
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
A novel theranostic activity-based probe targeting kallikrein 7 for the diagnosis and treatment of skin diseases†
Evangelos
Bisyris
,
Eleni
Zingkou
,
Golfo G.
Kordopati
 ,
Minos
Matsoukas
,
Plato A.
Magriotis
,
Georgios
Pampalakis‡
* and
Georgia
Sotiropoulou
*
,
Minos
Matsoukas
,
Plato A.
Magriotis
,
Georgios
Pampalakis‡
* and
Georgia
Sotiropoulou
*
Department of Pharmacy, School of Health Sciences, University of Patras, Rion-Patras, 26504, Greece. E-mail: gpampalakis@pharm.auth.gr; gdsotiro@upatras.gr
First published on 1st June 2021
Abstract
We applied a new in silico approach for using protease-substrate motifs to design a kallikrein 7 (KLK7)-specific phosphonate activity-based probe (ABP) to quantify the active KLK7 in situ. Epidermal application of the ABP-inhibitor on Spink5−/−Klk5−/− mice, a Netherton syndrome model, reversed disease hallmarks, providing preclinical proof-of-concept for using ABPs as theranostics.
KLK7 is a serine protease with well-established functions in pathological skin desquamation and inflammation. It is produced as a zymogen (proKLK7) that is assumingly activated by KLK5.1In vivo, the activity of KLK7 is further regulated by specific endogenous inhibitors such as the Lympho-Epithelial Kazal-Type related Inhibitor (LEKTI). Increased KLK7 expression has been found in the epidermis of Netherton syndrome (NS) and atopic dermatitis (AD) patients.2 NS is caused by loss-of-function mutations in the SPINK5 gene encoding LEKTI. It is a rare form of ichthyosis highlighted by abnormally elevated proteolysis, excessive desquamation and sustained inflammation in the epidermis associated with a severe barrier defect leading to dehydration that is often lethal.3 AD is a frequent disease for which predisposing gene polymorphisms in SPINK5 have been identified.4 Recently, we showed that targeting the KLK5 protease can rescue neonatal lethality in Spink5−/− mice that recapitulate NS5 but KLK7 must be additionally inhibited in Spink5−/−Klk5−/− epidermis to sustain low proteolysis, normal desquamation rates and suppressed inflammation.6 KLK7 could regulate the inflammatory pathways by various mechanisms such as activation of pro-IL-1β or cathelicidin antimicrobial peptide precursors.7 Thus, KLK7 emerged as a novel therapeutic target and synthetic inhibitors against KLK5 and KLK7 represent potential drug compounds for NS and AD. On the other hand, the expression of KLK7 is altered in various types of cancer for which it represents a new biomarker for molecular diagnosis/monitoring and a putative therapeutic target.8,9
It must be noted that the biological function of enzymes, including KLK7, and their implication in disease phenotypes depend on their activity, which is often not linearly proportional to protein abundance, since enzymes also occur in inactive forms, i.e. zymogen, in complex with inhibitors, truncated. However, the assays used in clinics and in research are mainly designed to determine enzyme concentrations independent of their activation status. So far, there has been no available method to determine the levels of active KLK7 in biological and/or clinical specimens. This significantly reduces the predictive value of diagnostic assays used to measure KLK7. In general, an assay to detect the active KLK7 would have a great impact for elucidation of its putative function(s) in various (patho)physiologies and assessment of its diagnostic and therapeutic potential in cancer and skin disorders.
Currently, inhibitors or ABPs specific for KLK7 are not available. Previous attempts have been made to generate reversible or suicide KLK7 inhibitors but their main drawback is their lack of specificity, while none of these compounds has ever been pharmacologically validated in preclinical models.10,11 Thus, we aimed to develop an ABP that (a) will specifically and selectively bind to active KLK7, and (b) will inhibit its enzymatic activity so that it could ideally be used for diagnosis and therapy combined, i.e. as a theranostic agent.
To design ABPs for KLK7, we first developed here a specific KLK7 synthetic inhibitor. We describe a novel strategy that entails MEROPS (http://www.ebi.ac.uk/merops) database mining to identify specific substrates for KLK7 based on mapped protease cleavage sites in various proteins, peptides and synthetic substrates.12 It was revealed that KLK7 can cleave after the Phe–Phe motif (Fig. S1, ESI†). Similarly, ABPs for monoamine oxidases (MAOs) were designed by modification of known MAO inhibitors.13 Reciprocal analysis showed that the ability of KLK7 to cleave after Phe–Phe is rather specific, thus, this motif was used to rationally design an inhibitor and a theranostic ABP (3b and 5b, respectively). Both have α-aminomethylphosphonate reactive groups and carry a modified Phe–Phe dipeptide as a recognition sequence. The ABP was further biotinylated at the Nα-terminus. Chemical syntheses of the compounds are shown in Schemes 1 and 2.
 | ||
| Scheme 1 Synthesis of the inhibitor and the ABP. Reagents and conditions: (a) HOAc/reflux for 2 hours; (b) 1,4-cyclohexadiene, 10% Pd/C in ethanol for 2 hours or triethylsilane, 10% Pd/C in MeOH/CH2Cl2 for 1 hour; and (c) Boc-L-Phe-OH, TBTU, HOBt, DIPEA in CH2Cl2 for 16 hours. | ||
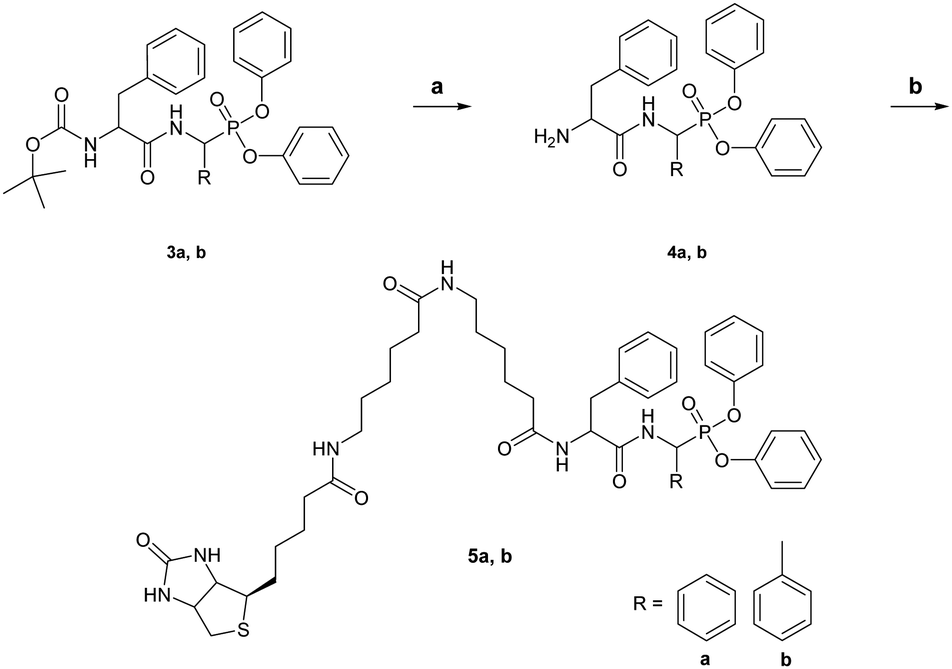 | ||
| Scheme 2 Synthesis of the KLK7-ABP biotin-FBP (5a) and biotin-FFP (5b). Reagents and conditions: (a) TFA/CH2Cl2 for 2 hours; and (b) biotinamidohexanoyl-6-aminohexanoic acid N-hydroxysuccinimide ester, HOBt, DIPEA in DMSO for 16 hours. | ||
For initial optimization, the α-carbon length at the P1-like position was varied in analogues 3a and 3c. Compound 3b (Boc-FFP) along with 3a (Boc-FBP) and 3c (Boc-FCP) was tested by casein gel zymography for specific inhibition of KLK7. Fig. S2 (ESI†) shows that only Boc-FFP inhibits the activity of KLK7, a finding supported by docking studies. Compound 3b (both R, S isomers at α-carbon) demonstrates the highest docking scores with 3b-R performing better (Table S1, ESI†). 3b-R interacts with KLK7 by inserting the benzyl group located between the two amide bonds deep in the catalytic pocket and specifically in the main S1 hydrophobic subpocket14 to interact with A190 and V213. The two phenyl groups attached to the phosphonic oxygens interact with the aromatic rings of H99 and W215 and the tert-butyl group of Boc interacts via sigma–pi positioning with the aromatic ring of H57 in subpocket S1’. One of the phosphonic oxygens forms a hydrogen bond with the main chain NH group of F218 and one of the amides forms hydrogen bond with the side chain of N192. Finally, the benzyl group at the α-carbon of 3b forms a pi–pi interaction with the aromatic ring of F218, providing an explanation for its better performance relative to 3a and 3c that do not allow for correct positioning of the aromatic ring and thus, strong interactions with F218 (Fig. 1).
 | ||
| Fig. 1 Docking pose of 3b-R bound to the catalytic site of KLK7. The protein in shown in teal color, compound 3b-R is depicted with brown sticks, and hydrogen bonds are highlighted with yellow dashed lines. | ||
Therefore, 3b was selected for validation in vitro and in vivo. 3b has an IC50 of 1.91 μM, and when tested against various serine proteases as shown in Fig. S3 (ESI†) it was found to be selective for KLK7. Then, we tested whether the Boc-group is required for inhibition. Following removal of the Boc-protective group we purified the free amino-terminated FFP (H2N-FFP, compound 4b) and tested its inhibitory activity against KLK7, as shown in Fig. S4A (ESI†). H2N-FFP inhibits KLK7 activity to the same extent as the Boc-FFP 3b indicating that modifications at the N-terminus do not affect recognition by the active KLK7 protease.
Subsequently, a biotin derivative was attached at the N-terminus to yield KLK7-ABP 5b (biotin-FFP). Fig. S4B (ESI†) shows that the KLK7-ABP (biotin-FFP) can detect active KLK7 by Western blotting. We further generated the biotin-FBP (5a) that also detected KLK7, albeit with significantly lower affinity than the biotin-FFP (Fig. S4B, ESI†) enforcing that the length of the α-carbon chain on the P1-like position is crucial for optimal recognition by KLK7. Next, an ELISA-type reaction was set up to investigate whether the new ABP can be used to determine the levels of active KLK7 in biological samples as shown in Fig. S5 (ESI†).
Human KLK7 and mouse Klk7 show 76% identity and 86% positivity, while all residues that determine the substrate specificity are highly conserved (Fig. S6, ESI†). Therefore, it is expected that the Boc-FFP and the biotin-FFP would likely react with the mouse Klk7 as well. To test whether biotin-FFP can indeed bind to mouse Klk7, skin extracts from Spink5−/−Klk5−/− mice obtained on P3 were incubated with 50 μM biotin-FFP and analyzed by Western blotting. On P3, Spink5−/−Klk5−/− mice develop symptoms of desquamation and inflammation, associated with increased epidermal Klk7 expression and chymotrypsin activity15 and about 70% of them succumb 7–8 days after birth. As shown in (Fig. 2A), Klk7 is highly expressed in Spink5−/−Klk5−/− skin, as expected. No other bands were detected indicating that biotin-FFP successfully labels active Klk7 without binding to other endogenous proteins. This further supports the specificity of the ABP (Fig. 2B). The biotin-FFP was used to detect and spatially localize by activography the active Klk7 induced by TPA in mouse skin16 (Fig. 2C–E). Consequently, Boc-FFP and biotin-FFP can be exploited for validation of the inhibition of Klk7 activity in Spink5−/−Klk5−/− mice.
 | ||
| Fig. 2 Detection of the endogenous mouse Klk7 protein. (A) Detection of Klk7 in Spink5−/−Klk5−/− mouse skin extracts by Western blotting. (B) The biotin-FFP detects only the Klk7 in the same sample. The nonspecific band of ∼75 kDa detected in the absence of ABP is likely due to endogenous biotinylated proteins. (C) TPA application on mice skin induces the expression of KLK7 as shown by western blotting (upper). α-Tubulin was used as a loading control (lower). (D) Quantification of western blots in C with ImageJ. Data shown are median ± s.e.m. (E) The TPA-induced Klk7 activity is quantified spatially in the epidermis by activography using the biotin-FFP. | ||
Using activography we also tested whether the biotin-FFP could be used to detect the active endogenous KLK7 in skin biopsy cryosections obtained from a healthy human donor and two NS patients. Fig. 3 shows that, in a healthy epidermis, active KLK7 is detectable only at the stratum corneum, as expected. Nevertheless, in NS epidermis it is highly upregulated at sites of stratum corneum rupture and at the junction of the stratum granulosum with the stratum corneum, thus providing evidence for in situ activation of the KLK7 target protease.
 | ||
| Fig. 3 Tissue localization of active KLK7. Skin cryosections were subjected to activography with the biotin-FFP. As expected, normal healthy skin was stained only at the superficial stratum corneum. In contrast, skin samples from NS patients were stained at the regions of stratum corneum rupture (red asterisks, Patient 1) or throughout the damaged SC (Patient 2), and staining was evident at the interface of SC with stratum granulosum (separated by a green dashed line) consistent with the known spatial localization of the KLK7 expression in the NS epidermis. The red dashed line indicates the epidermal–dermal junction. Due to large acanthosis (thickening of the living epidermal layer) the epidermal–dermal junction is not shown in NS samples. | ||
To assess the theranostic value of Boc-FFP and biotin-FFP, first, we examined their in vivo safety. No signs of dermal irritation or corrosion were observed following their application on the skin at 1 mM (Fig. S7, ESI†). The assay was conducted with the standard protocol for testing chemicals (https://ntp.niehs.nih.gov/iccvam/suppdocs/feddocsoecd/oecdtg404.pdf) with a slight modification in that the substances were not removed after application for 4 hours. Also, we observed no signs of skin irritation or corrosion in the epidermis of Klk5−/− mice. The rationale behind testing the compounds on the Klk5−/− background is that for the successful treatment of NS cocktails of KLK5 and KLK7 inhibitors will likely be required.6 The KLK7-ABP-inhibitor was applied on the epidermis of Spink5−/−Klk5−/− mice, which allowed validation of the effect of Klk7 inhibition following ablation of Klk5. Previous studies have established that KLK7 elimination alone is not sufficient to rescue the lethal phenotype of Spink5−/− but its combined inhibition with KLK5 is required for a sustained normalization of NS skin.6
Spink5 −/− Klk5 −/− mice were treated daily with 1 mM of Boc-FFP inhibitor from day 3 onwards when macroscopic signs of desquamation appeared. Fig. 4A and B show significant reduction of desquamation upon application of Boc-FFP. We investigated the expression of the proinflammatory cytokines Tslp, Tnfα, Il-1β, Il-18, Il-17α, and Il-23, which are known to be constitutively induced in the epidermis of Spink5−/− mice and, also, were shown to be reactivated when delayed desquamation and inflammation were manifested in Spink5−/−Klk5−/− mice.6,15Fig. 4C shows that the Boc-FFP inhibitor suppressed the expression of proinflammatory cytokines in Spink5−/−Klk5−/− epidermis uniformly, including Tslp which links inflammation and itching and drives the inflammatory phenotype and asthma in AD. Our study sets the basis for the putative usefulness of KLK7 inhibitors to treat AD, a disease characterized by intolerable itching. We propose that KLK7 inhibitors could represent an alternative way to treat epidermal inflammation with the added benefit of suppressed TNFα levels. Finally, to provide a rationale for the theranostic action, the biotin-FFP was applied onto the skin of Spink5−/−Klk5−/− mice following the application protocol of the Boc-FFP. It was macroscopically observed that biotin-FFP inhibited desquamation (Fig. S8, ESI†).
 | ||
| Fig. 4 (A) Topical application of the Boc-FFP on the epidermis of Spink5−/−Klk5−/− mice led to delayed desquamation rates as shown in this representative image. The area treated is marked with a black line. (B) Quantification of desquamation. Application of Boc-FFP results in almost 50% suppression of desquamation. (C) Application of the KLK7 inhibitor results in a strong suppression of proinflammatory cytokines in Spink5−/−Klk5−/− skin. The cytokine expression was quantified by RT-qPCR. Data shown are median ± s.e.m. (n = 4 mice, per genotype). | ||
Until now, quenched ABPs targeting multiple cathepsin cysteine proteases were assessed as theranostics for cancer and cardiovascular diseases.17 Our study expands the use of ABPs as theranostic agents by demonstrating their application to target the KLKs, the largest family of serine proteases in the human genome. In conclusion, we propose a rapid in silico strategy to identify enzyme specific substrates, which can be modified chemically to generate specific and selective inhibitors and ABPs. Development and validation of a novel ABP-inhibitor prototype that targets the therapeutically significant KLK7 protease is presented. Importantly, the therapeutic potential of the developed biotin-FFP was demonstrated in a preclinical mouse model of NS, which reinforces the use of ABPs as theranostic compounds.
We thank M. Valari, MD/PhD (Aghia Sofia Children's Hospital, Athens, Greece), for providing the human biopsies, Dr D. Darmoul (INSERM, Hôpital Saint Louis, Paris, France) for a sample of bacterial recombinant KLK7, and E. Tsiaousi for technical assistance. We also thank Prof. A. McKenzie (MRC LMB, Cambridge, UK) for kindly providing the Spink5−/− mice. We acknowledge support of this work by the project “BIOLUMINPD; code T1EDK-03884”, which is implemented under the call “Research-create-innovate” funded by the Operational Program “Competitiveness, Entrepreneurship and Innovation”: (NSRF 2014–2020) and co-financed by Greece and the European Union (European Regional Development Fund).
Conflicts of interest
There are no conflicts to declare.Notes and references
- G. Pampalakis and G. Sotiropoulou, Biochim. Biophys. Acta, 2007, 1776, 22 CAS.
- S. Morizane, K. Yamasaki, A. Kajita, K. Ikeda, M. Zhan, Y. Aoyama, G. L. Gallo and K. J. Iwatsuki, J. Allergy Clin. Immunol., 2012, 130, 259 CrossRef CAS PubMed.
- S. Chavanas, C. Bodemer, A. Rochat, D. Hamel-Teillac, M. Ali, A. D. Irvine, J. L. Bonafé, J. Wilkinson, A. Taïeb, Y. Barrandon, J. I. Harper, Y. de Prost and A. Hovnanian, Nat. Genet., 2000, 25, 141 CrossRef CAS PubMed.
- A. J. Walley, S. Chavanas, M. F. Moffatt, R. M. Esnouf, B. Ubhi, R. Lawrence, K. Wong, G. R. Abecasis, E. Y. Jones, J. I. Harper, A. Hovnanian and W. O. Cookson, Nat. Genet., 2001, 29, 175 CrossRef CAS PubMed.
- L. Furio, G. Pampalakis, I. P. Michael, A. Nagy, G. Sotiropoulou and A. Hovnanian, PLoS Genet., 2015, 11, e1005389 CrossRef PubMed.
- P. Kasparek, Z. Ileninova, O. Zbodakova, I. Kanchev, O. Benada, K. Chalupsky, M. Brattsand, I. M. Beck and R. Sedlacek, PLoS Genet., 2017, 13, e1006566 CrossRef PubMed.
- G. Sotiropoulou and G. Pampalakis, Biol. Chem., 2010, 391, 321 CAS.
- A. Tamir, U. Jag, S. Sarojini, C. Schindewolf, T. Tanaka, R. Gharbaran, H. Patel, A. Sood, W. Hu, R. Patwa, P. Blake, P. Chirina, J. Oh Jeong, A. Goy, A. Pecora and K. S. Suh, J. Ovarian Res., 2014, 7, 109 CrossRef PubMed.
- J. P. Du, L. Li, J. Zheng, D. Zhang, W. Liu, W. H. Zheng, X. S. Li, R. C. Yao, F. Wang, S. Liu and X. Tan, Oncotarget, 2018, 9, 12984 Search PubMed.
- G. Sotiropoulou and G. Pampalakis, Trends Pharmacol. Sci., 2012, 33, 623 CrossRef CAS PubMed.
- N. Masurier, D. P. Arama, C. El Amri and V. Lisowski, Med. Res. Rev., 2018, 38, 655 CrossRef CAS PubMed.
- N. D. Rawling, A. J. Barrett, P. D. Thomas, X. Huang, A. Bateman and R. D. Finn, Nucleic Acids Res., 2018, 46, D624 CrossRef PubMed.
- X. Wu, W. Shi, X. Li and H. Ma, Angew. Chem., Int. Ed., 2017, 56, 15319 CrossRef CAS PubMed.
- J. Maibaum, S. M. Liao, A. Vulpetti, N. Ostermann, S. Randl, S. Rudisser, E. Lorthiois, P. Erbel, B. Kinzel, F. A. Kolb, S. Barbieri, J. Wagner, C. Durand, K. Fettis, S. Dussauge, N. Hughes, O. Delgado, U. Hommel, T. Gould, A. Mac Sweeney, B. Gerhartz, F. Cumin, S. Flohr, A. Schubart, B. Jaffee, R. Harrison, A. M. Risitano, J. Eder and K. Anderson, Nat. Chem. Biol., 2016, 12, 1105 CrossRef CAS PubMed.
- E. Zingkou, G. Pampalakis and G. Sotiropoulou, Biochim. Biophys. Acta, Mol. Basis Dis., 2020, 1866, 165831 CrossRef CAS PubMed.
- G. Pampalakis, E. Zinkgou, K. Vekrellis and G. Sotiropoulou, Chem. Commun., 2017, 53, 3246 RSC.
- T. Weiss-Sadan, Y. Ben-Nun, D. Maimoun, E. Merquiol, I. Abd-Elrahman, I. Gotsman and G. Blum, Theranostics, 2019, 9, 5731 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1cc01673c |
| ‡ Current address: Department of Pharmacognosy-Pharmacology, School of Pharmacy, Aristotle University of Thessaloniki, Thessaloniki, 54124, Greece. |
| This journal is © The Royal Society of Chemistry 2021 |