
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Plug-and-play aqueous electrochemical atom transfer radical polymerization†
Boyu
Zhao
,
Mahir
Mohammed
,
Bryn
A. Jones
and
Paul
Wilson
 *
*
Department of Chemistry, University of Warwick, Coventry, CV4 7AL, UK. E-mail: p.wilson.1@warwick.ac.uk
First published on 17th March 2021
Abstract
A simplified ‘plug-and-play’ approach to aqueous electrochemical atom transfer radical polymerization (eATRP) has been developed. Well-controlled polymerization of PEGA480 (Đm = 1.17–1.31) is reported under potentiostatic (3-electrodes, undivided cell) and galvanostatic (2-electrodes, 6-steps) conditions.
Recent progress in polymerization methods has advanced to the point where we can exquisitely design and subsequently control polymer synthesis with respect to molecular weight (including dispersity), composition and topology.1,2 In reversible deactivation radical polymerization (RDRP) (bio)chemical, photochemical,3 electrochemical,4 mechanochemical5 and sonochemical6 methods have all been developed, enabling external control over the dynamic equilibrium between dormant and active (radical) species, ultimately allowing the overall radical concentration to be accurately controlled.
In Cu-mediated atom transfer radical polymerization (ATRP),7 the equilibrium (KATRP) is between dormant alkyl (R-X) or macromolecular (Pn-X) halides and propagating radicals (R˙/Pn˙) that undergo reversible redox reactions with Cu-complexes. The redox nature of the ATRP mechanism lends itself to electrochemical manipulation and control.4,8 The active, yet oxidatively labile CuIL complex can be formed in situ when a potential is applied (Eapp) to promote a one electron reduction of an inactive CuIIL precursor. The equilibrium between dormant and active species (KATRP), and the kinetics of polymerization, can therefore be controlled by tuning the potential of the system. Thus, electrochemical ATRP (eATRP) has been demonstrated to provide high fidelity on–off temporal control over polymer synthesis in solution for a variety of polymer architectures,9,10 in dispersed media11 and from surfaces (si-eATRP).12
A perceived limitation of the original eATRP reactions was the complexity of the reaction set-up. A 3-electrode divided electrochemical cell was used with reactions carried out under potentiostatic (constant potential) conditions to avoid unwanted side-reactions occurring at the counter electrode (anode). This was alleviated by the development of simplified eATRP (seATRP), which employs a sacrificial counter electrode as the anode.13 For example, aluminum wire has been employed for the synthesis of linear and star-like polymers via seATRP with the Al-wire, and the products of its oxidation (Al3+), being inert to the eATRP reaction conditions.14–21 Thus, seATRP reaction conditions allow undivided cells to be used in 3-electrode configurations (potentiostatic). Furthermore, reactions can be run in a 2-electrode configuration under galvanostatic conditions (constant current) using a simple (and cheaper) current generator in place of a potentiostat.
The complexity of electrosynthetic reaction set up is not unique to eATRP. In organic electrosynthesis and electrocatalysis the reaction set up, from electrode materials to cell configuration has historically been viewed as a black-box which has limited wide-spread application of electrolysis to solve synthetic problems.22 This perspective has dramatically changed recently due to the development and reporting of standardized hardware and reaction conditions.23,24 For example, the IKA Electrasyn device, which is a magnetic stirring plate with an in-built potentiostat allows both constant current and potential reactions to be performed.25 With a range of standardized electrodes and other accessories, this has transformed electrosynthesis into almost a ‘plug-and-play’ methodology that is accessible to the entire synthetic community.
Thus, we were interested to see if further simplification of eATRP using these ‘plug-and-play’ features could be achieved. Although a number of seATRP reactions in organic solvents14–21 have been reported, the number of aqueous reactions reported is more limited.26,27 With this in mind, we opted to investigate whether aqueous seATRP of poly(ethylene glycol) methyl ether acrylate (Mn = 480; PEGA480) could be achieved using the capabilities afforded by the Electrasyn device.
Initially, cyclic voltammetry (CV) of the Cu-complex (CuII/TPMA) was performed in solutions of the reaction mixture (10% (v/v) PEGA480 in H2O) in the absence and presence of the initiator, hydroxyethyl-2-bromoisobutyrate (HEBiB) (Fig. S1, ESI†). The expected [CuII/TPMA]/[CuI/TPMA] redox process was observed and the standard reduction potential (Eθ ≈ E1/2 = Epc + Epa/2) was found to be −0.197 V (vs. Ag/AgCl). Upon addition of HEBiB the intensity of Epc increased while the intensity of Epa decreased significantly indicative of electrochemical reduction of [CuII/TPMA] to [CuI/TPMA] followed by rapid activation of HEBiB by the [CuI/TPMA].
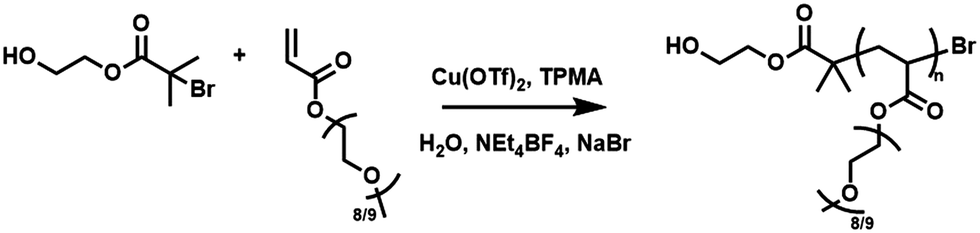
Potentiostatic reactions, performed in undivided cells, were investigated first using a Pt-coated working electrode (IKA), Al-wire counter electrode and a Ag/AgCl reference (Fig. S2 and S3, ESI†). Based on Eθ ≈ E1/2 = −0.197 V, seATRP was initially performed with using [PEGA480]![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :[HEBiB]:[Cu(OTf)2]:[TPMA]:[NaBr] = [38]:[1]:[0.15]:[0.45]:[0.15] with Eapp = −0.20 V (Table 1, entry 1). The polymerization was following by 1H NMR and conversion was determined via integration of one the vinyl protons (Hf) against the methylene protons (Hc) adjacent to the ester group of the PEGA480 monomer and emerging polymer (Fig. S4, ESI†). Kinetic analysis of the reaction revealed pseudo-first order kinetics (Fig. 1A), reaching 73% conversion after 8 h. The evolution of molecular weight with conversion was followed by SEC (Fig. 1C and Fig. S5, ESI†) and found to be linear, as expected for RDRP. The purified polymer (PPEGA480) had a number average molecular weight; Mn,SEC = 11000 g mol−1, which was in good agreement to the theoretical value (Mn,th = 13526 g mol−1), and a relatively low dispersity (Đm = 1.25).
:[HEBiB]:[Cu(OTf)2]:[TPMA]:[NaBr] = [38]:[1]:[0.15]:[0.45]:[0.15] with Eapp = −0.20 V (Table 1, entry 1). The polymerization was following by 1H NMR and conversion was determined via integration of one the vinyl protons (Hf) against the methylene protons (Hc) adjacent to the ester group of the PEGA480 monomer and emerging polymer (Fig. S4, ESI†). Kinetic analysis of the reaction revealed pseudo-first order kinetics (Fig. 1A), reaching 73% conversion after 8 h. The evolution of molecular weight with conversion was followed by SEC (Fig. 1C and Fig. S5, ESI†) and found to be linear, as expected for RDRP. The purified polymer (PPEGA480) had a number average molecular weight; Mn,SEC = 11000 g mol−1, which was in good agreement to the theoretical value (Mn,th = 13526 g mol−1), and a relatively low dispersity (Đm = 1.25).
:[HEBiB]:[Cu(OTf)2]:[TPMA]:[NaBr] = [38]:[1]:[0.15]:[0.45]:[0.15]
|
|
||||||
|---|---|---|---|---|---|---|
| Entry | E app/V | Time/h | Conv.e/% | M n,th /g mol−1 | M n,SEC /g mol−1 | Đ m |
| a [PEGA480] = 20% v/v. b [PEGA480] = 30% v/v. c [PEGA480] = 40% v/v. d [PEGA480] = 50% v/v. e Determined via1H NMR of reaction samples performed in D2O. f M n,th = [(conv./100 × DPn,th) × 480] + 221. g From THF SEC of purified polymers. | ||||||
| 1 | −0.2 | 8 | 73 | 13526 |
11000 |
1.25 |
| 2 | −0.15 | 8 | 54 | 10100 |
9200 | 1.17 |
| 3 | −0.25 | 8 | 63 | 11337 |
12900 |
1.33 |
| 4 | −0.3 | 4.5 | 49 | 9149 | 8800 | 1.52 |
| 5 | −0.35 | 4 | 50 | 9331 | 8900 | 1.71 |
| 6a | −0.2 | 8 | 84 | 15532 |
13700 |
1.41 |
| 7b | −0.2 | 8 | 86 | 15897 |
11910 |
1.61 |
| 8c | −0.2 | 8 | 50 | 9331 | 8600 | 2.56 |
| 9a | −0.15 | 8 | 65 | 12067 |
14900 |
1.23 |
| 10b | −0.15 | 8 | 83 | 15350 |
14500 |
1.31 |
| 11c | −0.15 | 8 | 76 | 14073 |
16500 |
1.41 |
| 12d | −0.15 | 8 | 23 | 4406 | 6100 | 3.28 |
 | ||
| Fig. 1 For [PEGA480]:[HEBiB]:[Cu(OTf)2]:[TPMA]:[NaBr] = [38]:[1]:[0.15]:[0.45]:[0.15]; (A) conversion and pseudo first order kinetic plots as a function of Eapp. (B) A zoom in the region between t = 0–2.5 h from which kappp was determined. (C) Evolution of the Mn,SEC and Đm with conversion (Eapp = −0.20 V). | ||
To investigate the effect of Eapp in our ‘plug-and-play’ configuration, reactions were repeated at ±0.05 V increments. At less reducing potentials (Eapp = −0.15 V, Table 1, entry 2) the polymerization was under better control furnishing PPEGA480 with lower final dispersity (Đm = 1.17). However, at more reducing potentials (Eapp = −0.25 to −0.35 V, Table 1, entries 3–5) the dispersities of the final polymers gradually increased (Đm = 1.33–1.72). These observations are in agreement with the previous literature28 and the proposed relationship between Eapp and ratio of [CuI] and [CuII] present in the reaction, as described by the Nernst equation.
Kinetic analysis was also performed for the polymerizations at Eapp = E1/2 ± 0.05 V. The gradient of the linear region of the kinetic plots revealed an apparent rate constant for propagation, kappp = 0.21 h−1 when Eapp = −0.15 V. This increased to kappp = 0.24 h−1 when Eapp = E1/2 = −0.20 V and kappp = 0.26 h−1 at Eapp = −0.25 V (Fig. 1B). All three polymerizations proceeded with a linear evolution of Mn,SEC with conversion (Fig. 1C). Deviations in Mn,SEC from Mn,th, are likely due to errors caused by overlap of the monomer and emerging polymer peaks in the SEC chromatographs of the reaction samples (Fig. S5, ESI†).
The effect of monomer concentration was then investigated at Eapp = −0.20 V and Eapp = −0.15 V. Increasing the [PEGA480] to 20% and 30% (v/v) resulted in increased conversions of 84% and 86% when Eapp = −0.20 V (Table 1, entries 6 and 7). However, the polymerizations were not as well controlled producing polymers with Đm = 1.41 and 1.61 respectively. Increasing [PEGA480] further to 40% v/v resulted in only 50% conversion and Đm > 2.5 indicating that the reaction was completely uncontrolled (Table 1, entry 8). When Eapp = −0.15 V, increasing [PEGA480] to 20% and 30% (v/v) had a similar effect. The conversion increased to 65% and 83% respectively (Table 1, entries 9 and 10). In this case the control over the polymerizations was retained (Đm < 1.31). Increasing the [PEGA480] further to 40% and 50% (v/v) led to the conversion decreasing to the 76% and 23% respectively, and control over the polymerizations being compromised (Đm = 1.41–3.28, Table 1, entries 11 and 12). The gradual increase in Đm can be attributed to the gradual increase in termination reactions, manifest as the emergence of high molecular weight shoulder peaks in the SEC chromatograms as [PEGA480] is increased from 20–40% (v/v) (Fig. S6, ESI†). This is exacerbated at 50% (v/v) where significant high molecular weight tailing was observed leading to Đm = 3.28.
Based on conversion, kinetics and Đm reported in Table 1, subsequent polymerizations targeting PPEGA480 with different DPn,th were performed at Eapp = −0.15 V for 2.5 h. Repeating the polymerization for [PEGA480]:[HEBiB] = [38]:[1] resulted in 53% conversion and final polymer with and Mn,SEC (11600 g mol−1) in good agreement with Mn,th (9878 g mol−1) and a low dispersity (Đm = 1.17) (Table 2, entry 2). Decreasing [PEGA480]:[HEBiB] to [19]:[1] led to higher conversion (68%) after 2.5 h (Table 2, entry 1).
:[HEBiB]:[Cu(OTf)2]:[TPMA]:[NaBr] = [38]:[1]:[0.15]:[0.45]:[0.15]
| Entry | [M]/[I] | Time/h | Conv.a/% | M n,th /g mol−1 | M n,SEC /g mol−1 | Đ m |
|---|---|---|---|---|---|---|
| a Determined via1H NMR of reaction samples performed in D2O. b M n,th = [(conv./100 × DPn,th) × 480] + 221. c From THF SEC of purified polymers. | ||||||
| 1 | 19 | 2.5 | 68 | 6412 | 9100 | 1.21 |
| 2 | 38 | 2.5 | 53 | 9878 | 11600 |
1.17 |
| 3 | 57 | 2.5 | 54 | 14985 |
16100 |
1.28 |
| 4 | 76 | 2.5 | 55 | 20275 |
18900 |
1.21 |
| 5 | 114 | 2.5 | 45 | 24835 |
19300 |
1.32 |
However, a slightly higher dispersity (Đm = 1.21) and deviation in Mn,SEC (9100 g mol−1) and Mn,th (6412 g mol−1) was observed. Increasing [PEGA480]:[HEBiB] to [57]:[1], [76]:[1] and [114]:[1] resulted in conversions between 45–55% within 2.5 h (Table 2, entries 3–5). The final polymers all exhibited good agreement between Mn,SEC and Mn,th and low Đm indicative of well controlled polymerizations. An overlay of the SEC chromatograms shows the gradual shift to higher molecular weight as a function of increasing [PEGA480]:[HEBiB] (Fig. S7, ESI†).
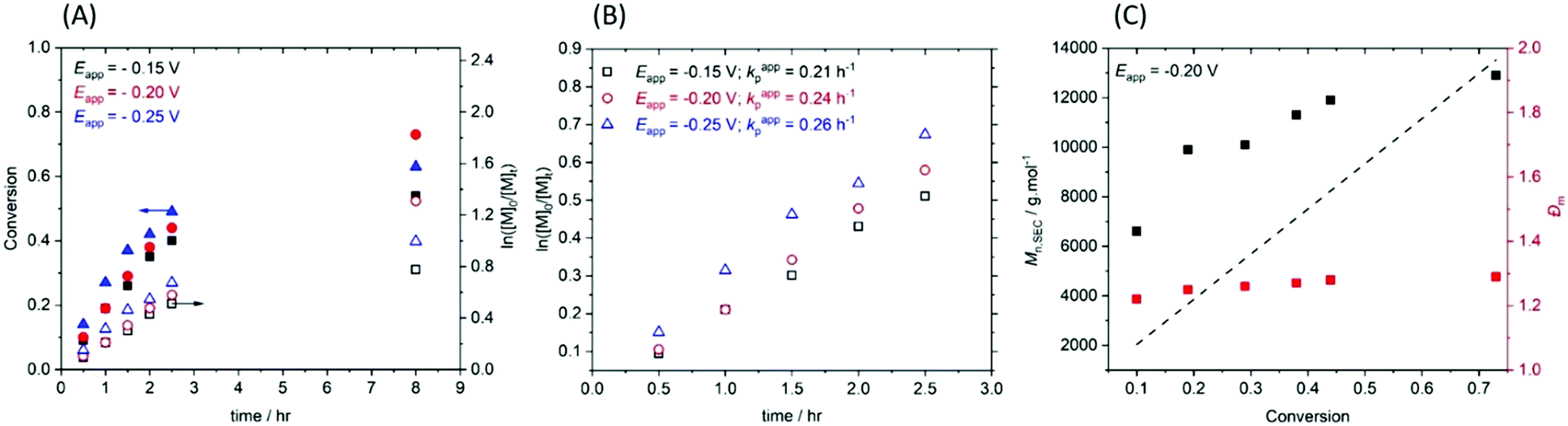
The temporal control afforded by standard eATRP reactions was then investigated in an experiment wherein Eapp = −0.15 V was switched on and off for 30 min intervals during the course of a reaction (Fig. 2). During step 1 (tON = 0.5 h, kapp,1p = 0.71 h−1) 30% conversion was obtained. At this point the Eapp was set to 0 V and the reaction was left for a further 30 min during which no further polymerization was observed. During step 2 (Eapp = −0.15 V) polymerization restarted, albeit at a slightly slower rate (kapp,2p = 0.48 h−1), reaching 45% conversion (tON = 1 h). Again polymerization was halted upon removal of Eapp and restarted again after 30 min. Conversion increased to 56% (Step 3, tON = 1.5 h, kappp = 0.45 h−1) before Eapp was switched off for a third and final 30 min period. Eapp was switched back on and the polymerization was allowed to proceed for a further 6.5 h (tON = 8 h in total), resulting in a final conversion of 73% (Fig. S8A, ESI†). Molecular weight analysis of the final polymer revealed a Mn,SEC = 22900 g mol−1 which is higher than expected (Mn,th = 13526 g mol−1), indicating that some active chain-ends were lost during these polymerizations. Evidence for this was not apparent in the symmetrical SEC chromatogram and the final dispersity (Đm = 1.28) indicated that good control was retained (Fig. S8B, ESI†).
Finally, the polymerization of PEGA480 was attempted in a 2-electrode set up under galvanostatic conditions (constant current). Conveniently, during potentiostatic reactions (vide supra), the IKA Electrasyn (via a bespoke mobile app) records current (I) vs. time graphs which can be integrated to determine the total charge passed during a reaction (Q). This can be used to determine the applied currents (Iapp) for subsequent galvanostatic reactions. The current vs. time plot (Fig. 3A) contains two distinct regimes. With reference to the mechanism, the initial current decay regime can be explained by the reduction of CuII/L to CuI/L at the Pt-cathode, coupled to activation of R-X/Pn-X by CuI/L to form R˙/Pn˙ and CuII/L in the bulk. The constant current regime that follows is therefore indicative of equilibrium being reached and the concentration of each component, particularly CuI/L, reaching steady state. Based on the this, and previous reports,13 a 4-step current profile (−3.3 mA to −0.3 mA, 180 min, Table S1, entry 1, ESI†) was attempted which yielded 60% conversion and a final PPEGA480 with good agreement between experimental (Mn,SEC = 10900 g mol−1) and theoretical (Mn,th = 11154 g mol−1) molecular weights.
 | ||
| Fig. 2 Conversion and pseudo first order kinetic plot demonstrating temporal control afforded by eATRP in the ‚plug-and-play configuration. [PEGA480]:[HEBiB]:[Cu(OTf)2]:[TPMA]:[NaBr] = [38]:[1]:[0.15]:[0.45]:[0.15]; Eapp = −0.15 V. | ||
However, a relatively high dispersity (Đm = 1.51) suggested that the polymerization was not well controlled. According to Ohm's law, in galvanostatic reactions, the potential changes in order to maintain constant current. During our reactions we observed, via the user interface, that the (unreferenced) potential gradually became more reducing during the course of these reactions. In our potentiostatic reactions, Eapp < −0.25 V had a detrimental effect on the outcome of the polymerizations (vide supra). With this in mind we developed a 6-step current profile (−3.3 mA to −0.3 mA, 120 min, Tables S1, entry 2 and S2, ESI†) to minimize the drift in the potential. These conditions gave an almost identical outcome to the potentiostatic reactions yielding PPEGA480 in 55% conversion with good agreement between experimental (Mn,SEC = 11600 g mol−1) and theoretical (Mn,th = 10243 g mol−1) molecular weights and low dispersity (Đm = 1.17, Fig. 3B)
 | ||
| Fig. 3 (A) I vs. time plot obtained during a potentiostatic reaction when [PEGA480]:[HEBiB]:[Cu(OTf)2]:[TPMA]:[NaBr] = [38]:[1]:[0.15]:[0.45]:[0.15]; Eapp = −0.15 V. (B) SEC chromatogram of the purified polymer obtained from the polymerization performed using the 2-electrode, galvanostatic configuration with [PEGA480]:[HEBiB]:[Cu(OTf)2]:[TPMA]:[NaBr] = [38]:[1]:[0.15]:[0.45]:[0.15]; Iapp can be found in Table S2 (ESI†). | ||
In conclusion, a new simplified approach to eATRP has been demonstrated using the ‘plug-and-play’ capabilities afforded by the IKA Electrasyn device. Well controlled PPEGA480 has been synthesized under potentiostatic conditions (3-electrodes, undivided cell) with conversions up to 83%, good agreement between Mn,SEC and Mn,th and relatively low dispersities (Đm = 1.17–1.31). Likewise, a polymerization performed under galvanostatic conditions (2-electrodes, 6-step profile) has been shown to afford very similar polymers, with good agreement between Mn,SEC and Mn,th and low dispersity (Đm = 1.17). The availability of a range of standardized, manufactured electrodes as well as accessories that support e.g. variable temperatures, parallel and flow reactions, offers great potential for optimization and further development of this seATRP methodology. Ultimately, it could make eATRP more accessible to the polymer chemistry and materials science communities and the simplicity of the hardware and reaction set-up is even amenable to translation into undergraduate teaching laboratories.
The authors would like to thank the Polymer Characterization and Microscopy Research Technology Platforms for maintenance, access and use of SEC, DLS and TEM facilities. P. W. thanks the Royal Society and Tata companies for the award of a University Research Fellowship (URF\R1\180274). P. W. also thanks the EPSRC for funding a DTP studentship (M. M. EP/R513374/1).
Conflicts of interest
There are no conflicts to declare.Notes and references
- K. Parkatzidis, H. S. Wang, N. P. Truong and A. Anastasaki, Chemistry, 2020, 6, 1575–1588 CrossRef CAS.
- N. Corrigan, K. Jung, G. Moad, C. J. Hawker, K. Matyjaszewski and C. Boyer, Prog. Polym. Sci., 2020, 111, 101311 CrossRef CAS.
- B. P. Fors and C. J. Hawker, Angew. Chem., Int. Ed., 2012, 51, 8850–8853 CrossRef CAS PubMed.
- A. J. D. Magenau, N. C. Strandwitz, A. Gennaro and K. Matyjaszewski, Science, 2011, 332, 81–84 CrossRef CAS PubMed.
- H. Y. Cho and C. W. Bielawski, Angew. Chem., Int. Ed., 2020, 59, 13929–13935 CrossRef CAS PubMed.
- H. Mohapatra, M. Kleiman and A. P. Esser-Kahn, Nat. Chem., 2017, 9, 135–139 CrossRef CAS.
- X. Pan, M. Fantin, F. Yuan and K. Matyjaszewski, Chem. Soc. Rev., 2018, 47, 5457–5490 RSC.
- P. Chmielarz, M. Fantin, S. Park, A. A. Isse, A. Gennaro, A. J. D. Magenau, A. Sobkowiak and K. Matyjaszewski, Prog. Polym. Sci., 2017, 69, 47–78 CrossRef CAS.
- S. Park, H. Y. Cho, K. B. Wegner, J. Burdynska, A. J. D. Magenau, H.-J. Paik, S. Jurga and K. Matyjaszewski, Macromolecules, 2013, 46, 5856–5860 CrossRef CAS.
- P. Chmielarz, eXPRESS Polym. Lett., 2017, 11, 140–151 CrossRef CAS.
- M. Fantin, S. Park, Y. Wang and K. Matyjaszewski, Macromolecules, 2016, 49, 8838–8847 CrossRef CAS PubMed.
- B. Li, B. Yu, W. T. S. Huck, F. Zhou and W. Liu, Angew. Chem., Int. Ed., 2012, 51, 5092–5095 CrossRef CAS PubMed.
- S. Park, P. Chmielarz, A. Gennaro and K. Matyjaszewski, Angew. Chem., Int. Ed., 2015, 54, 2388–2392 CrossRef CAS.
- P. Chmielarz, Chem. Pap., 2017, 71, 161–170 CrossRef CAS.
- P. Chmielarz, Polymer, 2016, 102, 192–198 CrossRef CAS.
- P. Chmielarz, S. Park, A. Sobkowiak and K. Matyjaszewski, Polymer, 2016, 88, 36–42 CrossRef CAS.
- P. Chmielarz, A. Sobkowiak and K. Matyjaszewski, Polymer, 2015, 77, 266–271 CrossRef CAS.
- P. Chmielarz, Polym. Adv. Technol., 2017, 28, 1787–1793 CrossRef CAS.
- P. Chmielarz, Polym. Adv. Technol., 2018, 29, 470–480 CrossRef CAS.
- P. Chmielarz, T. Pacześniak, K. Rydel-Ciszek, I. Zaborniak, P. Biedka and A. Sobkowiak, Beilstein J. Org. Chem., 2017, 13, 2466–2472 CrossRef CAS.
- P. Chmielarz and A. Sobkowiak, J. Polym. Res., 2017, 24, 77 CrossRef.
- M. Yan, Y. Kawamata and P. S. Baran, Angew. Chem., Int. Ed., 2018, 57, 4149–4155 CrossRef CAS PubMed.
- C. Schotten, T. P. Nicholls, R. A. Bourne, N. Kapur, B. N. Nguyen and C. E. Willans, Green Chem., 2020, 22, 3358–3375 RSC.
- M. Yan, Y. Kawamata and P. S. Baran, Chem. Rev., 2017, 117, 13230–13319 CrossRef CAS PubMed.
- C. Kingston, M. D. Palkowitz, Y. Takahira, J. C. Vantourout, B. K. Peters, Y. Kawamata and P. S. Baran, Acc. Chem. Res., 2020, 53, 72–83 CrossRef CAS PubMed.
- F. De Bon, S. Marenzi, A. A. Isse, C. Durante and A. Gennaro, ChemElectroChem, 2020, 7, 1378–1388 CrossRef CAS.
- M. Fantin, F. Lorandi, A. A. Isse and A. Gennaro, Macromol. Rapid Commun., 2016, 37, 1318–1322 CrossRef CAS PubMed.
- A. Michieletto, F. Lorandi, F. De Bon, A. A. Isse and A. Gennaro, J. Polym. Sci., 2020, 58, 114–123 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1cc01312b |
| This journal is © The Royal Society of Chemistry 2021 |