
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Modulating photoswitch performance with halogen, coordinative and hydrogen bonding: a comparison of relative bond strengths†
Sofia
Lindblad
 a,
Daniel
Sethio
a,
Orion B.
Berryman
*b and
Máté
Erdélyi
*a
a,
Daniel
Sethio
a,
Orion B.
Berryman
*b and
Máté
Erdélyi
*a
aDepartment of Chemistry–BMC, Uppsala University, Uppsala SE-751 23, Sweden. E-mail: Mate.Erdelyi@kemi.uu.se
bDepartment of Chemistry and Biochemistry, 32 Campus Drive, University of Montana, Missoula, Montana 59812, USA. E-mail: Orion.Berryman@mso.umt.edu
First published on 25th May 2021
Abstract
The behavior of an enediyne photoswitch is modulated with halogen bonding, coordinative bonding and hydrogen bonding. Through NMR and computational studies we demonstrate that the relative strength of the secondary bonding directly influences the rate of photoisomerization and the photostationary state.
Controlling molecular conformation is critical to the function of both synthetic and biological systems. For example, molecular machines have received enormous recognition for their unique ability to adopt multiple conformations that can access different functions.1,2 As such, using external stimuli to control the conformation of molecular machines and functional materials has been critical to their development.3,4 Using light as an external stimulus is especially desirable as it is a clean reagent that does not leave residues in a reaction mixture, allowing for simple work up and purification.5,6 Additionally, light can be used to control isomerization in both a temporal and spatial manner. Thus, understanding new ways to control molecular switches will enable the further refinement of the function of molecular machines. Herein, we illustrate that halogen bonding, coordinative bonding and hydrogen bonding can be used to modulate an enediyne photoswitch (Fig. 1). To study the photophysical properties, the systems were non-selectively irradiated with a Xe ARC light source, and the relative cis–trans isomerization rates and photostationary states were compared using NMR spectroscopy.
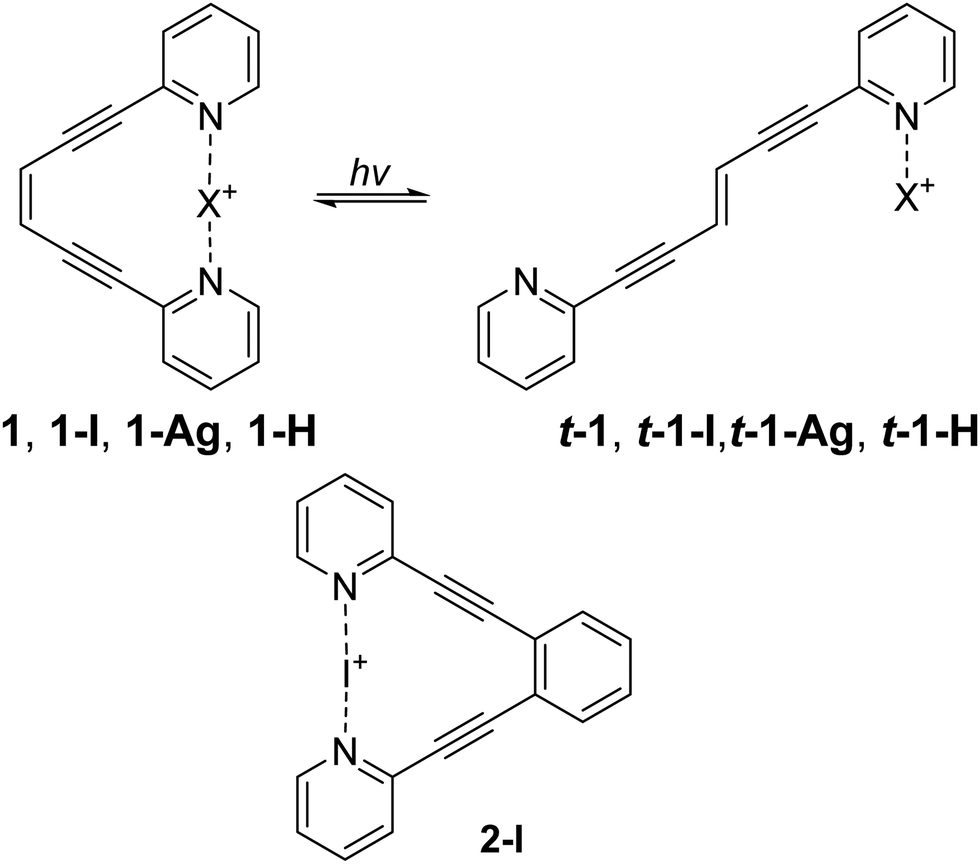 | ||
Fig. 1 The enediyne ligand 1 was used to compare how halogen bonding, hydrogen bonding, and metal coordination modulate photoisomerization (top). The halogen bonded 1,2-bis(pyridine-2-ylethynyl)benzene 2-I was used as a control, to demonstrate that the halogen bond remains intact upon photo irradiation (bottom). Here, X is iodine(I) for 1-I and t-1-I, silver(I) for 1-Ag and t-1-Ag, proton (hydrogen(I)) for 1-H and t-1-H. The counterion tetrafluoroborate, is identical for these complexes (apart from 1 that has no counterion) and is omitted for clarity. The depiction of t-1-I and t-1-Ag is schematic here, these complexes do not form (vide infra, Fig. S1, ESI†). Protonation alters the photostationary state of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :t-1 (1:0.76) to 1-H:t-1-H (1:0.20), whereas silver coordination makes the isomerisation unidirectional t-1-H → 1-H. The 1-I complex quickly decomposes upon isomerization to form t-1-H. :t-1 (1:0.76) to 1-H:t-1-H (1:0.20), whereas silver coordination makes the isomerisation unidirectional t-1-H → 1-H. The 1-I complex quickly decomposes upon isomerization to form t-1-H. | ||
For experimental evaluation of the ability of weak interactions to influence photoisomerization, we synthesized ligand 1 following a previously described route.7 This ligand contains an enediyne photoswitch with two appended pyridine rings, appropriately spaced to interact with a halogen(I), silver(I) or a proton via a three-center, four electron bond.8 From 1, the halogen bonded system 1-I, and the metal coordinated system 1-Ag, were obtained following a literature protocol.9–11
Specifically, ligand 1 was mixed with AgBF4 to obtain 1-Ag. Sequential addition of I2 in dry conditions produced 1-I. To obtain the hydrogen bonded system 1-H, 1-I was reacted with water, which typically results in rapid decomposition of three-center [N–X–N]+ halogen bonds12,13 to the corresponding [N–H–N]+ complex. The halogen bond of 1-I decomposed after 4 days, and thus, appears unusually stable. Adding trifluoroacetic acid to 1-I immediately produced some 1-H, and after a day almost full conversion was noted. The formation of 1-H was validated by LCMS analysis. Observation of the major peak corresponding to the mass of the ligand (M + 1, 231 g mol−1) revealed that the I+ had not added covalently to the enediyne moiety. The formation of 1-H was further validated by raising the pH with NaOH to produce 1. The control 2-I (Fig. 1) was synthesized as previously described.10,14
Hydrogen15–18 and halogen bonding19–22 as well as metal coordination23,24 have been used to control photoisomerization processes. However, they have not yet been evaluated for controlling enediyne photoswitches, and neither have been directly compared within a similar system. Additionally, incorporating three-center bonds12,13,25 into photoswitches has not been reported, despite the strong nature of these halogen bonds that makes them useful to prepare spectacular supramolecular complexes.26,27 With the cis-compounds 1, 1-I, 1-Ag and 1-H in hand, we were interested in assessing how the [N–X–N]+ halogen, [N–Ag–N]+ coordination and [N–H–N]+ hydrogen bonds, respectively, influence the kinetics and thermodynamics of cis to trans enediyne photoisomerization. For the photoirradiation experiments, ∼0.02 mmol compound was dissolved in 0.6 mL of CD2Cl2 in an NMR tube, and the solution was irradiated with non-selective light from a 1000 W Xe ARC light source (Fig. S2, ESI†).
Ligand 1 does not isomerize at room temperature, or upon heating. However, photo-irradiating the noncoordinated enediyne 1 caused the ligand to reach its photostationary state (1:0.76, cis:trans) within ≤15 min (Fig. 2). In comparison, the parent enediyne with phenyl rings instead of pyridines have a nearly equal mixture of cis and trans isomers at the photostationary state, when irradiated with 366 nm light.28
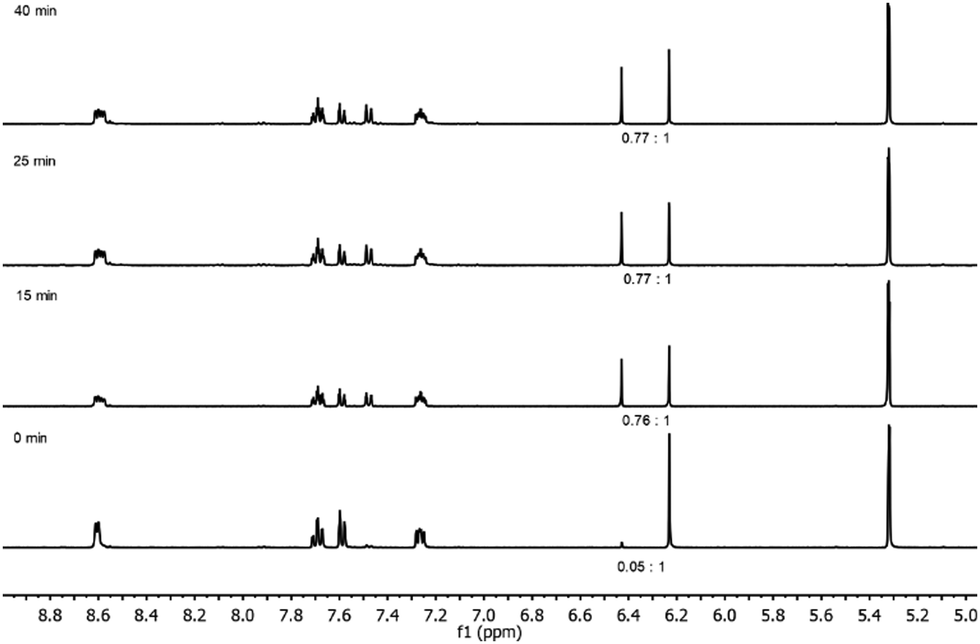 | ||
| Fig. 2 The stacked 1H NMR spectra (400 MHz, 25 °C, CD2Cl2) of 1 with indicated trans:cis ratios before (bottom), after 15 min (middle) and 25 min (top), using non-selective irradiation (Xe Arc 1000 W). | ||
The hydrogen bond in 1-H caused the enediyne to photoisomerize at a significantly slower rate than 1, taking 54 minutes to reach full conversion. Additionally, the photostationary state deviated substantially from the parent ligand 1, reaching a cis:trans ratio of 1:0.2 (Fig. S38 and S39, ESI†). Thus, the hydrogen bond retards photoisomerization and biases 1 towards its cis conformation at the photostationary state.
Remarkably, the silver(I) complex 1-Ag did not isomerize at all upon irradiation (Fig. S35, ESI†). Some precipitate was formed during the experiment, but re-dissolving it in CD3CN proved this to be the cis-isomer 1-Ag (Fig. S36, ESI†). Further investigation showed that adding AgBF4 to a mixture of isomers 1 and t-1 produced 1-Ag and t-1, and irradiating this new mixture converted t-1 into 1-Ag (Fig. 3). Thus, while hydrogen bonding provides a means to modulate conformational equilibria and photoisomerization rate, silver(I) coordination affectively turns off photoisomerization for the 1-Ag complex. This, strategy could also be used as a method for isomeric resolution in future applications.
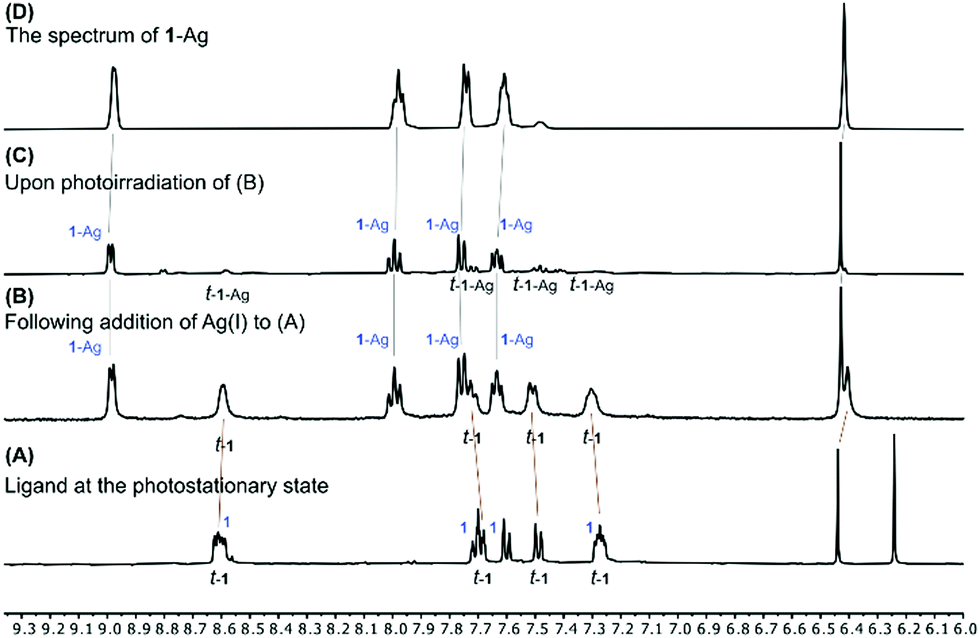 | ||
| Fig. 3 Stacked 1H NMR spectra (500 MHz, 25 °C, CD2Cl2) of ligand 1 (A) after irradiation to reach its photostationary state, a mixture of its cis (1) and trans (t-1) isomers (bottom), then (B) after addition of AgBF4 to that same sample (second from bottom), and (C) after sequential irradiation for 10 min (second from top). Also shown is a reference spectrum of 1-Ag (D, top). Complex t-1 is marked by brown lines, showing that the trans-isomer does not interact with silver(I); the cis-isomer, however, coordinates silver(I) forming 1-Ag (marked by grey lines). Upon irradiation, the signals of t-1 disappear, leaving behind only those of 1-Ag. | ||
Photoirradiation of the halogen bonded complex 1-I highlighted the sensitivity of this complex to conformation. When 1-I was photoirradiated, it was converted to the hydrogen bonding 1-H complex over the course of ∼31 minutes. This can be rationalized: photoisomerization to the t-1-I complex would produce an unstable dimer, owing to the instability of N-iodopyridinum cation.29,30 The fact that 1-I was converted to the 1-H complex, however, indicates that isomerization was occurring, since control experiments with the halogen bonded complex 2-I proved that irradiation alone does not cause decomposition of an [N–X–N]+ complex to any detectable extent (Fig. S34, ESI†).
To estimate the rotational barriers, and to understand how the different bond strengths influence photoisomerization, we performed calculations at the DLPNO-CCSD(T)/def2-TZVP//ω-B97X-D//aug-cc-pVDZ level of theory. The aug-cc-pVDZ basis set was chosen as it is known to provide a good balance between accuracy and cost in predicting geometries.31–33 We used the one nuclear coordinate with respect to torsional angle approach (see ESI† for details) that singles out the torsional motion around the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bond that is isomerized, and allows the one-dimensional description of the decay from the excited state and the fate of the molecule in the ground state following irradiation,34 providing an approximation of the photoisomerization process for a reasonable computational cost. Topological analysis of electron density shows that the three-center [N–Ag–N]+ and [N–I–N]+ bonds are covalent in nature, whereas the hydrogen bonded complex possess one covalent and one noncovalent interaction (see ESI†).8,10 This observation is in agreement with the previous literature.12,29 The free ligand 1 showed the lowest barrier to rotation, 269 kJ mol−1, whereas the 1-Ag complex the highest barrier, 530 kJ mol−1, 26% higher than the barrier of the 1-I complex, 422 kJ mol−1. In good agreement with the experiments, the barrier of the hydrogen bonding 1-H complex was found to be lower, 347 kJ mol−1. This trend indicates that the rotational barrier for isomerization tracks with the relative strengths of the three-center, four-electron bonds, namely 1-H < 1-I < 1-Ag. Here it should be noted that 1-H does not form a true three-center bond; this has been discussed in detail in ref. 8. Whereas the computed barrier heights are high, due to the use of the simple one nuclear coordinate with respect to torsional angle approach, the trends in energies fit well to the experimental observations.
C bond that is isomerized, and allows the one-dimensional description of the decay from the excited state and the fate of the molecule in the ground state following irradiation,34 providing an approximation of the photoisomerization process for a reasonable computational cost. Topological analysis of electron density shows that the three-center [N–Ag–N]+ and [N–I–N]+ bonds are covalent in nature, whereas the hydrogen bonded complex possess one covalent and one noncovalent interaction (see ESI†).8,10 This observation is in agreement with the previous literature.12,29 The free ligand 1 showed the lowest barrier to rotation, 269 kJ mol−1, whereas the 1-Ag complex the highest barrier, 530 kJ mol−1, 26% higher than the barrier of the 1-I complex, 422 kJ mol−1. In good agreement with the experiments, the barrier of the hydrogen bonding 1-H complex was found to be lower, 347 kJ mol−1. This trend indicates that the rotational barrier for isomerization tracks with the relative strengths of the three-center, four-electron bonds, namely 1-H < 1-I < 1-Ag. Here it should be noted that 1-H does not form a true three-center bond; this has been discussed in detail in ref. 8. Whereas the computed barrier heights are high, due to the use of the simple one nuclear coordinate with respect to torsional angle approach, the trends in energies fit well to the experimental observations.
In conclusion, the photoisomerization of an enediyne photoswitch is modulated by secondary interactions at the periphery of the molecule. Hence, the photoisomerization rate is retarded using a hydrogen bond, and is eliminated using silver(I) coordination. The halogen bonded complex photoisomerizes, but is simultaneously converted to the hydrogen bonded complex. Additionally, the photostationary state of the system can be adjusted away from the trans isomer with hydrogen bonding, or removed with silver(I) coordination. Computations highlight that the propensity for the ligand to isomerize is directly related to the bond strength in the secondary interaction. Thus, we demonstrate that three-center, four-electron bonding offers an attractive strategy to further control molecular machines that utilize photoisomerization to access different conformations. In time, this approach could be used to advance molecular machines and information storage.
This project made use of the NMR Uppsala infrastructure, which is funded by the Department of Chemistry–BMC and the Disciplinary Domain of Medicine and Pharmacy. We thank Ignacio Fernández Galván for fruitful discussions, and the Swedish Research Council (2020-03431), FORMAS (2017-01173), the Wenner-Gren Foundation (F2020-0003), and the National Science Foundation (CHE-1555324) for financial support. Computations were performed on resources provided by Swedish National Infrastructure for Computing (SNIC) through the National Supercomputer Center (NSC) at Linköping University under Project SNIC2020/5-435.
Conflicts of interest
There are no conflicts to declare.References
- K. Ariga, Chem. Sci., 2020, 11, 10594–10604 RSC.
- V. Balzani, A. Credi, F. M. Raymo and J. F. Stoddart, Angew. Chem., Int. Ed., 2000, 39, 3348–3391 CrossRef CAS PubMed.
- M. H.-Y. Chan, S. Y.-L. Leung and V. W.-W. Yam, J. Am. Chem. Soc., 2019, 141, 12312–12321 CrossRef CAS PubMed.
- V. Garcia-Lopez, F. Chen, L. G. Nilewski, G. Duret, A. Aliyan, A. B. Kolomeisky, J. T. Robinson, G. Wang, R. Pal and J. M. Tour, Nature, 2017, 548, 567–572 CrossRef CAS PubMed.
- J. Lee, H. Lee and C. Kim, New J. Chem., 2020, 44, 14177–14780 Search PubMed.
- L. Yin, H. Tang, K. H. Kim, N. Zheng, Z. Song, N. P. Gabrielson, H. Lu and J. Cheng, Angew. Chem., Int. Ed., 2013, 52, 9182–9186 CrossRef CAS PubMed.
- C. F. Lin and M. J. Wu, Aryl-substituted acyclic enediyne compounds, Google patents, US 7332623 B2, 2008 Search PubMed.
- A. C. Reiersolmoen, S. Battaglia, S. Oien-Odegaard, A. K. Gupta, A. Fiksdahl, R. Lindh and M. Erdelyi, Chem. Sci., 2020, 11, 7979–7990 RSC.
- J. Barluenga, J. M. Gonzalez, P. J. Campos and G. Asensio, Angew. Chem., Int. Ed. Engl., 1985, 97, 341–342 CrossRef CAS.
- A.-C. C. Carlsson, J. Gräfenstein, A. Budnjo, J. L. Laurila, J. Bergquist, A. Karim, R. Kleinmaier, U. Brath and M. Erdelyi, J. Am. Chem. Soc., 2012, 134, 5706–5715 CrossRef CAS PubMed.
- A.-C. C. Carlsson, J. Gräfenstein, J. L. Laurila, J. Bergquist and M. Erdelyi, Chem. Commun., 2012, 48, 1458–1460 RSC.
- L. Turunen and M. Erdelyi, Chem. Soc. Rev., 2020, 49, 2688–2700 RSC.
- S. B. Hakkert and M. Erdelyi, J. Phys. Org. Chem., 2015, 28, 226–233 CrossRef CAS.
- E. Bosch and C. L. Barnes, Inorg. Chem., 2001, 40, 3097–3100 CrossRef CAS PubMed.
- O. B. Berryman, A. C. Sather and J. Rebek, Pacifichem 2010, International Chemical Congress of Pacific Basin Societies, Honolulu, HI, United States, 2010, ORGN-76.
- O. B. Berryman, A. C. Sather and J. Rebek, Jr., Abstracts of Papers, 243rd ACS National Meeting & Exposition, San Diego, CA, United States, 2012, ORGN-439.
- A. Kwiatkowski, B. Jedrzejewska, M. Jozefowicz, I. Grela and B. Osmialowski, RSC Adv., 2018, 8, 23698–23710 RSC.
- Z. Ye, Z. Yang, L. Wang, L. Chen, Y. Cai, P. Deng, W. Feng, X. Li and L. Yuan, Angew. Chem., Int. Ed., 2019, 58, 12519–12523 CrossRef CAS PubMed.
- M. Saccone, F. F. Palacio, G. Cavallo, V. Dichiarante, M. Virkki, G. Terraneo, A. Priimagi and P. Metrangolo, Faraday Discuss., 2017, 203, 407–422 RSC.
- M. Saccone, A. Siiskonen, F. Fernandez-Palacio, A. Priimagi, G. Terraneo, G. Resnati and P. Metrangolo, Acta Crystallogr., Sect. B: Struct. Sci., Cryst. Eng. Mater., 2017, 73, 227–233 CrossRef CAS PubMed.
- M. Saccone, G. Cavallo, P. Metrangolo, G. Resnati and A. Priimagi, Top. Curr. Chem., 2015, 359, 147–166 CrossRef CAS PubMed.
- M. Saccone, G. Terraneo, T. Pilati, G. Cavallo, A. Priimagi, P. Metrangolo and G. Resnati, Acta Crystallogr., Sect. B: Struct. Sci., Cryst. Eng. Mater., 2014, 70, 149–156 CrossRef CAS PubMed.
- G. Markiewicz, A. Walczak, F. Perlitius, M. Piasecka, J. M. Harrowfield and A. R. Stefankiewicz, Dalton Trans., 2018, 47, 14254–14262 RSC.
- B. Tylkowski, R. Jastrzab and M. Skrobanska, New-Generation Bioinorganic Complexes, 2016, pp. 41–68 Search PubMed.
- J. Emsley, Chem. Soc. Rev., 1980, 9, 91–124 RSC.
- L. Turunen, A. Peuronen, S. Forsblom, E. Kalenius, M. Lahtinen and K. Rissanen, Chem. – Eur. J., 2017, 23, 11714–11718 CrossRef CAS PubMed.
- L. Turunen, U. Warzok, R. Puttreddy, N. K. Beyeh, C. A. Schalley and K. Rissanen, Angew. Chem., Int. Ed., 2016, 55, 14033–14036 CrossRef CAS PubMed.
- B. Koenig, E. Schofield, P. Bubenitschek and P. G. Jones, J. Org. Chem., 1994, 59, 7142–7143 CrossRef CAS.
- A. Karim, M. Reitti, A.-C. C. Carlsson, J. Gräfenstein and M. Erdelyi, Chem. Sci., 2014, 5, 3226–3233 RSC.
- S. Lindblad, K. Mehmeti, A. X. Veiga, B. Nekoueishahraki, J. Gräfenstein and M. Erdelyi, J. Am. Chem. Soc., 2018, 140, 13503–13513 CrossRef CAS PubMed.
- S. Ghosh, S. Bhattacharyya and S. Wategaonkar, J. Phys. Chem. A, 2015, 119, 10863–10870 CrossRef CAS PubMed.
- Z. L. Seeger and E. I. Izgorodina, J. Chem. Theory Comput., 2020, 16, 6735–6753 CrossRef CAS PubMed.
- H. Kruse, R. Szabla and J. Sponer, J. Chem. Phys., 2020, 152, 214104 CrossRef CAS PubMed.
- N. J. Turro, Modern Molecular Photochemistry, Benjamin/Cummings, Menlo Park, CA, 1978 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures, NMR spectra, computational data. See DOI: 10.1039/d1cc01827b |
| This journal is © The Royal Society of Chemistry 2021 |