
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCreating porosity in a trianglimine macrocycle by heterochiral pairing†
Donglin
He
 ,
Rob
Clowes
,
Marc A.
Little
*,
Ming
Liu
* and
Andrew I.
Cooper
*
,
Rob
Clowes
,
Marc A.
Little
*,
Ming
Liu
* and
Andrew I.
Cooper
*
Materials Innovation Factory and Chemistry Department, University of Liverpool, Liverpool, L7 3NY, UK. E-mail: aicooper@liverpool.ac.uk
First published on 21st May 2021
Abstract
Macrocycles are usually non-porous or barely porous in the solid-state because of their small intrinsic cavity sizes and tendency to close-pack. Here, we use a heterochiral pairing strategy to introduce porosity in a trianglimine macrocycle, by co-crystallising two macrocycles with opposing chiralities. The stable racemic trianglimine crystal contains an interconnected pore network that has a Brunauer–Emmett–Teller (BET) surface area of 355 m2 g−1.
Porous materials are widely useful in applications such as gas storage, molecular separations, and sensing of gases or vapours.1–4 Synthetic control over pore structure and topology has been achieved for extended framework materials such as zeolites,5 metal–organic frameworks (MOFs)6 and covalent organic frameworks (COFs).7 There is also a growing interest in porous molecular solids; one such example is porous organic cages (POCs),8–10 where solid state function can be intrinsic to the molecular building units. Porous molecular crystals have some potential advantages compared with their extended framework cousins, such as their improved processibility.11,12 In general, however, it is also more challenging to design structure and hence function for porous molecular solids because their crystal packing is often dictated by the sum of a variety of relatively weak and often non-directional intermolecular forces.
Macrocycles, whose solution-phase host–guest chemistry has been studied extensively,13 have been explored recently for a range of molecular separations using the macrocycles in the solid, crystalline state. For example, Janiak and co-workers reported a trianglamine macrocycle crystal with 1-D channels that absorbed ethanol.14 We reported a trianglimine macrocycle crystal that can separate ethyl acetate from its azeotropic mixture with ethanol.15 Eddaoudi and co-workers reported a triangleamine-based supramolecular organic framework that showed permanent porosity and high affinity for CO2.16 We also reported formally non-porous pillar[n]arenes that selectively adsorbed styrene from ethylbenzene17 and para-xylene from its structural isomers.18 However, a challenge to the practical use of macrocycles as adsorbents for separations is their limited adsorption capacities and (often) slow adsorption kinetics. One strategy to solve these kinetic and capacity problems is to increase the porosity in the molecular system. For example, 3-D POCs, with larger cavities can exhibit Brunauer–Emmett–Teller surface areas (SABET) as high as 3758 m2 g−1.19,20 It is more challenging to introduce significant porosity into macrocycles. This is because macrocycles have lower dimensional intrinsic porosity; they are also prone to close packing, minimizing any void space in the solid state.21,22 Hence, unlike POCs with 3-D intrinsic connected pores in the solid state, macrocycle solids rarely have interconnected pore networks that facilitate the rapid diffusion of guest molecules. To date, examples of macrocycles that have been reported with a specific surface area higher than 100 m2 g−1 are still rare.18,23 There is also potential benefit in accessing porous macrocycle crystals with porosity levels that are high enough to permit good diffusion kinetics while retaining the small and size/shape-specific cavity of the macrocycle.
One approach to create porosity in macrocycle structures is to introduce “extrinsic” pores that connect to the small intrinsic macrocycle pores.24 Modular co-crystallization strategies have proven to be effective here.22 For example, the modular assembly of POCs of opposing chiralities has made it possible to control the size and shape of pores in POC crystals.25,26 The same approach also allowed ‘gating POCs’ to be combined with a second POC to exclude a competitive guest and to achieve high guest selectivity.27 However, to our knowledge, this method has not been applied to control the solid state porosity of intrinsically porous molecules, other than POCs.
Isotrianglimine [3+3] macrocycles, formed by reacting isophthalaldehyde with aliphatic diamines, were first reported by Gawronski and co-workers in 2000.28 Subsequently, a series of chiral isotrianglimines were developed, formed by the condensation of trans-1,2-cyclohexanediamine with substituted isophthalaldehydes were reported,29,30 including 1-R (Fig. 1a) that is synthesised by reacting trans-1,2-cyclohexanediamine with isophthalaldehyde. The structure of 1-R is reminiscent of the window motif in the chiral POC CC3-R (Fig. 1a), which we showed to direct the crystal packing of POCs by generating energetically favourable heterochiral window-to-window packing motifs in racemic crystals.26
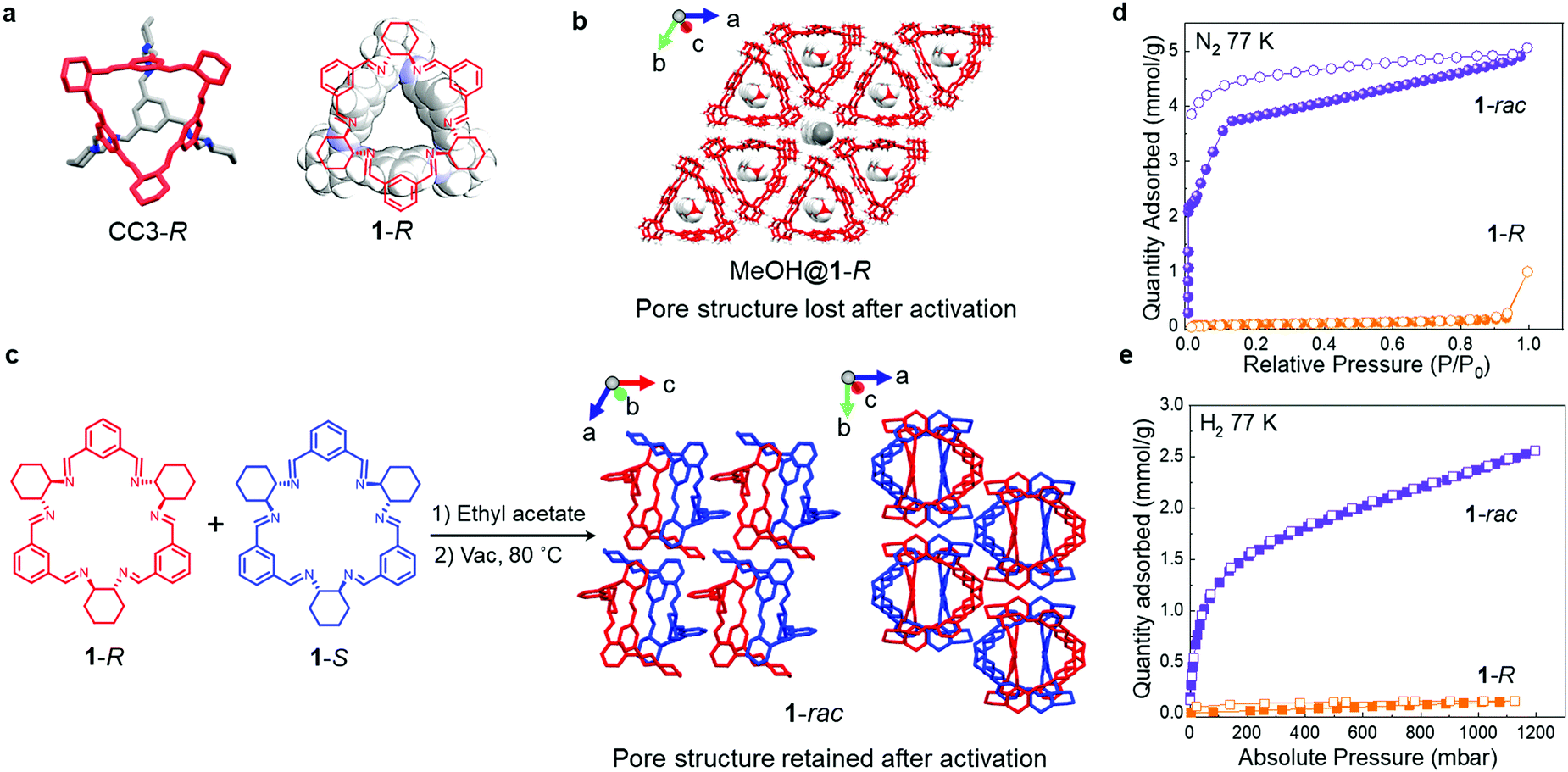 | ||
| Fig. 1 (a) Single crystal structures of the chiral POC, CC3-R, which contains a structural fragment equivalent to 1-R (left), and 1-R (red) displayed in space filling mode (right). (b) Crystal packing in MeOH@1-R, MeOH guest displayed in space filling mode. (c) Single crystal structure of 1-rac. 1-R (red) and 1-S (blue); H atoms are omitted for clarity. (d) N2 and (e) H2 sorption isotherms at 77 K for activated 1-R (orange) and 1-rac (purple). Solid symbols: adsorption; hollow symbols: desorption. | ||
Here, we obtained solvated single crystals of 1-R from methanol (MeOH@1-R, Fig. 1b) that revealed the 1-R molecules were stacked in an eclipsed fashion along the crystallographic c-axis. This motif gave rise to pillars of 1-R that contained MeOH in their intrinsic cavities. In this structure, the neighbouring 1-R pillars are linked by intermolecular hydrogen bonding interaction via the methanol solvent molecules. It was therefore unsurprising that the packing of 1-R changed when the MeOH molecules were removed from the MeOH@1-R crystals (Fig. S7, ESI†) to afford a polycrystalline 1-R sample that was confirmed to be non-porous to both N2 and H2 (Fig. 1d and e).
Solvated racemic co-crystals of 1 (EA@1-rac) were obtained by recrystallising an equimolar ratio of 1-R and 1-S from ethyl acetate (EA, Fig. 1c). These crystals were found to remain suitable for single crystal analysis after activation under a dynamic vacuum at 80 °C. In the activated structure, 1-rac, the racemic window-to-window packing between macrocycles is stabilised by π–π stacking and C–H⋯π interactions, and this was retained after removing the EA solvent (Fig. 1c and Fig. S8, S12, ESI†). The assembly of neighbouring 1-rac heterochiral pairings along the c-axis generates interconnected pores in the crystal structure that occupy 11.6% of the unit cell, as calculated using Platon with a probe radii of 1.2 Å (Fig. 1c), with the largest free sphere (Df) of 2.31 Å calculated by Zeo++.31 It should be noted that the 1-R structure crystallised from EA was also barely porous to N2 and H2 after activation (Fig. S7, S17a and b, ESI†), highlighting the importance of the racemic pairing motif for generating porosity in 1-rac.
To evaluate the gas sorption properties of 1-R and 1-rac, we used the probe gases, N2, H2, and CO2. The N2 isotherms (Fig. 1d) show that 1-rac is porous to N2, with an apparent SABET of 355 m2 g−1, and that it undergoes low-pressure adsorption step at 0.66 mbar. By contrast, 1-R is essentially non-porous to N2 and has a much lower apparent SABET of 4 m2 g−1.321-rac also has a much higher H2 uptake at 1 bar and 77 K (2.56 mmol g−1 for 1-rac vs. 0.12 mmol g−1 for 1-R) with no hysteresis found for either sample (Fig. 1e). The gas sorption isotherms confirm that we successfully created porosity in macrocycle 1 by using the heterochiral pairing strategy to stabilise a porous crystal packing.
For materials containing very small or disconnected pores, which N2 molecules cannot access at cryogenic temperatures, CO2 isotherms are often used to probe porosity.33 For 1-R, the rapid onset of CO2 adsorption isotherm at very low pressure at 195 K indicates the presence of ultra-fine pores that cannot be accessed by N2 molecules at 77 K (Fig. S17a, ESI†).34 CO2 uptake for 1-rac shows a typical type-I isotherm with no hysteresis loop at 195 K; at 1 bar, 1-rac absorbed significantly more CO2 than 1-R (3.80 mmol vs. 2.17 mmol) (Fig. 2a). This result is consistent with the N2 isotherms and confirms that substantial additional porosity has been created in 1-rac. The high pressure CO2 adsorption isotherm of 1-rac at 273 K shows two successive ‘plateaus’ (Fig. 2b), which corresponds to CO2 accessing the different pores in the flexible 1-rac structure as it expands, and similar behaviour has been observed in other porous solids.35 At 273 K, 283 K, and 298 K, 1-R adsorbs more CO2 than 1-rac in the low relative pressure range (0–1 bar). To understand this initially counterintuitive phenomenon, the isosteric heats of adsorption (Qst) were calculated from the CO2 isotherms (273–298 K, 0–1 bar) for 1-rac and 1-R. As shown in Fig. S17d (ESI†), the calculated isosteric heats of adsorption (Qst) for 1-R and 1-rac were less than 30 kJ mol−1, excluding chemisorption by either adsorbent.36 The Qst for CO2 on 1-R remained constant over a larger adsorbate loading range (0–1 mmol g−1) indicating an energetically homogeneous surface.371-rac had a linear decrease in Qst with CO2 loading over the range of 0–0.5 mmol g−1, indicating that 1-rac is more energetically heterogeneous for the adsorption of CO2.37 The higher CO2 uptake of 1-R at lower pressures can be attributed to ultra-fine pores in 1-R that can adsorb CO2 as a monolayer. By contrast, the larger interconnected pores of 1-rac lead to the multilayer adsorption of CO2 but with lower uptakes at lower pressures.
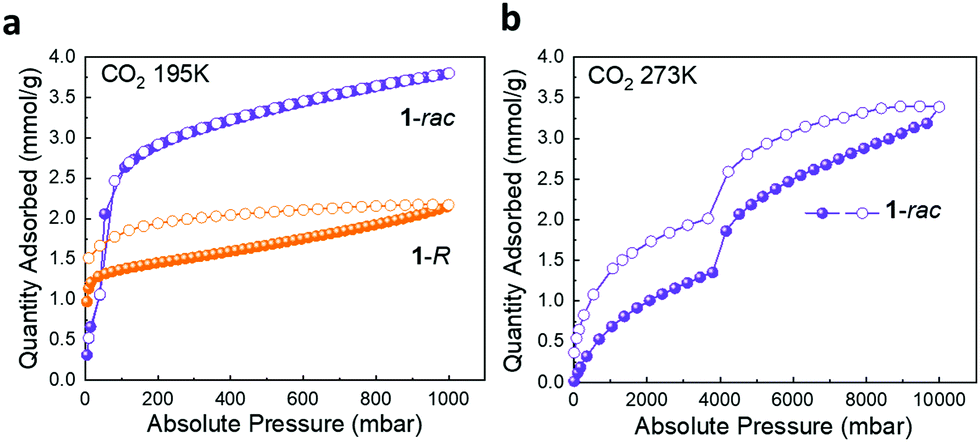 | ||
| Fig. 2 CO2 isotherms of 1-rac (violet) and 1-R (orange): (a) CO2 isotherms at 195 K form 0–1 bar. (b) CO2 isotherms at 273 K from 0 to 10 bar. Adsorption isotherms as closed symbols; desorption isotherms as open symbols. | ||
The separation of xylene isomers is challenging because they have similar molecular structures and physical properties. Recently, molecular materials such as cucurbit[7]uril38 and the polymorphic azobenzene cage39 have been studied as adsorbents for the separation of xylene isomers. The largest included sphere along the free sphere path (Dif) calculated by Zeo++31,40 in activated 1-rac is 4.26 Å, which is close to the molecular size of para-xylene (pX) (4.2 × 6.8 Å) (Table S1, ESI†). This close size match suggested that 1-rac might be a good host for pX over its structural isomer, meta-xylene (mX, Table S1, ESI†). We initially crystallised 1-R and 1-S from pX and found the resulting inclusion complex, 1pX@1-rac (Fig. 3a). The crystal structure of 1pX@1-rac revealed that one pX molecule crystallised in the centre of the cavity created between 1-R and 1-S molecules packed in a window-to-window arrangement. A second pX molecule in the structure was located in an extrinsic void created between four 1 molecules. Compared with the guest-free structure of 1-rac, the inclusion of pX does not significantly change the packing of 1 (Fig. 3a), as confirmed by the crystal packing overlay shown in Fig. 3b. However, 1-rac does expand by around 8% to accommodate 1 mol mol−1 of pX in its structure. By contrast, the inclusion complex, 3mX@2(1-rac), has a different packing mode compared to activated 1-rac (Fig. S9a, S14, and S16, ESI†). These results suggest that a racemic mixture of 1 can form inclusion complexes with both pX and mX isomers, but that the crystal packing in guest-free 1-rac is more closely matched for pX inclusion. When we activated 1pX@1-rac and 3mX@2(1-rac) they transformed into the 1-rac structure at 80 °C and 140 °C, respectively (Fig. 3c and Fig. S9a, ESI†), indicating that 1-rac is the energetically favourable phase. However, the lower activation temperature for 1pX@1-rac indicates that pX desorbs more easily from 1pX@1-rac (Fig. S6, ESI†), which is likely due to the more interconnected 1-D porosity in 1pX@1-rac (Fig. 3b) and the narrower dimensions of pX.
 | ||
Fig. 3 (a) Reversible capture of pX from 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 pX–mX vapour mixture illustrated by single-crystal structures for 1-rac (left) and 1pX@1-rac (right). 1-R and 1-S are coloured red and blue, respectively; H atoms are omitted for clarity. (b) Crystal packing overlay for 1pX@1-rac (green) and activated 1-rac (yellow). (c) PXRD patterns of 1-rac after being exposed to pX vapour and then dried under vacuum at elevated temperatures. (d) Time-dependent 1-rac solid–vapour sorption plot for pX and mX equimolar vapour mixture. (e) The capacity of 1-R and 1-rac, determined using a 1:1 pX–mX vapour mixture. :1 pX–mX vapour mixture illustrated by single-crystal structures for 1-rac (left) and 1pX@1-rac (right). 1-R and 1-S are coloured red and blue, respectively; H atoms are omitted for clarity. (b) Crystal packing overlay for 1pX@1-rac (green) and activated 1-rac (yellow). (c) PXRD patterns of 1-rac after being exposed to pX vapour and then dried under vacuum at elevated temperatures. (d) Time-dependent 1-rac solid–vapour sorption plot for pX and mX equimolar vapour mixture. (e) The capacity of 1-R and 1-rac, determined using a 1:1 pX–mX vapour mixture. | ||
To determine if 1-rac could separate pX from mX, we performed time-dependent solid–vapour sorption experiments using the vapours generated from a physical mixture of the two xylene isomers. As shown in Fig. 3d, porous 1-rac captures pX selectively from a 1:1 (vol:vol) mixture of pX and mX. The maximum uptake of pX with 1-rac was 0.83 mol mol−1 after 10 h, which is close to the ideal ratio of 1 in 1pX@1-rac. The capacity of 1-rac for pX in 1:1 pX–mX vapour mixture is about 1.5 times higher than for 1-R, which we attribute to the increased porosity in 1-rac (Fig. 3e). A larger difference, however, is in the adsorption kinetics. For example, a formally non-porous pillar[6]arene macrocycle that adsorbs pX over mX with similar selectivity was found to reach saturation after 20 hours.181-rac performs much better than this, reaching saturation after only 3 hours under the same conditions, with a selectivity coefficient of KpX:mX = 15.7.41 The greatly improved adsorption kinetics of 1-rac compared to the pillar[6]arene system is a direct result of its increased porosity, which results from the chiral pairing strategy.
In conclusion, we have introduced porosity into a trianglimine macrocycle system by using a heterochiral pairing strategy. Porosity was created by co-crystallising two macrocycles with the opposing chiralities such that they pack in a window-to-window arrangement to connect the intrinsic macrocycle voids. This generates an interconnected pore network with an apparent SABET of 355 m2 g−1. This is the highest reported surface area for the trianglimine macrocycle,16,23 which are usually barely porous in the solid state. Because of its increased porosity, the 1-rac co-crystal has greatly improved adsorption kinetics and shows the potential to separate xylene isomers, exhibiting much higher selectivity toward pX, by a factor of 15.7 vs. mX, outperforming related macrocyclic systems for the same separation.18 As well as introducing porosity, the heterochiral pairing strategy could also enrich the functionality of these macrocycle systems by enabling hybrid mixing of macrocycles with different functions that would otherwise not co-crystallise, as demonstrated with POCs for quantum sieving applications.42
The authors acknowledge the EPSRC (EP/N004884/1) and the Leverhulme Trust via the Leverhulme Research Centre for Functional Materials Design for funding. D. H. thanks the Oversea Study Program of Guangzhou Elite Project provided by Guangzhou City, China for financial support.
Conflicts of interest
There are no conflicts to declare.Notes and references
- C. Sapsanis, H. Omran, V. Chernikova, O. Shekhah, Y. Belmabkhout, U. Buttner, M. Eddaoudi and K. N. Salama, Sensors, 2015, 15, 18153–18166 CrossRef CAS PubMed.
- M. G. Rabbani and H. M. El-Kaderi, Chem. Mater., 2012, 24, 1511–1517 CrossRef CAS.
- J.-R. Li, R. J. Kuppler and H.-C. Zhou, Chem. Soc. Rev., 2009, 38, 1477–1504 RSC.
- M. Brutschy, M. W. Schneider, M. Mastalerz and S. R. Waldvogel, Adv. Mater., 2012, 24, 6049–6052 CrossRef CAS PubMed.
- V. M. Georgieva, E. L. Bruce, M. C. Verbraeken, A. R. Scott, W. J. Casteel, S. Brandani and P. A. Wright, J. Am. Chem. Soc., 2019, 141, 12744–12759 CrossRef CAS PubMed.
- S. Ma, X.-S. Wang, D. Yuan and H.-C. Zhou, Angew. Chem., Int. Ed., 2008, 47, 4130–4133 CrossRef CAS PubMed.
- R.-Q. Wang, X.-B. Wei and Y.-Q. Feng, Chem. – Eur. J., 2018, 24, 10979–10983 CrossRef CAS PubMed.
- T. Tozawa, J. T. Jones, S. I. Swamy, S. Jiang, D. J. Adams, S. Shakespeare, R. Clowes, D. Bradshaw, T. Hasell, S. Y. Chong, C. Tang, S. Thompson, J. Parker, A. Trewin, J. Bacsa, A. M. Slawin, A. Steiner and A. I. Cooper, Nat. Mater., 2009, 8, 973–978 CrossRef CAS PubMed.
- T. Mitra, K. E. Jelfs, M. Schmidtmann, A. Ahmed, S. Y. Chong, D. J. Adams and A. I. Cooper, Nat. Chem., 2013, 5, 276–281 CrossRef CAS PubMed.
- A. G. Slater and A. I. Cooper, Science, 2015, 348, aaa8075 CrossRef PubMed.
- J. R. Holst, A. Trewin and A. I. Cooper, Nat. Chem., 2010, 2, 915–920 CrossRef CAS PubMed.
- A. I. Cooper, ACS Cent. Sci., 2017, 3, 544–553 CrossRef CAS PubMed.
- F. Vögtle and E. Weber, Host Guest Complex Chemistry Macrocycles: Synthesis, Structures, Applications, Springer Science & Business Media, 2012 Search PubMed.
- A. Janiak, M. Bardziński, J. Gawroński and U. Rychlewska, Cryst. Growth Des., 2016, 16, 2779–2788 CrossRef CAS.
- D. He, C. Zhao, L. Chen, M. Little, S. Chong, R. Clowes, K. McKie, M. Roper, G. Day, M. Liu and A. Cooper, Chem. – Eur. J., 2021 DOI:10.1002/chem.202101510.
- A. Chaix, G. Mouchaham, A. Shkurenko, P. Hoang, B. Moosa, P. M. Bhatt, K. Adil, K. N. Salama, M. Eddaoudi and N. M. Khashab, J. Am. Chem. Soc., 2018, 140, 14571–14575 CrossRef CAS PubMed.
- K. Jie, M. Liu, Y. Zhou, M. A. Little, S. Bonakala, S. Y. Chong, A. Stephenson, L. Chen, F. Huang and A. I. Cooper, J. Am. Chem. Soc., 2017, 139, 2908–2911 CrossRef CAS PubMed.
- K. Jie, M. Liu, Y. Zhou, M. A. Little, A. Pulido, S. Y. Chong, A. Stephenson, A. R. Hughes, F. Sakakibara, T. Ogoshi, F. Blanc, G. M. Day, F. Huang and A. I. Cooper, J. Am. Chem. Soc., 2018, 140, 6921–6930 CrossRef CAS PubMed.
- M. Mastalerz, Acc. Chem. Res., 2018, 51, 2411–2422 CrossRef CAS PubMed.
- G. Zhang, O. Presly, F. White, I. M. Oppel and M. Mastalerz, Angew. Chem., Int. Ed., 2014, 53, 1516–1520 CrossRef CAS PubMed.
- N. B. McKeown, J. Mater. Chem., 2010, 20, 10588–10597 RSC.
- M. A. Little and A. I. Cooper, Adv. Funct. Mater., 2020, 30, 1909842 CrossRef CAS.
- E. Sanna, E. C. Escudero-Adán, A. Bauzá, P. Ballester, A. Frontera, C. Rotger and A. Costa, Chem. Sci., 2015, 6, 5466–5472 RSC.
- M. J. Bojdys, M. E. Briggs, J. T. A. Jones, D. J. Adams, S. Y. Chong, M. Schmidtmann and A. I. Cooper, J. Am. Chem. Soc., 2011, 133, 16566–16571 CrossRef CAS PubMed.
- J. T. A. Jones, T. Hasell, X. Wu, J. Bacsa, K. E. Jelfs, M. Schmidtmann, S. Y. Chong, D. J. Adams, A. Trewin, F. Schiffman, F. Cora, B. Slater, A. Steiner, G. M. Day and A. I. Cooper, Nature, 2011, 474, 367–371 CrossRef CAS PubMed.
- T. Hasell, S. Y. Chong, K. E. Jelfs, D. J. Adams and A. I. Cooper, J. Am. Chem. Soc., 2012, 134, 588–598 CrossRef CAS PubMed.
- T. Hasell and A. I. Cooper, Nat. Rev. Mater., 2016, 1, 16053 CrossRef CAS.
- J. Gawroński, H. Kołbon, M. Kwit and A. Katrusiak, J. Org. Chem., 2000, 65, 5768–5773 CrossRef PubMed.
- N. Kuhnert, G. M. Rossignolo and A. Lopez-Periago, Org. Biomol. Chem., 2003, 1, 1157–1170 RSC.
- A. Troć, J. Gajewy, W. Danikiewicz and M. Kwit, Chem. – Eur. J., 2016, 22, 13258–13264 CrossRef PubMed.
- T. F. Willems, C. H. Rycroft, M. Kazi, J. C. Meza and M. Haranczyk, Microporous Mesoporous Mater., 2012, 149, 134–141 CrossRef CAS.
- M. Thommes, K. Kaneko, A. V. Neimark, J. P. Olivier, F. Rodriguez-Reinoso, J. Rouquerol and K. S. Sing, Pure Appl. Chem., 2015, 87, 1051–1069 CAS.
- P. I. Ravikovitch, A. Vishnyakov, R. Russo and A. V. Neimark, Langmuir, 2000, 16, 2311–2320 CrossRef CAS.
- B. D. Chandler, D. T. Cramb and G. K. Shimizu, J. Am. Chem. Soc., 2006, 128, 10403–10412 CrossRef CAS PubMed.
- P. Lama and L. J. Barbour, J. Am. Chem. Soc., 2018, 140, 2145–2150 CrossRef CAS PubMed.
- K.-J. Chen, D. G. Madden, T. Pham, K. A. Forrest, A. Kumar, Q.-Y. Yang, W. Xue, B. Space, J. J. Perry Iv, J.-P. Zhang, X.-M. Chen and M. J. Zaworotko, Angew. Chem., Int. Ed., 2016, 55, 10268–10272 CrossRef CAS PubMed.
- S. Sircar and D. V. Cao, Chem. Eng. Technol., 2002, 25, 945–948 CrossRef CAS.
- G. Zhang, A.-H. Emwas, U. F. Shahul Hameed, S. T. Arold, P. Yang, A. Chen, J.-F. Xiang and N. M. Khashab, Chem, 2020, 6, 1082–1096 CAS.
- B. Moosa, L. O. Alimi, A. Shkurenko, A. Fakim, P. M. Bhatt, G. Zhang, M. Eddaoudi and N. M. Khashab, Angew. Chem., Int. Ed., 2020, 59, 21367–21371 CrossRef CAS PubMed.
- M. D. Foster, I. Rivin, M. M. J. Treacy and O. Delgado Friedrichs, Microporous Mesoporous Mater., 2006, 90, 32–38 CrossRef CAS.
- A. M. Pivovar, K. T. Holman and M. D. Ward, Chem. Mater., 2001, 13, 3018–3031 CrossRef CAS.
- M. Liu, L. Zhang, M. A. Little, V. Kapil, M. Ceriotti, S. Yang, L. Ding, D. L. Holden, R. Balderas-Xicohténcatl, D. He, R. Clowes, S. Y. Chong, G. Schütz, L. Chen, M. Hirscher and A. I. Cooper, Science, 2019, 366, 613–620 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2073632–2073637. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d1cc01650d |
| This journal is © The Royal Society of Chemistry 2021 |