Parameswaran and C. W. So, Angew. Chem., Int. Ed., 2017, 56, 7573–7578 CrossRef CAS PubMed.
- S. C. Bart, E. Lobkovsky and P. J. Chirik, J. Am. Chem. Soc., 2004, 126, 13794–13807 CrossRef CAS PubMed.
- J. Flock, A. Suljanovic, A. Torvisco, W. Schoefberger, B. Gerke, R. Pöttgen, R. C. Fischer and M. Flock, Chem. – Eur. J., 2013, 19, 15504–15517 CrossRef CAS PubMed.
- T. Sasamori, T. Sugahara, T. Agou, K. Sugamata, J. D. Guo, S. Nagase and N. Tokitoh, Chem. Sci., 2015, 6, 5526–5530 RSC.
- Y. Xiong, S. Yao, S. Inoue, E. Irran and M. Driess, Angew. Chem., Int. Ed., 2012, 51, 10074–10077 CrossRef CAS PubMed.
- Y. Xiong, S. Yao, S. Inoue, A. Berkefeld and M. Driess, Chem. Commun., 2012, 48, 12198–12200 RSC.
- Y. Wang, Y. Xie, P. Wei, R. B. King, H. F. Schaefer, P. von, R. Schleyer and G. H. Robinson, Science, 2008, 321, 1069–1071 CrossRef CAS PubMed.
- A. Sidiropoulos, C. Jones, A. Stasch, S. Klein and G. Frenking, Angew. Chem., Int. Ed., 2009, 48, 9701–9704 CrossRef CAS PubMed.
- K. A. Kreisel, G. P. A. Yap and K. H. Theopold, Organometallics, 2006, 25, 4670–4679 CrossRef CAS.
- Y. Mizuhata, T. Sasamori and N. Tokitoh, Chem. Rev., 2009, 109, 3479–3511 CrossRef CAS PubMed.
- M. Asay, C. Jones and M. Driess, Chem. Rev., 2011, 111, 354–396 CrossRef CAS PubMed.
- S. Yao, Y. Xiong and M. Driess, Organometallics, 2011, 30, 1748–1767 CrossRef CAS.
- C. Shan, S. Yao and M. Driess, Chem. Soc. Rev., 2020, 49, 6733–6754 RSC.
- C. W. So, H. W. Roesky, J. Magull and R. B. Oswald, Angew. Chem., Int. Ed., 2006, 45, 3948–3950 CrossRef CAS PubMed.
- Y.-L. Shan, W.-L. Yim and C.-W. So, Angew. Chem., Int. Ed., 2014, 53, 13155–13158 CrossRef CAS PubMed.
- B. Blom, M. Stoelzel and M. Driess, Chem. – Eur. J., 2013, 19, 40–62 CrossRef CAS PubMed.
- B. Blom, D. Gallego and M. Driess, Inorg. Chem. Front., 2014, 1, 134–148 RSC.
- S. Raoufmoghaddam, Y. P. Zhou, Y. Wang and M. Driess, J. Organomet. Chem., 2017, 829, 2–10 CrossRef CAS.
- Y.-P. Zhou and M. Driess, Angew. Chem., Int. Ed., 2019, 58, 3715–3728 CrossRef CAS PubMed.
- Z. Benedek and T. Szilvási, RSC Adv., 2015, 5, 5077–5086 RSC.
- D. Gallego, S. Inoue, B. Blom and M. Driess, Organometallics, 2014, 33, 6885–6897 CrossRef CAS.
- Y. Wang, A. Kostenko, S. Yao and M. Driess, J. Am. Chem. Soc., 2017, 139, 13499–13506 CrossRef CAS PubMed.
- Y.-P. Zhou, S. Raoufmoghaddam, T. Szilvási and M. Driess, Angew. Chem., Int. Ed., 2016, 55, 12868–12872 CrossRef CAS PubMed.
- R. N. Grimes, Carboranes, Elsevier Science and Technology, Amsterdam, 2nd edn, 2011 Search PubMed.
- R. Núñez, M. Tarrés, A. Ferrer-Ugalde, F. F. De Biani and F. Teixidor, Chem. Rev., 2016, 116, 14307–14378 CrossRef PubMed.
- A. C. Filippou, Y. N. Lebedev, O. Chernov, M. Straßmann and G. Schnakenburg, Angew. Chem., Int. Ed., 2013, 52, 6974–6978 CrossRef CAS PubMed.
- Y. Xiong, S. Yao, R. Müller, M. Kaupp and M. Driess, Angew. Chem., Int. Ed., 2015, 54, 10254–10257 CrossRef CAS PubMed.
- Y. Xiong, S. Yao, M. Karni, A. Kostenko, A. Burchert, Y. Apeloig and M. Driess, Chem. Sci., 2016, 7, 5462–5469 RSC.
- B. Su, R. Ganguly, Y. Li and R. Kinjo, Chem. Commun., 2016, 52, 613–616 RSC.
- A. Burchert, R. Müller, S. Yao, C. Schattenberg, Y. Xiong, M. Kaupp and M. Driess, Angew. Chem., Int. Ed., 2017, 56, 6298–6301 CrossRef CAS PubMed.
- A. V. Zabula, T. Pape, A. Hepp, F. M. Schappacher, U. C. Rodewald and R. Po, J. Am. Chem. Soc., 2008, 130, 5648–5649 CrossRef CAS PubMed.
- G. Thiele, Y. Franzke, F. Weigend and S. Dehnen, Angew. Chem., Int. Ed., 2015, 54, 11283–11288 CrossRef CAS PubMed.
- L. C. Siemes, J. Keuter, A. Hepp and F. Lips, Inorg. Chem., 2019, 58, 13142–13149 CrossRef CAS PubMed.
- C. Mohapatra, K. C. Mondal, P. P. Samuel, H. Keil, B. Niepötter, R. Herbst-Irmer, D. Stalke, S. Dutta, D. Koley and H. W. Roesky, Chem. – Eur. J., 2015, 21, 12572–12576 CrossRef CAS PubMed.
- K. Chandra Mondal, S. Roy, B. Dittrich, B. Maity, S. Dutta, D. Koley, S. K. Vasa, R. Linser, S. Dechert and H. W. Roesky, Chem. Sci., 2015, 6, 5230–5234 RSC.
- Y. C. Chan, B. X. Leong, Y. Li, M. C. Yang, Y. Li, M. Der Su and C. W. So, Angew. Chem., Int. Ed., 2017, 56, 11565–11569 CrossRef CAS PubMed.
- R. Rodriguez, D. Gau, J. Saouli, A. Baceiredo, N. Saffon-Merceron, V. Branchadell and T. Kato, Angew. Chem., Int. Ed., 2017, 56, 3935–3939 CrossRef CAS PubMed.
- A. Burchert, S. Yao, R. Müller, C. Schattenberg, Y. Xiong, M. Kaupp and M. Driess, Angew. Chem., Int. Ed., 2017, 56, 1894–1897 CrossRef CAS PubMed.
- S. Roy, K. C. Mondal, L. Krause, P. Stollberg, R. Herbst-Irmer, D. Stalke, J. Meyer, A. C. Stückl, B. Maity, D. Koley, S. K. Vasa, S. Q. Xiang, R. Linser and H. W. Roesky, J. Am. Chem. Soc., 2014, 136, 16776–16779 CrossRef CAS PubMed.
- G. D. Frey, V. Lavallo, B. Donnadieu, W. W. Schoeller and G. Bertrand, Science, 2007, 316, 439–441 CrossRef CAS PubMed.
- A. Jana, C. Schulzke and H. W. Roesky, J. Am. Chem. Soc., 2009, 131, 4600–4601 CrossRef CAS PubMed.
- J. P. Moerdyk, G. A. Blake, D. T. Chase and C. W. Bielawski, J. Am. Chem. Soc., 2013, 135, 18798–18801 CrossRef CAS PubMed.
- A. V. Protchenko, K. H. Birjkumar, D. Dange, A. D. Schwarz, D. Vidovic, C. Jones, N. Kaltsoyannis, P. Mountford and S. Aldridge, J. Am. Chem. Soc., 2012, 134, 6500–6503 CrossRef CAS PubMed.
- A. V. Protchenko, A. D. Schwarz, M. P. Blake, C. Jones, N. Kaltsoyannis, P. Mountford and S. Aldridge, Angew. Chem., Int. Ed., 2013, 52, 568–571 CrossRef CAS PubMed.
- D. Wendel, A. Porzelt, F. A. D. Herz, D. Sarkar, C. Jandl, S. Inoue and B. Rieger, J. Am. Chem. Soc., 2017, 139, 8134–8137 CrossRef CAS PubMed.
- D. Wendel, T. Szilvási, C. Jandl, S. Inoue and B. Rieger, J. Am. Chem. Soc., 2017, 139, 9156–9159 CrossRef CAS PubMed.
- R. Rodriguez, D. Gau, T. Kato, N. Saffon-Merceron, A. De Cõzar, F. P. Cossío and A. Baceiredo, Angew. Chem., Int. Ed., 2011, 50, 10414–10416 CrossRef CAS PubMed.
- F. Lips, J. C. Fettinger, A. Mansikkamäki, H. M. Tuononen and P. P. Power, J. Am. Chem. Soc., 2014, 136, 634–637 CrossRef CAS PubMed.
- J. S. Han, T. Sasamori, Y. Mizuhata and N. Tokitoh, J. Am. Chem. Soc., 2010, 132, 2546–2547 CrossRef CAS PubMed.
- D. W. Stephan and G. Erker, Angew. Chem., Int. Ed., 2015, 54, 6400–6441 CrossRef CAS PubMed.
- D. W. Stephan, Science, 2016, 354, 1248 CrossRef CAS PubMed.
- A. Schäfer, M. Reißmann, A. Schäfer, M. Schmidtmann and T. Müller, Chem. – Eur. J., 2014, 20, 9381–9386 CrossRef PubMed.
- Z. Mo, T. Szilvási, Y. P. Zhou, S. Yao and M. Driess, Angew. Chem., Int. Ed., 2017, 56, 3699–3702 CrossRef CAS PubMed.
- Z. Dong, Z. Li, X. Liu, C. Yan, N. Wei, M. Kira and T. Müller, Chem. – Asian J., 2017, 12, 1204–1207 CrossRef CAS PubMed.
- N. Del Rio, M. Lopez-Reyes, A. Baceiredo, N. Saffon-Merceron, D. Lutters, T. Müller and T. Kato, Angew. Chem., Int. Ed., 2017, 56, 1365–1370 CrossRef CAS PubMed.
- K. C. Mondal, P. P. Samuel, H. W. Roesky, R. R. Aysin, L. A. Leites, S. Neudeck, J. Lu, B. Dittrich, N. Holzmann, M. Hermann and G. Frenking, J. Am. Chem. Soc., 2014, 136, 8919–8922 CrossRef CAS PubMed.
- S. S. Sen, A. Jana, H. W. Roesky and C. Schulzke, Angew. Chem., Int. Ed., 2009, 48, 8536–8538 CrossRef CAS PubMed.
- S. Inoue and C. Eisenhut, J. Am. Chem. Soc., 2013, 135, 18315–18318 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2021 |