
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePd-catalyzed C(sp3)–C(sp) bond formation in iodocyclobutenes†
Carlos
Lázaro-Milla
 a,
M. Teresa
Quirós
bc,
Diego J.
Cárdenas
*b and
Pedro
Almendros
*d
a,
M. Teresa
Quirós
bc,
Diego J.
Cárdenas
*b and
Pedro
Almendros
*d
aGrupo de Lactamas y Heterociclos Bioactivos, Departamento de Química Orgánica, Unidad Asociada al CSIC, Facultad de Química, Universidad Complutense de Madrid, Madrid 28040, Spain
bDepartamento de Química Orgánica, Facultad de Ciencias, Institute for Advanced Research in Chemical Sciences, IadChem, Universidad Autónoma de Madrid, Av. Francisco Tomás y Valiente 7, Cantoblanco, Madrid 28049, Spain. E-mail: diego.cardenas@uam.es
cDepartamento de Química Orgánica y Química Inorgánica, Facultad de Farmacia, Universidad de Alcalá, Alcalá de Henares, Madrid 28805, Spain
dInstituto de Química Orgánica General, IQOG-CSIC, Juan de la Cierva 3, Madrid 28006, Spain. E-mail: palmendros@iqog.csic.es
First published on 27th July 2021
Abstract
A synthesis of skipped 1,4-enynes through functionalization of the cyclobutene core with alkynes has been achieved, suggesting an unusual pathway of oxidative addition in tertiary iodoalkanes.
The preparation of alkyl alkynes through irradiation or metal-catalyzed reactions of alkyl halides with alkynes is attractive.1 Lei and others have recently reported the Sonogashira-type alkynylation with alkanes,2 while Park's group described the addition of an alkyne to an allenamide.3 Nonetheless, there is absence of protocol for the direct incorporation of alkynyl groups into existing cyclobutene motifs through C(sp3)–C(sp) bond formation.
Having in hand a convenient method for the direct preparation of starting halocyclobutenes 1a–e from iodoalkynes,4 we intended to study the feasibility of their palladium-catalyzed reactions with terminal alkynes 2. Bis(triflyl)iodocyclobutene 1a was selected as model substrate and was reacted with phenylacetylene 2a using a Pd–Cu bimetallic catalytic system. After extensive experimentation, the adoption of 5 mol% of Pd(PPh3)2Cl2 as the palladium complex and 5 mol% of CuI as the copper salt gave the best performance (Table 1, entry 3). Serendipitously, 4-alkynyl-2-(triflyl)cyclobutene 3aa was obtained in a reasonable 70% yield instead of the expected Sonogashira product, namely, 2-alkynyl-4,4-bis(triflyl)cyclobutene 4aa. The screening of different palladium complexes such as Pd(PPh3)4 and copper salts produced inadequate conversions and less gratifying yields (Table 1, entries 1–5). The screening of solvents and bases revealed that the use of triethylamine as both base and solvent was superior to other combinations (Table 1, entry 8). Temperature studies disclosed that 35 °C was the optimal T (Table 1, entries 6 and 7). The cooperative presence of the palladium complex and the copper salt was required (Table 1, entries 9 and 10).
|
|
|||
|---|---|---|---|
| Entry | Catalysta | Reaction conditions | Yieldb (%) |
| a The reactions were run using 1a (0.1 mmol) and catalyst (5 mol%). b Yield of pure isolated product. c 1,4-Dioxane was used as solvent. | |||
| 1 | Pd(PPh3)4, CuI | Et3N, 35 °C, 3 h | 55 |
| 2 | Pd(AcO)2, CuI | Et3N, 35 °C, 3 h | 43 |
| 3 | Pd(PPh3)2Cl2, CuI | Et3N, 35 °C, 1 h | 70 |
| 4 | Pd(PPh3)2Cl2, CuBr | Et3N, 35 °C, 3 h | 47 |
| 5 | Pd(PPh3)2Cl2, CuOTf | Et3N, 35 °C, 4 h | 16 |
| 6 | Pd(PPh3)2Cl2, CuI | Et3N, 20 °C, 14 h | 58 |
| 7 | Pd(PPh3)2Cl2, CuI | Et3N, 60 °C, 2 h | 51 |
| 8 | Pd(PPh3)2Cl2, CuI | K2CO3, 35 °C, 3 hc | 24 |
| 9 | CuI | Et3N, 35 °C, 12 h | — |
| 10 | Pd(PPh3)2Cl2 | Et3N, 35 °C, 12 h | — |

When we studied the halogen effect it was observed that the nature of the halide affected the efficiency of the reaction. (Triflyl)cyclobutene 3aa was obtained at 80 °C in poor yield (34%) starting from bis(triflyl)bromocyclobutene 1a-Br, while no reaction was observed from the chloride counterpart 1a-Cl (Scheme 1A). In order to see the generality of the process in the presence of different alkynes, bis(triflyl)iodocyclobutene 1a was reacted with a series of terminal alkynes 2a–g. As shown in Scheme 1B, aryl–alkynes bearing on the aryl ring both electron-donating (MeO) as well as electron-withdrawing (CF3) substituents were well accommodated. Heterocyclic (thienyl) and heteroatomic (trimethylsilyl) functionalities were well tolerated in the alkyne. Likewise, alkyl and alkynyl-substituted alkynes were also successful coupling partners. The resulting (triflyl)cyclobutenes 3aa–ag were smoothly obtained in good isolated yields after flash chromatography in silica gel (Scheme 1B).
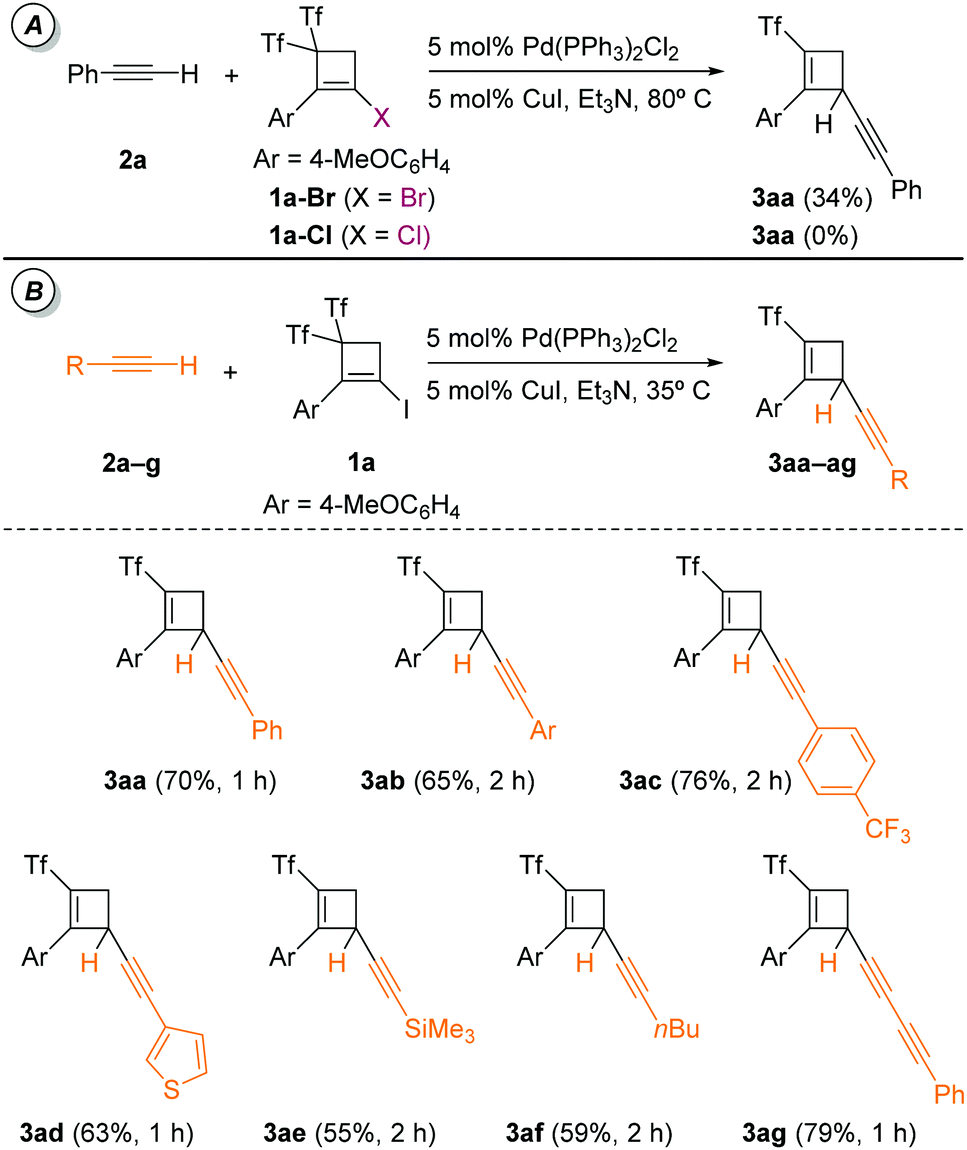 | ||
| Scheme 1 Palladium–copper-catalyzed alkylation of terminal alkynes 2a–g with bis(triflyl)iodocyclobutene 1a. | ||
The scope of the C(sp3)–C(sp) bond formation in cyclobutenes was investigated with respect to the bis(triflyl)iodocyclobutene component. As depicted in Scheme 2, a range of substituents are amenable for this Pd-catalyzed conditions. Both, aryl (phenyl and naphthyl) and hetaryl (thienyl) moieties are tolerated. Besides, the bulky naphthyl ring provided the corresponding (triflyl)cyclobutene 3da in good yield. Noteworthy, the presence of deuterium atoms in the cyclobutene framework is compatible with our protocol, which allows a convenient access to D-labelled adduct [D2]-3aa. The synthetic utility of alkynyl-cyclobutenes 3 was demonstrated by the preparation of chromanone-like compound 4 and alkenyl iodide 5 (Scheme 3).
 | ||
| Scheme 2 Palladium–copper-catalyzed alkylation of phenylacetylene 2a with bis(triflyl)iodocyclobutenes [D2]-1a and 1b–e. | ||
 | ||
| Scheme 3 Transformations of alkynyl–cyclobutene 3aa. | ||
The addition of TEMPO to the reaction between 1a and phenylacetylene 2a resulted in insignificant differences in the obtained yields of the final product 3aa, which exclude a radical mechanism. At least, two possible mechanisms may be postulated for this Pd-catalyzed alkylation reaction of terminal alkynes (Scheme 4). A Sonogashira coupling followed by an allylic substitution with water acting as a hydrogen source5 may be postulated in the first possible route (Scheme 4, path a). Alternative incorporation of H from Et3N by β-elimination and reductive elimination can be ruled out since the formation of 3aa in the presence of d15-Et3N gave no deuteration. The second plausible pathway (Scheme 4, path b) may involve as initial step an allylic substitution with the participation of the alkyne as the nucleophile, and a subsequent oxidative addition of the alkyl iodide. To assess the above proposed pathways, we used the cyclization reaction of 1-methoxy-4-(octa-1,3-diyn-1-yl)benzene with 2-(2-fluoropyridin-1-ium-1-yl)-1,1-bis[(trifluoromethyl)sulfonyl]ethan-1-ide (Yanai's reagent)6 to prepare the formal Sonogashira adduct 4af which may be considered as an intermediate. Under the standard reaction conditions bis(triflyl)-1,3-enyne 4af could not be converted into (triflyl)-1,4-enyne 3af (Scheme 5), which should point to Path a as the less likely pathway.
 | ||
| Scheme 4 Possible pathways of the C(sp3)–C(sp) bond formation reactions. | ||
 | ||
| Scheme 5 Control experiment. | ||
Product 3aa did not undergo H/D exchange in presence of D2O under the reaction conditions. Next, we decided to test the reaction between 1a and 2a under the standard reaction conditions but with the inclusion of 2 equiv. of D2O (Scheme 6A). Intriguingly, the above experiment did not form exclusively (triflyl)cyclobutene 3aa. Instead, labelled (triflyl)cyclobutene [D]-3aa was formed as very major (67% deuterium content) adduct. Unexpectedly, the combination of labelled alkyne [D]-2a and bis(triflyl)iodocyclobutene 1a using the optimized Pd–Cu bimetallic catalytic system was capable of promoting the formation of partially deuterated (triflyl)cyclobutene [D]-3aa (66% deuterium content) (Scheme 6B). This should be attributed to the fact that the new hydrogen atom at the skipped 1,4-enyne moiety may arise from the terminal alkyne, and also may point to a competence in the final protonation step when external water (e.g. D2O, Scheme 6A) is added to the reaction. A kinetic isotopic effect (KIE) was not clearly detected in the standard reaction when deuterated phenylacetylene [D]-2a was used as alkyne in the standard reaction with 1a in parallel experiments (Scheme 6C), which may suggest that the breakage of the C(sp)–H bond is not implicated in the rate-determining step. Consequently, the above control experiment should call into question Path b (Scheme 4) as a plausible pathway.
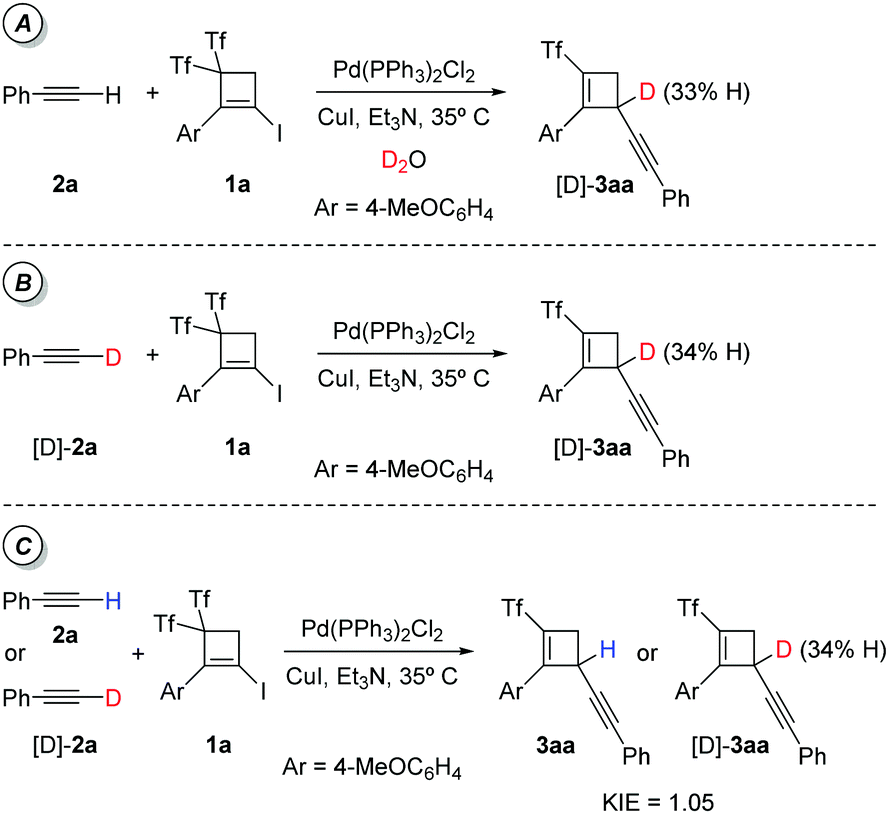 | ||
| Scheme 6 Labelling experiments and determination of the KIE for the alkyne partner. Reactions were run using 1a (0.1 mmol) and catalyst (5 mol%). | ||
Based on the results of the above-mentioned control experiments, a reasonable mechanistic proposal for the generation of cyclobutene-embedded skipped 1,4-enynes is depicted in Scheme 7. In this catalytic cycle, activation of the substrate takes place after Pd(0) coordination to the cyclobutene, by C–O allylic oxidative addition involving one of the triflyl groups to give complex I.7 Then, following the steps of a usual Sonogashira coupling, transmetallation with Cu-acetylide affords II which undergoes subsequent reductive elimination to furnish species IIIa. A facile equilibration by decoordination-coordination of Pd(0) to cyclobutene would render IIIb from IIIa. Complex IIIb, would evolve by C–I oxidative addition to form π-allyl palladium species IV. In turn, the early released Et3NH+ (in the formation of the Cu acetylide) plays an essential role in promoting an electrophilic cleavage of the Pd–C bond of intermediate IV, to give functionalized products 3 and a Pd(II) salt.8 Finally, the active palladium(0) catalyst is regenerated by reductive elimination of palladium(II) assisted by triethylamine, closing the catalytic cycle.
 | ||
| Scheme 7 Mechanistic proposal for the Pd-catalyzed formation of 3. | ||
The feasibility of this proposal was explored by DFT calculations (Scheme 8 and ESI†) to corroborate whether the proposed steps were energetically preferred over other secondary pathways.9 Bis(triflyl)iodocyclobutene 1b and phenylacetylene 2a, which react to give product 3ba, were used as model substrates, and (PPh3)Pd(0) was considered as the catalytically active species. The energy profile for the transformation (Scheme 8) shows that the overall process exhibits a downhill energetic trend and all the computed energies are compatible with a temperature of 36 °C. Although the first oxidative addition is an endergonic step (ΔG = 2.8 kcal mol−1), it occurs through TS1Pd-I with a relatively low activation energy of 14.5 kcal mol−1. The coordination of the copper from the copper acetylide to one of the oxygen atoms of the triflyl group bonded to Pd decreases the energy by 1.3 kcal mol−1 and generates the association complex I–Cu, which facilitates a fast transmetallation step viaTSI-II (ΔG = −2.8 kcal mol−1, ΔG‡ = 3.1 kcal mol−1). The consequent reductive elimination step is highly exergonic (ΔG = −27.8 kcal mol−1) and has a ΔG‡ = 17.2 kcal mol−1. This step constitutes the driving force of the reaction, since it falls into a highly stable energy minimum IIIa. As commented before, species IIIa and IIIb are related through a low energy equilibrium, from which IIIb, would react via an oxidative addition though TSIII-IV. This step has a low activation energy (ΔG‡ = 3.5 kcal mol−1) and is energetically very favored (ΔG = −23.0 kcal mol−1). It is worth noting that this oxidative addition takes place in a concerted manner, through a three-membered transition state, as it usually occurs for C(sp2)–I activations of aryl- and alkenyl-iodides. Although alkyl halides have been proposed to react with Pd(0) from the backside, following a SN2 type mechanism,10 this tertiary halide undergoes a facile oxidative addition by the metal atom bound to the same face of the cyclobutene. Interestingly, previous calculations showed that this activation mode has a lower activation energy compared to the SN2 type reaction.11 The experimental results in the presence of radical inhibitors allow disregarding radical activation pathways. Finally, Et3NH+ would act as a proton source to depalladate IV and furnish the reaction product 3ba.5b
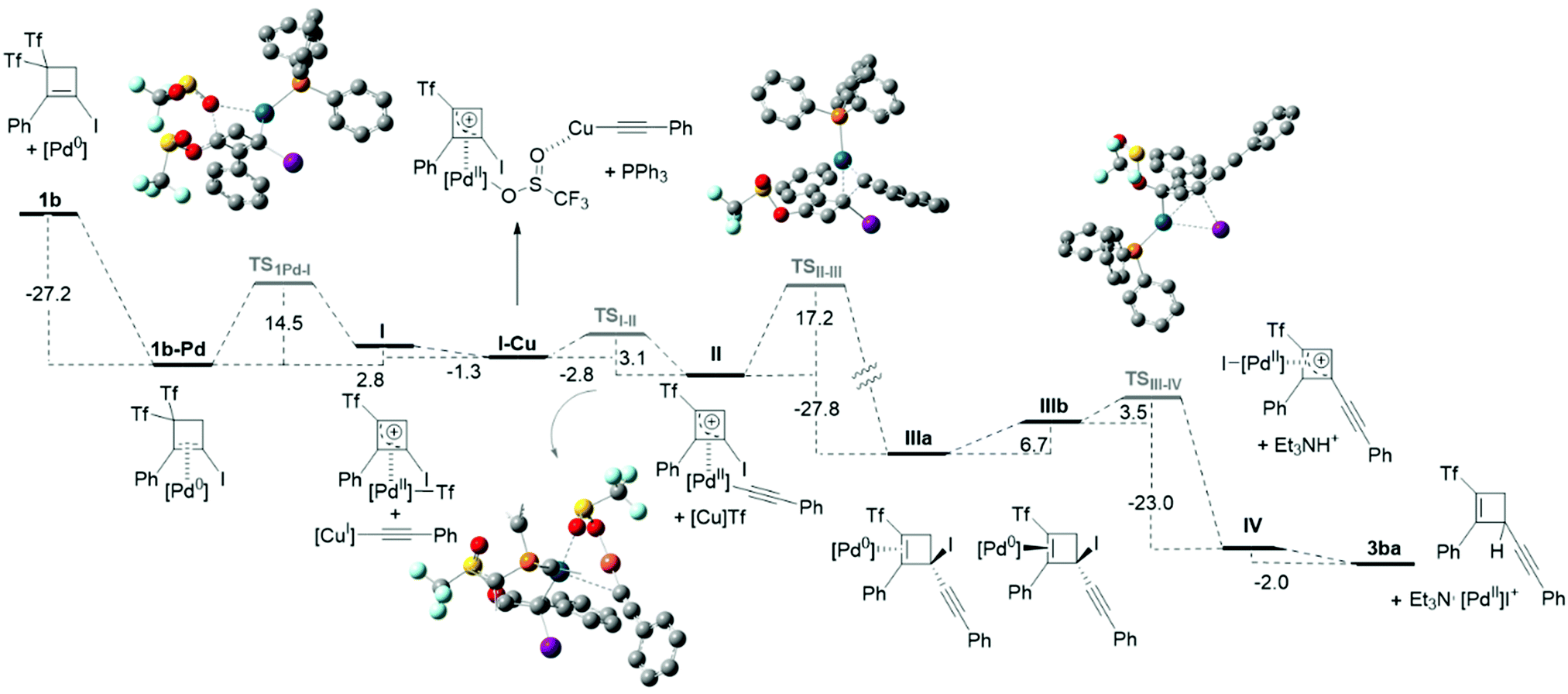 | ||
| Scheme 8 Energy profile for the Pd-catalyzed formation of 4-alkynyl-2-(triflyl)cyclobutenes 3. Values of reaction free energies and activation energies (kcal mol−1) are calculated M06/6-311+G** (C,H,O,S,F,P,N), SDD (Pd,Cu,I), SMD(triethylamine)/ B3LYP/6-31G(d) (C,H,O,S,F,P,N), LANL2DZ (Pd,Cu,I) with [Pd] = (PPh3)Pd and [Cu] = (PPh3)Cu. Phenyl substituents in the PPh3 ligand are omitted in the ball-and-stick model of TSI–II for clarity. | ||
In conclusion, we have developed a metal-catalyzed direct alkylation reaction of terminal alkynes with bis(triflyl)iodocyclobutenes for the generation of (triflyl)cyclobutene-embedded skipped 1,4-enynes. A pathway which contrasts to the widespread activation of the C(sp2)–I bond by the Pd catalyst has been suggested with the aid of control and labelled experiments as well as DFT computations.
This work was supported in part by AEI (MICIU) and FEDER (Projects PGC2018-095025-B-I00, CTQ2016-79826-R, and PID2019-109088GB-I00). M. T. Q. thanks MICIU for a Juan de la Cierva contract. C. L.-M. thanks MICIU and UCM for a postdoctoral contract. We thank the Centro de Computación Científica-UAM for computational time.
Conflicts of interest
There are no conflicts of interest to declare.Notes and references
- (a) V. G. Landge, A. Parveen, A. Nandakumar and E. Balaraman, Chem. Commun., 2018, 54, 7483 RSC; (b) L. Chen, Y. Kametani, K. Imamura, T. Abe, Y. Shiota, K. Yoshizawa, Y. Hisaeda and H. Shimakoshi, Chem. Commun., 2019, 55, 13070 RSC; (c) T. Adak, M. Hoffmann, S. Witzel, M. Rudolph, A. Dreuw and A. S. K. Hashmi, Chem. – Eur. J., 2020, 26, 15573 CrossRef CAS PubMed; (d) E. Tan, M. Zanini and A. M. Echavarren, Angew. Chem., Int. Ed., 2020, 59, 10470 CrossRef CAS PubMed.
- (a) S. Tang, P. Wang, H. Li and A. Lei, Nat. Commun., 2016, 7, 11676(1) Search PubMed; (b) B. Zhao, Y. Wu, Y. Yuanc and Z. Shi, Chem. Commun., 2020, 56, 4676 RSC.
- T. R. Pradhan, H. W. Kim and J. K. Park, Angew. Chem., Int. Ed., 2018, 57, 9930 CrossRef CAS PubMed.
- B. Alcaide, P. Almendros and C. Lázaro-Milla, Adv. Synth. Catal., 2017, 359, 2630 CrossRef CAS The preparation of starting cyclobutenes 1 having aliphatic substituents or just proton is not feasible.
- For Pd-catalyzed reactions using water as a hydrogen source, see: (a) C.-Q. Zhao, Y.-G. Chen, H. Qiu, L. Wei, P. Fang and T.-S. Mei, Org. Lett., 2019, 21, 1412 CrossRef CAS PubMed; (b) C. Lázaro-Milla, M. T. Quirós, D. J. Cárdenas and P. Almendros, Chem. Commun., 2020, 56, 6070 RSC.
- H. Yanai, R. Takahashi, Y. Takahashi, A. Kotani, H. Hakamata and T. Matsumoto, Chem. – Eur. J., 2017, 23, 8203 CrossRef CAS PubMed.
- For selected reports on allylic substitution reaction, see: (a) B. M. Trost and C. A. Kalnmals, Chem. – Eur. J., 2020, 26, 1906 CrossRef CAS PubMed; (b) Y.-X. Li, Q.-Q. Xuan, L. Liu, D. Wang, Y. Chen and C.-J. Li, J. Am. Chem. Soc., 2013, 135, 12536 CrossRef CAS PubMed; (c) B. M. Trost and M. L. Crawley, Chem. Rev., 2003, 103, 2921 CrossRef CAS PubMed.
- Perhaps, final protonation at the α position of alkyne is competitive with protonation at the α position of Tf group, but it could be possible that the isomerization of the double bond leads to a single product, favoured by the basic reaction conditions and the natural acidity of the protons in α to sulfones.
- Other feasible reaction pathways such as the initial oxidative addition of the C−I bond instead of the C−Tf bond, have been explored and disregarded in view of the higher energies (see ESI† for details).
- J. K. Stille and K. S. Y. Lau, Acc. Chem. Res., 1977, 10, 434 CrossRef CAS.
- G. T. de Jong and F. M. Bickelhaupt, J. Chem. Theory Comput., 2006, 2, 322 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Computational details, experimental procedures, characterization data of new compounds, and copies of NMR spectra for all new compounds. See DOI: 10.1039/d1cc03087f |
| This journal is © The Royal Society of Chemistry 2021 |