
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
EPR evidence for α-triphenylstannylvinyl radicals in the O-directed hydrostannation of dialkylacetylenes with Ph3SnH/cat. Et3B/O2†
Hamish A.
Watson
a,
Alistair J.
Fielding
 *b and
Karl J.
Hale
*a
*b and
Karl J.
Hale
*a
aThe School of Chemistry and Chemical Engineering, Queen's University Belfast, Stranmillis Road, Belfast BT9 5AG, Northern Ireland, UK. E-mail: k.j.hale@qub.ac.uk
bThe School of Pharmacy and Biomolecular Sciences, Liverpool John Moores University, Byrom Street, Liverpool L3 3AF, UK. E-mail: A.J.Fielding@ljmu.ac.uk
First published on 8th July 2021
Abstract
Here we provide definitive EPR evidence for the existence of α-triphenylstannylvinyl radicals in the low temperature O-directed free radical hydrostannation of dialkyl propargylic alcohols with Ph3SnH/cat. Et3B and O2 in PhMe. Isotropic hyperfine splitting patterns and spectral simulations confirm the assignments made. In the case of the α-triphenylstannylvinyl radical (Z)-2, an isotopic 119/117Sn hyperfine coupling constant of 9.5 mT (95 G) was measured along with a 1Hβ hyperfine coupling constant of 1.1 mT.
Although the mechanism of the O-directed free radical hydrostannation of propargylically-oxygenated dialkyl acetylenes with stannanes1 has been the subject of considerable debate2 over the past decade, quite recently this controversy was unambiguously resolved1a,3 in favor of the proposals of Hale and Manaviazar,1a,4 and Alabugin,5 who collectively provided strong experimental support for such reactions proceeding by way of an entirely free radical mechanism that operates under strict O–Sn coordinative control.1,4–7 In an effort to further validate these conclusions, we recently undertook an examination of the O-directed hydrostannation1 of several dialkyl acetylenic alcohols by electron paramagnetic resonance (EPR) spectroscopy, and here we present the first readily interpretable EPR spectra of two α-stannylvinyl radical intermediates (Z)-2 and (Z)-9, generated from the alkynols 1 and 8 by application of our standard, chemically-initiated, Ph3SnH/cat. Et3B/O2 reaction conditions1 at high substrate concentration (0.5 M) and low reaction temperature (−10 to −50 °C).
Now with respect to the alkynol 1, our previous mechanistic work3,4 had already confirmed that three different radicals 2, 4, and 5 are produced to varying degrees in the room temperature hydrostannation of 1 at different stannane concentrations (see Scheme 1). Our finding, also, that the proportion of α-vinylstannane 3 increased steadily as the concentration of stannane increased provided strong mechanistic evidence for these hydrostannations operating under O–Sn coordinative control,1,3–7 since a purely electronically-controlled Sn-radical addition mechanism2 would give rise to an invariable regiochemical outcome as the stannane concentration increased, provided the reactions were run at identical temperatures.
 | ||
| Scheme 1 O–Sn coordinative control with probe 1 is favored by high stannane concentrations and low temperatures. | ||
Indeed, our 2020 work3a very clearly demonstrated the significant influence that temperature could have on the regiochemical outcome of the hydrostannation of 1 with Ph3SnH/cat. Et3B, with the proportion of the α-tin adduct 3 increasing progressively as the reaction temperature decreased, due to lower temperatures promoting a much greater degree of O–Sn coordination between the propargyl O-atom and the stannane, and the newly generated stannyl radical. It was reasoned, therefore, that if the reaction temperature could be decreased further, to −50 °C let us say, this might allow the desired α-vinylstannane 3 to be obtained almost exclusively, and we might thus be able to observe the α-triphenylstannylvinyl radical (Z)-2 as essentially a single regioisomer by EPR. If this proved to be so, then (Z)-2 would, of course, be expected to resonate as an apparent “doublet” with attendant long–range, low-intensity, 117,119Sn hyperfine splittings, although more formally it would be classified as a double doublet.
With this in mind, we initially tried to generate the α-stannylvinyl radical (Z)-2 by photoirradiation (30 W Hg-lamp) of solutions of 1 and Ph3SnH (1 equiv.) in dry cyclohexane, THF, and PhMe respectively, at room temperature and below (−50 °C), but we were never able to obtain a satisfactory EPR spectrum of (Z)-2, even when di-t-butylperoxide (DTBP) was used as a reaction initiator for some of the runs. In light of these problems, we resolved to generate (Z)-2 by treatment of 1 with Ph3SnH (1 equiv.), cat. Et3B (0.1 equiv.) and O2 in dry THF at −10 °C inside a quartz capillary EPR tube. Under such circumstances, a most encouraging EPR spectrum of predominantly (Z)-2 was obtained, and it is shown in Fig. 1.
 | ||
| Fig. 1 The EPR spectrum of the α-stannylvinyl radical (Z)-2 generated from the O-directed hydrostannation of a THF solution of 1, Et3B (0.1 equiv.), Ph3SnH (1 equiv.) at 263 K (−10 °C). | ||
It consisted of an apparent “doublet”, due to the newly created vinyl radical coupling to its β-CH. However, because of the rather weak intensity of this signal, we were unable to satisfactorily detect the requisite 117,119Sn hyperfine splittings, because of the high level of background noise. In fact, it was only after we completely abandoned the use of THF and quartz capillary EPR tubes, and we effected the hydrostannation of 1 in PhMe, in a 4 mm EPR tube at −50 °C (223 K), that we were able to finally observe the characteristic “doublet” and accompanying 117,119Sn hyperfine splittings that unambiguously confirmed the identity of the α-stannylvinyl radical (Z)-2. Importantly, the new signal observed (see Fig. 2) once more indicated that we were dealing with predominantly a single Boltzmann conformer/invertomer. However, before we could be absolutely certain of our assignment, we felt that we needed to simulate the EPR spectrum of the radical (Z)-2 with the computer programme EasySpin.8
 | ||
| Fig. 2 EPR spectrum of the α-stannylvinyl radical (Z)-2 generated from treatment of 1 with Ph3SnH (1 equiv.) and Et3B (0.1 equiv.)/O2 in PhMe at 223 K (−50 °C). The simulated signal is overlayed in red. | ||
After inputting the experimentally determined g and a values for (Z)-2, and parameterizing the programme for the presence of C, H and Sn, our simulation of the α-stannylvinyl radical (Z)-2 produced the EPR spectrum shown in Fig. 2 (in red) which has been superimposed upon our experimentally-determined spectrum at 223 K. Clearly there is an excellent correlation, right down to the 117,119Sn hyperfine couplings which, although unresolved, have similar hyperfine splitting values. The aforementioned simulation enabled the g value (g = 2.0014) and the hyperfine splittings, a(Hβ) = 1.1 mT and a(119/117)Sn = 9.5 mT, to be accurately determined.
Having secured this excellent result in PhMe at −50 °C, we next decided to use near identical experimental conditions to acquire the α-stannylvinyl radical 9 from 3-hexyn-2-ol (8). In this instance, the 230 K (−43 °C) EPR spectrum in PhMe gave rise to the anticipated “apparent” triplet (Fig. 3) for the α-stannylvinyl radical (Z)-9. Following EasySpin analysis8 of the radical (Z)-9, the simulated EPR spectrum in Fig. 3 was obtained; it is again shown in red. It overlaid perfectly with the experimentally-derived spectrum of (Z)-9 at 230 K, whose “triplet” at g = 2.0021 had a hyperfine splitting of 0.43 mT (4.3 G) due to the radical coupling with the neighboring –CH2– of the Et. Significantly, the simulated spectrum highlighted how the weak intensity of the signal and the strong background noise were both preventing the Sn hyperfine couplings from being seen.
 | ||
| Fig. 3 EPR spectrum of the α-triphenylstannylvinyl radical (Z)-9 generated from 3-hexyn-2-ol (8) by hydrostannation with Ph3SnH (1 equiv.) and Et3B (0.1 equiv.)/O2 in PhMe at 230 K. Simulation in red. | ||
On this basis, we repeated the aforementioned hydrostannation reaction multiple times at progressively lower temperatures on each occasion, in an attempt to reduce the background noise and reveal the 117,119Sn hyperfine splittings.
Although these efforts did not provide the requisite information, they did lead to us observing additional lines of steadily increasing intensity and partial asymmetry as we moved closer to 200 K (see Fig. 4). We believe that these additional signals are due to the rate of inversion of the two interconverting (E)- and (Z)-α-stannylvinyl radical invertomers being markedly slowed down, to the extent where both conformers of 9 are now observable. According to Galli and Rappoport,9 the barriers to inversion of alkyl-bearing σ-vinyl radicals are typically low, and of the order of 1–3 kcal mol−1. It is thus not at all surprising that a decrease in reaction temperature could now be making this process observable on the EPR timescale.
 | ||
| Fig. 4 The multiple EPR spectra obtained from hydrostannation of 3-hexyn-2-ol (8) with Ph3SnH (1 equiv.) and Et3B (0.1 equiv.) and O2 in PhMe over a range of temperatures between 200–230 K. | ||
The quite different g values observed for these signals would appear to corroborate this proposal, since quite profound structural differences would exist between the two radical conformers. In this connection, the carbon side chains would be positioned anti- to one another in (Z)-9, while in (E)-9 they would be oriented syn to each other, and in quite different steric and electronic environments. While this is one possible explanation of these intriguing effects, equally well, one can offer a rotational isomer rationale of these phenomena.
Given that continuous wave EPR spectroscopy cannot detect radical intermediates with lifetimes less than 10−6 seconds, and we have been able to continuously acquire these EPR spectra for the α-stannylvinyl radicals (Z)-2 and (Z)-9 over timeframes of more than 40 min at −43 to −50 °C, our observations do not align well with Organ's stannylvinyl cation mechanistic theory2a–d for how such alkyne hydrostannation reactions proceed. According to this proposal (Scheme 2), a non-regiocontrolled stannyl radical addition occurs to the acetylene.2a,b A fast, near instantaneous (10−15 s), non-bonded outer-sphere electron-transfer is then suggested to take place from the resulting mixture of α- and β-stannylvinyl radical intermediates 13α and 13β to O2, at essentially the diffusion-controlled limit, followed by a cationic equilibration to the all α-stannylvinyl cation superoxide ion pair 14α, which is then ionically reduced by the stannane to give (Z)-15 and 16. The latter is then reduced by O2− to R3Sn˙, to allow yet another round of addition.2a–d
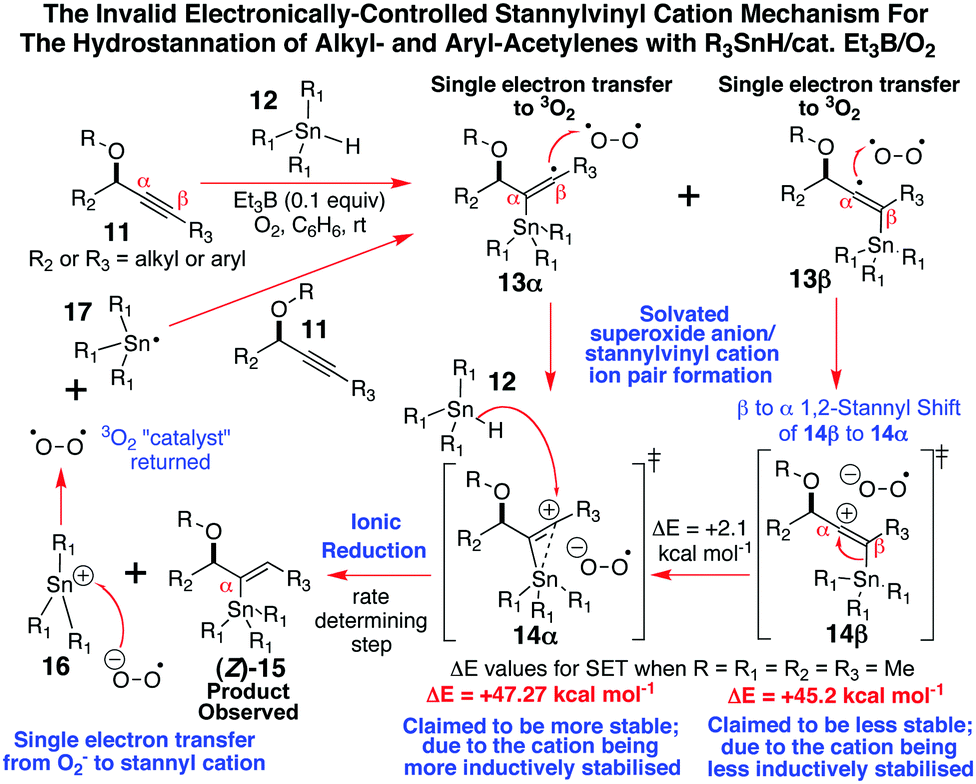 | ||
| Scheme 2 Organ's stannylvinyl cation mechanistic proposal.2a–d | ||
The fact that we have been unable to detect the requisite singlet10 of gav = 2.03110 for the postulated superoxide radical in the EPR spectra of (Z)-2 or (Z)-9 at −50 °C, and we are able to spectroscopically observe fairly long-lived α-stannylvinyl radicals, once more calls into question1,3,5 the validity of this mechanistic theory,2a–d as does a wide range of other experimental evidence.3–5 Clearly the low temperature EPR spectra that we have gathered here for the α-stannylvinyl radicals (Z)-2 and (Z)-9, plus Alabugin's5a computational uncovering of a stabilizing nO→σ*Sn–C interaction of 2.5 kcal mol−1 in such radical additions, alongside our own earlier work of Scheme 1,3,4 each collectively leave very little room for mechanistic alternatives other than the entirely free radical, O–Sn coordinately-controlled, hydrostannation reaction mechanism of Scheme 3 controlling the outcome of these additions.
 | ||
| Scheme 3 A summary of the primary mechanism of the O-directed free radical hydrostannation of disubstituted alkyl acetylenes. | ||
Our success in obtaining these EPR spectra for the α-stannylvinyl radicals (Z)-2 and (Z)-9 is almost certainly due to both radicals being very strongly stabilized by the powerful C–Sn–σ-bond/SOMO radical hyperconjugative interactions5 that can arise. Electronic effects that can involve the C–Sn–σ-bond donating electron density into the partially-filled SOMO of the radical, and the vinyl radical itself delocalising into the empty orbitals associated with the tin. According to Alabugin,5 strong hyperconjugative interactions of the former type are typically associated with large stabilization energies of the order of 26–31 kcal mol−1. Our observation of a large 117,119Sn β-hyperfine coupling constant of 9.5 mT (95 Gauss) between the Ph3Sn unit and the vinylic radical in (Z)-2 seemingly supports11,12 the existence of such a computed5 stabilizing electronic interaction. It also supports the idea that the stannylvinylic radicals (Z)-2 and (Z)-9 significantly delocalise their unpaired electron into the empty low-lying orbitals associated with the tin. Presumably, the simultaneous manifestation of these two effects leads to the high degree of longevity and persistence12 that is seen for (Z)-2 and (Z)-9 when they are generated at −50 °C; a persistence that diminishes as the radicals are warmed above −10 °C, when fast H-atom abstraction occurs from the Ph3SnH.13
How we assigned the bent σ-vinyl radical geometry to (Z)-2 and (Z)-9 was through comparison of their g values (2.0014 and 2.0021 respectively) with the g values reported for other bent alkyl-bearing σ-vinyl radicals, which are generally below the value of g for a free electron (g = 2.0023).14 By way of contrast, linear π-vinyl radicals typically have g values that lie well above this number.14
So, to summarize, we have identified a steady-state set of conditions where rapid stannyl radical addition can proceed near exclusively at the propargyloxy α-acetylenic carbon, and where the rate of H-atom abstraction by the α-stannylvinyl radical from the stannane is slowed sufficiently to allow these intermediates to be spectroscopically observed by EPR.
We thank the EPSRC National EPR Service (U of Manchester) for instrument time funded by EPSRC Grant NS/A000055/1, and Adam Brookfield for his expert technical assistance. HAW also thanks the DfE NI for a PG studentship.
Conflicts of interest
There are no conflicts to declare.Notes and references
- (a) Review: K. J. Hale, S. Manaviazar and H. A. Watson, Chem. Rec., 2019, 19, 238 CrossRef CAS PubMed; (b) P. Dimopoulos, A. Athlan, S. Manaviazar, J. George, M. Walters, L. Lazarides, A. E. Aliev and K. J. Hale, Org. Lett., 2005, 7, 5369 CrossRef CAS PubMed; (c) S. Manaviazar, K. J. Hale and A. LeFranc, Tetrahedron Lett., 2011, 52, 2080 CrossRef CAS; (d) K. J. Hale, M. Grabski, S. Manaviazar and M. Maczka, Org. Lett., 2014, 16, 1164 CrossRef CAS; (e) K. J. Hale, M. Maczka, A. Kaur, S. Manaviazar, M. Ostovar and M. Grabski, Org. Lett., 2014, 16, 1168 CrossRef CAS PubMed.
- (a) M. S. Oderinde, R. D. J. Froese and M. G. Organ, Angew. Chem., Int. Ed., 2013, 52, 11334 CrossRef CAS PubMed; (b) M. S. Oderinde, R. D. J. Froese and M. G. Organ, Chem. – Eur. J., 2014, 20, 8579 CrossRef CAS PubMed; (c) M. S. Oderinde and M. G. Organ, Chem. – Eur. J., 2013, 19, 2615 CrossRef CAS PubMed; (d) M. S. Oderinde, H. N. Hunter, R. D. J. Froese and M. G. Organ, Chem. – Eur. J., 2012, 18, 10821 CrossRef CAS PubMed; (e) D. P. Curran and T. McFadden, J. Am. Chem. Soc., 2016, 138, 7741 CrossRef CAS; (f) M. Alami, A. Hamze and O. Provot, ACS Catal., 2019, 9, 3437 CrossRef CAS.
- (a) H. A. Watson, S. Manaviazar, H. G. Steeds and K. J. Hale, Tetrahedron, 2020, 76, 131061 CrossRef CAS; (b) H. A. Watson, S. Manaviazar, H. G. Steeds and K. J. Hale, Chem. Commun., 2019, 55, 14454 RSC.
- P. Dimopoulos, J. George, D. A. Tocher, S. Manaviazar and K. J. Hale, Org. Lett., 2005, 7, 5377 CrossRef CAS PubMed.
- (a) K. Pati, G. dos Passos Gomes, T. Harris, A. Hughes, H. Phan, T. Banerjee, K. Hanson and I. V. Alabugin, J. Am. Chem. Soc., 2015, 137, 1165 CrossRef CAS PubMed; (b) E. Gonzalez-Rodriguez, M. A. Abdo, G. dos Passos Gomes, S. Ayad, F. D. White, N. P. Tsvetkov, K. Hanson and I. V. Alabugin, J. Am. Chem. Soc., 2020, 142, 8352 CrossRef CAS PubMed.
- C. Nativi and M. Taddei, J. Org. Chem., 1988, 58, 820 CrossRef.
- R. Willem, A. Delmotte, I. De Borger, M. Biesemans, M. Gielen, F. Kayser and E. R. T. Tiekink, J. Organomet. Chem., 1994, 480, 255 CrossRef CAS.
- S. Stoll and A. Schweiger, J. Magn. Reson., 2006, 178, 42 CrossRef CAS PubMed.
- C. Galli, A. Guarnieri, H. Koch, P. Mencarelli and Z. Rappoport, J. Org. Chem., 1997, 62, 4072 CrossRef CAS.
- R. N. Bagchi, A. M. Bond, F. Scholz and R. Stösser, J. Am. Chem. Soc., 1989, 111, 8270 CrossRef CAS.
- A. G. Davies, J. Chem. Soc., Perkin Trans. 2, 1999, 2461 RSC.
- A. G. Davies, Organotin Chemistry, Wiley-VCH, 2004, p. 35 Search PubMed.
- For two other rare reports of vinyl radical EPR spectra having been recorded, see: (a) D. Griller, J. W. Cooper and K. U. Ingold, J. Am. Chem. Soc., 1975, 97, 4269 CrossRef CAS (here β-silylvinyl radicals were generated with half-lives of 0.1 sec to h at 25 °C); (b) T. Ozawa and T. Kwan, J. Chem. Soc., Chem. Commun., 1983, 80 RSC.
- H. Rubin and H. Fischer, Helv. Chim. Acta, 1996, 79, 1670 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1cc01702k |
| This journal is © The Royal Society of Chemistry 2021 |