
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceEvolution of catalytic machinery: three-component nanorotor catalyzes formation of four-component catalytic machinery†
Abir
Goswami‡
,
Merve S.
Özer‡
,
Indrajit
Paul
and
Michael
Schmittel
 *
*
Center of Micro and Nanochemistry and Engineering, Organische Chemie I, Universität Siegen, Adolf-Reichwein-Str. 2, Siegen D-57068, Germany. E-mail: schmittel@chemie.uni-siegen.de; Tel: +49(0) 2717404356
First published on 24th June 2021
Abstract
The three-component nanorotor [Cu2(S)(R)]2+ (k298 = 46.0 kHz) that is a catalyst for a CuAAC reaction binds the click product at each of its copper centers thereby creating a new platform and a dynamic slider-on-deck system. Due to this sliding motion (k298 = 65.0 kHz) the zinc-porphyrin bound N-methylpyrrolidine is efficiently released into solution and catalyzes a follow-up Michael addition.
For many chemists, the unmatched performance of Nature's multicomponent machinery has been an inspiration for developing supramolecular machines that exhibit mechanical motion and perform useful tasks.1 However, one aspect of Nature's road to machinery has received little attention so far: new enzymes with alternative architectures and new functions are constantly being evolved from their ancestral enzymes.2,3 Evolution of new artificial machinery from a dynamic information–handling mixture4 or directly from multicomponent machines5 is thus a persuasive vision. However, despite remarkable developments in the field of useful molecular machines,6 only an outstanding paper by Leigh reports on a manmade machine building another functional device, in this case, a homooligomeric peptide for asymmetric catalysis.7
Knowledge of adaptive dynamic supramolecular devices8 is continuously growing and will eventually help to approach the complexity and seamless organization of biological systems.9 Recently, we have reported catalytic multicomponent machinery10 like rotors, walker, and sliders based on heteroleptic metal–ligand complexation and demonstrated how emergent functionality evolved from well-conceived self-sorting.11 Yet, transformation of a manmade machine to new dynamic catalytic machinery remains without precedent. Here, we demonstrate how the three-component nanorotor [Cu2(S)(R)]2+ (S = stator; R = rotator/slider) via CuAAC chemistry catalyzes formation of the four-component slider-on-deck [Cu2(S)(R)(P)2]2+ (Scheme 1), where the latter acts as catalytic machinery turning ON a Michael addition reaction.
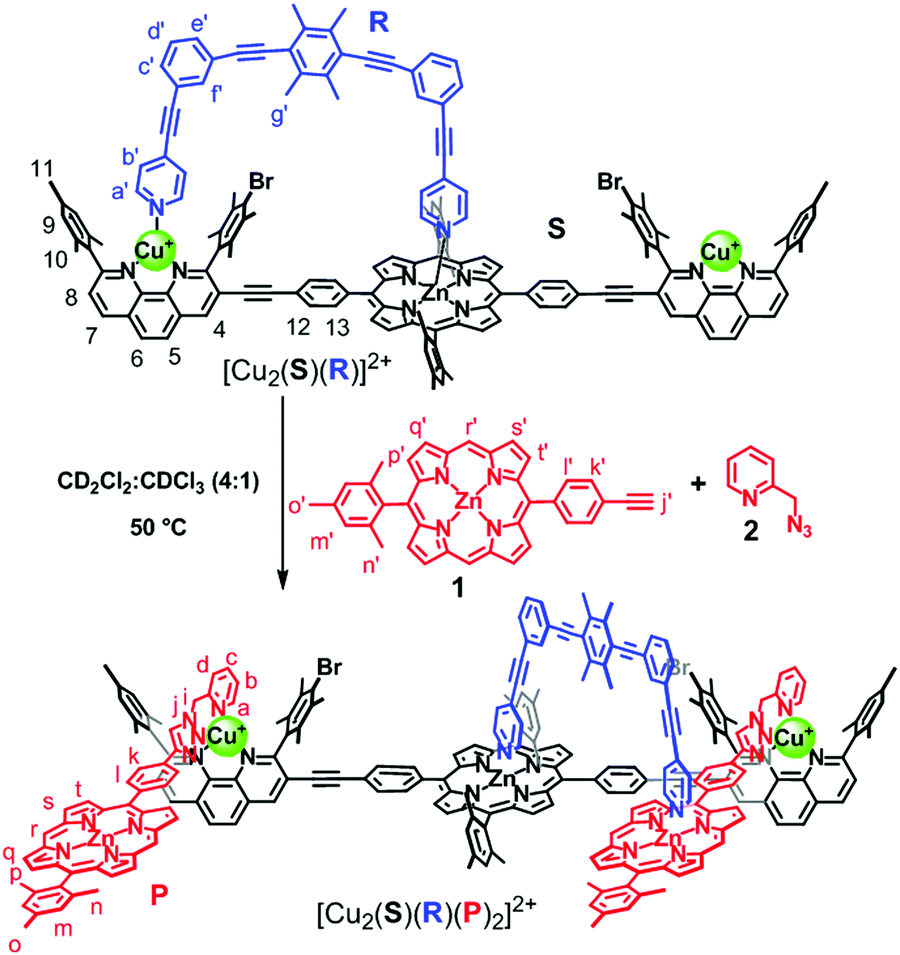 | ||
| Scheme 1 Transformation of the nanorotor [Cu2(S)(R)]2+ to the slider-on-deck [Cu2(S)(R)(P)2]2+. | ||
The design of this unique transformation is based on the nanorotor [Cu2(S)(R)]2+ (Scheme 1) whose catalytic activity for azide–alkyne cycloadditions has been established.10a In that work, we demonstrated that the click product was expelled from the catalytic site by the nanomechanical motion in the nanorotor.12 For the present project, we chose the reactants 1 and 2 in such a way that their bidentate click product P would block the catalytic site of the nanorotor by a strong HETPHEN-type (HETeroleptic bis-PHENanthroline) complexation.1b Since product P possesses a zinc porphyrin (ZnPor) site, the binding of P should establish a new coordination site for the rotary biped in the nanorotor. With two ZnPor units coordinated to [Cu2(S)(R)]2+, another type of nanomechanical motion is expected in [Cu2(S)(R)(P)2]2+ because it is formally a slider-on-deck system (Scheme 1), where the biped R should move between altogether three zinc porphyrins.10c
At first, we carried out a model catalytic study (Scheme 2) with the alkyne-terminated zinc(II) porphyrin 1 and 2-(azidomethyl)pyridine (2). They were allowed to react in a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio in CD2Cl2:CDCl3 (4:1) at 50 °C in the presence of an equimolar amount of [Cu(Phen)]+, the copper(I)-loaded 2,9-dimesityl-1,10-phenanthroline. Progress of the reaction was followed by the signals of protons r-H and j′-H in the 1H NMR (Fig. 1).
:1 ratio in CD2Cl2:CDCl3 (4:1) at 50 °C in the presence of an equimolar amount of [Cu(Phen)]+, the copper(I)-loaded 2,9-dimesityl-1,10-phenanthroline. Progress of the reaction was followed by the signals of protons r-H and j′-H in the 1H NMR (Fig. 1).
 | ||
| Scheme 2 Model study for self-sorting of platform P after formation with Click reaction. | ||
 | ||
| Fig. 1 Comparison of the partial 1H NMR spectra (400 MHz, 298 K) in CD2Cl2 of (A) [Cu(Phen)]+, (B) 1, (C) [Cu(Phen)(P)]+. | ||
Disappearance of the signal of proton C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) CH (j′-H) and a single set of zinc porphyrin r-H, s-H, q-H, t-H and p-H signals in the 1H NMR as well as the clean formation of [Cu(Phen)(P)]+ indicated completion of the azide–alkyne cycloaddition reaction after ∼24 h (ESI,† Fig. S19). Moreover, the signal of phenanthroline proton 2′-H experienced a diagnostic upfield shift from 7.01 to 6.74 ppm, showing that the copper(I)-loaded ligand was now coordinated to platform P (Fig. 1A and C). The emergence of a broad signal for the methylene protons i-H confirmed formation of the click product P. Finally, the single peak at m/z = 1205.3 in the ESI-MS proved the integrity of the heteroleptic complex [Cu(Phen)(P)]+ (ESI,† Fig. S30).
CH (j′-H) and a single set of zinc porphyrin r-H, s-H, q-H, t-H and p-H signals in the 1H NMR as well as the clean formation of [Cu(Phen)(P)]+ indicated completion of the azide–alkyne cycloaddition reaction after ∼24 h (ESI,† Fig. S19). Moreover, the signal of phenanthroline proton 2′-H experienced a diagnostic upfield shift from 7.01 to 6.74 ppm, showing that the copper(I)-loaded ligand was now coordinated to platform P (Fig. 1A and C). The emergence of a broad signal for the methylene protons i-H confirmed formation of the click product P. Finally, the single peak at m/z = 1205.3 in the ESI-MS proved the integrity of the heteroleptic complex [Cu(Phen)(P)]+ (ESI,† Fig. S30).
After these exemplary catalytic investigations, we wanted to test the analogous click reaction with the rotor system as catalyst (Scheme 1). First of all, nanorotor [Cu2(S)(R)]2+ was prepared by mixing S, R and 2.0 equiv. of Cu+ in CD2Cl2.10a The data unambiguously demonstrated that one pyridine foot of rotator R (= biped) was attached to the ZnPor unit of stator S by axial coordination while the other foot toggled between the Cu+-loaded phenanthroline stations at a frequency k298 = 46.0 kHz. Activation parameters of the nanorotor [Cu2(S)(R)]2+ were reported earlier as ΔH‡ = 49.1 ± 0.4 kJ mol−1 and ΔS‡ = 9.5 ± 1.7 J mol−1 K−1.10a
Finally, reactants 1 and 2 were heated with nanorotor [Cu2(S)(R)]2+ as a CuAAC13 catalyst in a 2:2:1 ratio in CD2Cl2:CDCl3 (4:1) at 50 °C. Formation of [Cu2(S)(R)(P)2]2+ was monitored by 1H NMR spectroscopy and ESI-MS. Similar to the model reaction, the formation of platform P was followed by the disappearance of proton signals j′-H and meso r′-H of 1 and appearance of the meso r-H signal of P in 1H NMR (Fig. 2C). Parallel, the peaks at m/z = 1970.8, 1606.0 and 1358.9 in the ESI-MS showed the expected mass signals for [Cu2(S)(R)(P)2]2+, [Cu2(S)(R)(P)]2+ and [Cu2(S)(P)(CH3CN)]2+, respectively (ESI,† Fig. S32). The upfield shift of proton signal 9-H from 7.03 to 6.79 ppm indicated that platform P was coordinated to the Cu+-loaded phenanthroline stations of stator S (Fig. 2A and C). Furthermore, the proton signal b′-H of rotator R shifted from 6.89 to 5.77 ppm as a result of the pyridine arm moving from the Cu+-loaded phenanthroline station to the ZnPor platform. After 24 h, all the signals corresponding to 1 had disappeared and [Cu2(S)(R)(P)]2+ was furnished quantitatively.
 | ||
| Fig. 2 Partial comparison 1H NMR (400 MHz, 298 K) in CD2Cl2 of (A) nanorotor [Cu2(S)(R)]2+, (B) deck [Cu2(S)(P)2]2+, (C) slider-on-deck [Cu2(S)(R)(P)2]2+. | ||
To clearly establish the Npy → ZnPor coordination in slider-on-deck [Cu2(S)(R)(P)2]2+, we separately prepared deck [Cu2(S)(P)2]2+ by mixing S, P and [Cu(CH3CN)4]PF6 in a 1:2:2 ratio in CD2Cl2 and compared it with [Cu2(S)(R)(P)2]2+. In the 1H NMR, the proton signal r-H resonating at 10.30 ppm experienced a notable upfield shift to 10.22 ppm when rotator R was added due to the axial pyridine coordination in slider-on-deck [Cu2(S)(R)(P)2]2+. Furthermore, the DOSY spectrum yielded a single diffusion coefficient (D = 3.3 × 10−10 m2 s−1) indicating that the multi-component slider-on-deck [Cu2(S)(R)(P)2]2+ was a single species. The experimental hydrodynamic radius was derived as 16.1 Å (ESI,† Fig. S17) in agreement with the theoretical value (ESI,† Fig. S34, S35). The system was further analyzed by UV-vis. In the final slider-on-deck, there are two different types of ZnPor units present; one emerging from stator S and two from both platforms P. The Q-band of S at 550 nm was reported earlier to undergo a red shift upon formation of rotor [Cu2(S)(R)]2+.10a In order to assign the ZnPor absorptions in [Cu2(S)(R)(P)2]2+, the position of the ZnPor Q-band of 1 (536 nm) was determined in 1,1,2,2-tetrachloroethane (ESI,† Fig. S33). The Q-band absorption of the multicomponent deck [Cu2(S)(P)2]2+ without rotator R (Fig. 3A) was observed at λmax = 539 nm with a shoulder at 549 nm. The former absorption belongs to the ZnPor chromophore of P and the shoulder to the ZnPor unit of S. The Q-band absorption of the slider-on-deck system exhibited a λmax = 550 nm with a hump at 542 nm (Fig. 3). As expected, upon coordination of R to the deck, the Q-bands experienced a red shift in agreement with our previous reports.14
 | ||
| Fig. 3 (A) Comparison of the UV-vis spectra of [Cu2(S)(P)2]2+ and [Cu2(S)(R)(P)2]2+ in 1,1,2,2-tetrachloroethane (c = 1.00 mM, 298 K) in 1.0 mm cuvette. (B) Partial VT 1H NMR (600 MHz) of slider-on-deck [Cu2(S)(R)(P)2]2+ in CD2Cl2. | ||
After establishing the slider-on-deck system [Cu2(S)(R)(P)2]2+, we examined the sliding motion of R across the three ZnPor units of deck [Cu2(S)(P)2]2+ by VT (variable temperature) 1H NMR (Fig. 3B and ESI,† Fig. S27, S28). The meso proton r-H (10.22 ppm) appearing as a singlet at 25 °C due to dynamic sliding showed coalescence at −30 °C and split at −50 °C into two singlets at a ratio of ca. 2:1. At −70 °C, splitting to 2:1 ratio can be seen more clearly. The signal at 10.28 ppm represents the Npy → ZnPor complexed site and the one at 10.22 ppm the pyridine-free ZnPor site. The software WinD-NMR15 was used to simulate the VT 1H NMR for the determination of the motion. The rate of exchange at room temperature was determined as k25 = 65.0 kHz and the corresponding activation parameters were calculated as ΔH‡ = 49.4 ± 0.2 kJ mol−1 and ΔS‡ = 13.1 ± 0.8 J mol−1 K−1 (Table 1). The kinetic barrier for exchange ΔG‡ = 45.5 kJ mol−1 in [Cu2(S)(R)(P)2]2+ is almost identical to that of a nanoslider that undoubtedly operates via a single Npy → ZnPor dissociation (ΔG‡ = 47.3 kJ mol−1).14
Finally, we coupled the slider-on-deck system with a base-catalyzed reaction (Scheme 3). From former work it had been established that the N-methylpyrrolidine (5) → ZnPor binding was strong enough to prevent catalytic activity of the nitrogen heterocycle whereas sliding motion in slider-on-deck systems was found to liberate the bound catalyst (N-methylpyrrolidine → ZnPor-based sliders) into the solution thus turning ON catalysis.10c Hence, we expected that the catalyst N-methylpyrrolidine would be initially bound at the ZnPor unit of 1 thus being inactive for catalysis. However, after formation of the slider-on-deck [Cu2(S)(R)(P)2]2+, the pyridyl end of the slider R would move from the Cu+-loaded phenanthroline stations to the newly bound ZnPor platform and should liberate catalyst 5. If this concept was correct, one would see that the N-methylpyrrolidine catalyzed reaction was turned ON.
 | ||
| Scheme 3 (A) Catalyst liberation due to the sliding motion enables catalysis. (B) N-methylpyrrolidine (5) catalyzed Michael addition. Binding of 5 to ZnPor prevents catalysis. | ||
To first check the ability of the slider-on-deck to act as a catalytic machinery in combination with organocatalyst 5, we mixed S (1.9 mM), P, R, [Cu(CH3CN)4]PF6, 3, 4 and 5 in 1:2:1:2:20:100:2 ratio in CD2Cl2:CDCl3 (4:1) and heated at 50 °C. Indeed, 46% of the product 6 was formed after 4 h as evidenced by 1H NMR (Scheme 3A). The reference reaction itself, the mixture of 3, 4 and free catalyst 5 (3.8 mM) in 10:50:1 ratio in CD2Cl2:CDCl3 (4:1), furnished 54% of product 6 after heating at 50 °C for 4 h (Scheme 3B). This indicated that the dynamic slider-on-deck liberated most of the available catalyst 5 into solution as its activity is close to that of free 5.
In the next step, the in situ evolution of the catalytic slider-on-deck and its effect on the Michael addition were tested. For that purpose, we mixed 1, nanorotor [Cu2(S)(R)]2+ (≈ 2.0 mM) as well as 3, 4 and catalyst 5 in 2:1:20:100:2 ratio in CD2Cl2:CDCl3 (4:1) and heated at 50 °C. No Michael addition product 6 was formed after 4 h because the static N-methylpyrrolidine (5) → ZnPor binding of 1 was strong enough to prevent catalytic activity (ESI,† Fig. S25).
To the same mixture, we added 2.0 equiv of azide 2 (with respect to the nanorotor) and heated at 50 °C. Now, 25% of the Michael addition product 6 formed after 4 h (ESI,† Fig. S24). The reduced yield of 6 may readily be rationalized by the fact that only 47% of the slider-on-deck was furnished after 4 h of heating. This yield agrees well with that of a control experiment (44% of slider-on-deck formed in absence of the substrates after 4 h at 50 °C, see ESI,† Fig. S21), demonstrating that the formation of [Cu2(S)(R)(P)2]2+ was itself not affected by the presence of compounds 3–5. In summary, the findings show that catalyst 5 is liberated due to the formation of the slider-on-deck and as a result turns ON the Michael addition.
While supramolecular transformations are well established,16 we demonstrate here how catalytic supramolecular machinery transforms itself into new catalytic machinery. In the key step a multicomponent machine catalyzes formation of an additional component that adds to its architecture.
In detail, the protocol involves the transformation of a nanorotor into a slider-on-deck system via copper(I)-catalyzed azide–alkyne cycloaddition (CuAAC).13 While the motion of the nanorotor was dictated by the cleavage the coordinative Npy→ [Cu(phenAr2)]+ bond, after click reaction that of the slider-on-deck was guided by the Npy → ZnPor interaction. Consequently, the exchange speed changed, i.e. from 46.0 kHz (nanorotor) to 65.0 kHz (slider-on-deck). Moreover, the sliding motion was utilized to liberate a catalyst into the solution to turn ON a Michael addition. The formation of new catalytic machinery from a catalytically active nanorotor is a lucid example of an adaptive evolutionary process leading to new properties.
We are indebted to the Deutsche Forschungsgemeinschaft (Schm 647/20-2) for financial support and Dr. T. Paululat for VT NMR measurements.
Conflicts of interest
There are no conflicts to declare.Notes and references
- (a) J. M. Abendroth, O. S. Bushuyev, P. S. Weiss and C. J. Barrett, ACS Nano, 2015, 9, 7746 CrossRef CAS PubMed; (b) M. Schmittel, Chem. Commun., 2015, 51, 14956 RSC; (c) A. J. McConnell, C. S. Wood, P. P. Neelakandan and J. R. Nitschke, Chem. Rev., 2015, 115, 7729 CrossRef CAS PubMed; (d) L. Zhang, V. Marcos and D. A. Leigh, Proc. Natl. Acad. Sci. U. S. A., 2018, 115, 9397 CrossRef CAS PubMed; (e) A. Goswami, S. Saha, P. K. Biswas and M. Schmittel, Chem. Rev., 2020, 120, 125 CrossRef CAS PubMed.
- P. J. O’Brien and D. Herschlag, Chem. Biol., 1999, 6, R91 CrossRef.
- Y. Yoshikuni, T. Ferrin and J. Keasling, Nature, 2006, 440, 1078 CrossRef CAS PubMed.
- (a) J.-M. Lehn, Angew. Chem., Int. Ed., 2013, 52, 2836 CrossRef CAS PubMed; (b) A. Goswami, T. Paululat and M. Schmittel, J. Am. Chem. Soc., 2019, 141, 15656 CrossRef CAS PubMed; (c) J. Andréasson and U. Pischel, Coord. Chem. Rev., 2021, 429, 213695 CrossRef; (d) F. Nicoli, E. Paltrinieri, M. Tranfić Bakić, M. Baroncini, S. Silvi and A. Credi, Coord. Chem. Rev., 2021, 428, 213589 CrossRef CAS.
- (a) S. Silvi, A. Arduini, A. Pochini, A. Secchi, M. Tomasulo, F. M. Raymo, M. Baroncini and A. Credi, J. Am. Chem. Soc., 2007, 129, 13378 CrossRef CAS PubMed; (b) S. Hiraoka, Y. Hisanaga, M. Shiro and M. Shionoya, Angew. Chem., Int. Ed., 2010, 49, 1669 CrossRef CAS PubMed; (c) Y. Takara, T. Kusamoto, T. Masui, M. Nishikawa, S. Kume and H. Nishihara, Chem. Commun., 2015, 51, 2896 RSC.
- (a) R. Herges, Chem. Sci., 2020, 11, 9048–9055 RSC; (b) S. Corra, M. Curcio, M. Baroncini, S. Silvi and A. Credi, Adv. Mater., 2020, 32, e1906064 CrossRef PubMed; (c) A. W. Heard and S. M. Goldup, ACS Cent. Sci., 2020, 6, 117 CrossRef CAS PubMed; (d) S. Krause and B. L. Feringa, Nat. Chem. Rev., 2020, 4, 550 CrossRef CAS; (e) J. Echavarren, M. A. Y. Gall, A. Haertsch, D. A. Leigh, J. T. J. Spence, D. J. Tetlow and C. Tian, J. Am. Chem. Soc., 2021, 143, 5158 CrossRef CAS PubMed; (f) Y. Feng, M. Ovalle, J. S. W. Seale, C. K. Lee, D. J. Kim, R. D. Astumian and J. F. Stoddart, J. Am. Chem. Soc., 2021, 143, 5569 CrossRef CAS PubMed.
- G. de Bo, M. A. Y. Gall, S. Kuschel, J. de Winter, P. Gerbaux and D. A. Leigh, Nat. Nanotechnol., 2018, 13, 381 CrossRef CAS PubMed.
- (a) J.-M. Lehn, Angew. Chem., Int. Ed., 2013, 52, 2836 CrossRef CAS PubMed; (b) R. Merindol and A. Walther, Chem. Soc. Rev., 2017, 46, 5588 RSC.
- M. Raynal, P. Ballester, A. Vidal-Ferran and P. W. N. M. van Leeuwen, Chem. Soc. Rev., 2014, 43, 1734 RSC.
- (a) A. Goswami, S. Pramanik and M. Schmittel, Chem. Commun., 2018, 54, 3955 RSC; (b) N. Mittal, M. S. Özer and M. Schmittel, Inorg. Chem., 2018, 57, 3579 CrossRef CAS PubMed; (c) I. Paul, A. Goswami, N. Mittal and M. Schmittel, Angew. Chem., Int. Ed., 2018, 57, 354 CrossRef CAS PubMed; (d) A. Goswami and M. Schmittel, Angew. Chem., Int. Ed., 2020, 59, 12362 CrossRef CAS PubMed.
- Z. He, W. Jiang and C. A. Schalley, Chem. Soc. Rev., 2015, 44, 779 RSC.
- P. K. Biswas, S. Saha, T. Paululat and M. Schmittel, J. Am. Chem. Soc., 2018, 140, 9038 CrossRef CAS PubMed.
- (a) M. Meldal and C. W. Tornøe, Chem. Rev., 2008, 108, 2952 CrossRef CAS PubMed; (b) M. Meldal and F. Diness, Trends Chem., 2020, 2, 569 CrossRef CAS.
- A. Goswami, I. Paul and M. Schmittel, Chem. Commun., 2017, 53, 5168 Search PubMed.
- H. J. Reich, NMR Spectrum Calculations: WinDNMR, Version 7.1, Department of Chemistry, University of Wisconsin Search PubMed.
- (a) W. Wang, Y.-X. Wang and H.-B. Yang, Chem. Soc. Rev., 2016, 45, 2656 RSC; (b) W. M. Bloch, J. J. Holstein, W. Hiller and G. H. Clever, Angew. Chem., Int. Ed., 2017, 56, 8285 CrossRef CAS PubMed; (c) G. Li, Z. Zhou, C. Yuan, Z. Guo, Y. Liu, D. Zhao, K. Liu, J. Zhao, H. Tan and X. Yan, Angew. Chem., Int. Ed., 2020, 59, 10013 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental procedures, compound characterizations, spectral data, UV-vis titrations data. See DOI: 10.1039/d1cc02805g |
| ‡ Abir Goswami and Merve S. Özer contributed equally. |
| This journal is © The Royal Society of Chemistry 2021 |