
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Enantioselective nickel-catalyzed anti-arylmetallative cyclizations onto acyclic electron-deficient alkenes†
Simone M.
Gillbard
ab,
Harley
Green
ab,
Stephen P.
Argent‡
 b and
Hon Wai
Lam
*ab
b and
Hon Wai
Lam
*ab
aThe GlaxoSmithKline Carbon Neutral Laboratories for Sustainable Chemistry, University of Nottingham, Jubilee Campus, Triumph Road, Nottingham, NG7 2TU, UK. E-mail: hon.lam@nottingham.ac.uk
bSchool of Chemistry, University of Nottingham, University Park, Nottingham, NG7 2RD, UK
First published on 6th April 2021
Abstract
Enantioselective nickel-catalyzed reactions of (hetero)arylboronic acids or alkenylboronic acids with substrates containing an alkyne tethered to various acyclic electron-deficient alkenes are described.
The metal-catalyzed addition of an arylboron reagent to an alkyne, followed by enantioselective intramolecular nucleophilic addition of the resulting alkenylmetal species onto a tethered electrophile, is a versatile domino reaction sequence for the synthesis of diverse chiral carbo- and heterocycles.1 We2 and others3 have recently described nickel-catalyzed variants of these reactions in which reversible E/Z isomerization of the intermediate alkenylnickel species enables enantioselective arylative cyclizations to proceed that would otherwise be impossible because of geometric constraints. Variants of these reactions that give achiral products,4 and several related processes,5–7 have also been described.
We have previously described enantioselective desymmetrizing nickel-catalyzed arylative cyclizations onto cyclohexa-2,5-dienones, which give fused bicyclic products with high diastereo- and enantioselectivities (Scheme 1A).2a However, the use of a broader range of acyclic electron-deficient, conjugated alkenes in cyclizations would be valuable in providing less complex, non-fused products, and would substantially increase the synthetic utility of this methodology. Herein, we demonstrate that acyclic enones, nitroalkenes, α,β-unsaturated esters, and α,β-unsaturated nitriles can be used as electrophiles in the enantioselective preparation of various non-fused chiral carbo- and heterocycles (Scheme 1B). Collectively, these results represent a substantial increase in the scope of nickel-catalyzed anti-carbometallative cyclizations.
 | ||
| Scheme 1 Enantioselective nickel-catalyzed arylative cyclizations onto electron-deficient alkenes. | ||
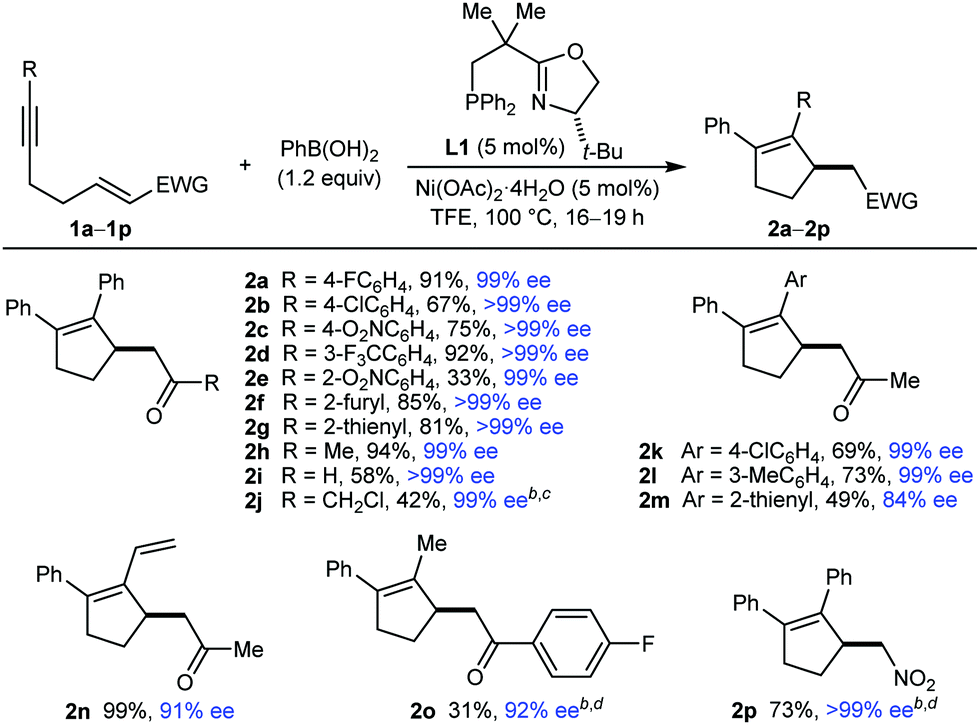
This study began with the reactions of PhB(OH)2 with substrates 1a–1p (Table 1). An evaluation of conditions8 led to the finding that heating the substrate 1, PhB(OH)2 (1.2 equiv.), and 5 mol% each of Ni(OAc)2·4H2O and (S)-t-Bu-NeoPHOX (L1)2b,9 in TFE at 100 °C for 16–19 h gave the desired products 2 in generally good yields and high enantioselectivities.10 In some cases (2j, 2o, and 2p), using 2.0 equivalents of PhB(OH)2 and increasing the catalyst loading were required for acceptable yields. Aromatic ketones with halide (2a and 2b), nitro (2c and 2e), or trifluoromethyl (2d) substituents at various positions of the benzene are tolerated, as are 2-furyl (2f), 2-thienyl (2g), and methyl ketones (2h and 2k–2n). Notably, an α,β-unsaturated aldehyde underwent arylative cyclization to give 2i in 58% yield and >99% ee. An α-chloroketone, containing a potentially labile carbon–chlorine bond, is also tolerated (2j). The alkynyl group can be changed from phenyl (2a–2j) to 4-chlorophenyl (2k), 3-methylphenyl (2l), 2-thienyl (2m), and vinyl (2n), although 2m was formed in lower yield and enantioselectivity. Aryl- and alkenyl-substituted alkynes are usually required for high regioselectivities in the initial arylnickelation step, presumably because the resulting alkenylnickel intermediates are better stabilized by an adjacent sp2-hybridized group. Therefore, it was of interest to evaluate the reaction of methyl-substituted alkyne 1o, which gave 2o in 31% yield and 92% ee. This reaction also gave a mixture of other unidentified products, presumably because of low regioselectivity in the initial arylnickelation. A nitroalkene can also be used as the electrophile (2p).
| a Reactions were conducted using 0.30 mmol of 1 in TFE (3 mL). Yields are of isolated products. Enantiomeric excesses were determined by HPLC analysis on a chiral stationary phase. b Using 2.0 equivalents of PhB(OH)2. c Using 20 mol% each of Ni(OAc)2·4H2O and L1. d Using 10 mol% each of Ni(OAc)2·4H2O and L1. |
|---|
|
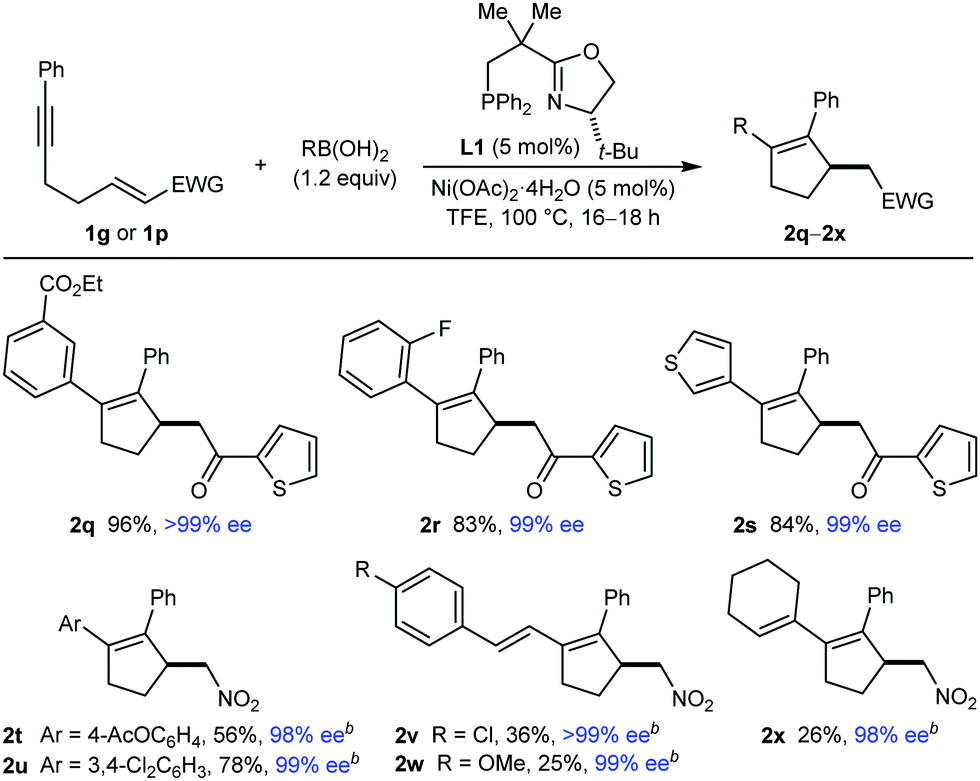
The results of evaluating different boronic acids in reactions with substrates 1g or 1p are shown in Table 2. Substituted phenylboronic acids with various groups at the para (2t), meta (2q), or ortho (2r) positions successfully underwent the reaction to give products with reasonable to high yields and high enantioselectivities, as did 3,4-dichlorophenylboronic acid (2u) and 3-thienylboronic acid (2s). Various alkenylboronic acids also reacted with 1p to give products 2v–2x in >99% ee but in low yields because of competitive protodeboronation.
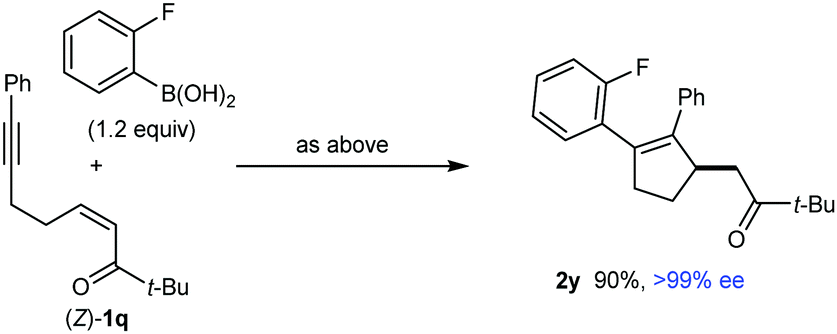
Further investigations into the scope of these reactions revealed some interesting findings. For example, the reaction of 2-fluorophenylboronic acid with substrate (E)-1q, which contains an α,β-unsaturated t-butyl ketone, gave the arylative cyclization product 2y in only 25% yield but in >99% ee (eqn (1)). This reaction also gave the alkyne hydroarylation products 3 in 30% yield, which were isolated as a 0.7![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mixture of inseparable E- and Z-isomers, respectively. Evidently, the steric hindrance imparted by the t-butyl group had a negative effect on the efficiency of arylative cyclization. Interestingly, however, the analogous reaction with the stereoisomeric substrate (Z)-1q gave 2y in 90% yield and >99% ee (eqn (2)). The markedly different propensity of (E)-1q and (Z)-1q to undergo the desired reaction is reminiscent of our prior work in enantioselective nickel-catalyzed intramolecular allylic alkenylations, where Z-allylic phosphates gave arylative cyclization products but the corresponding E-isomers did not.2b The reasons for the differing results obtained from (E)-1q and (Z)-1q are not clear, but perhaps the lower thermodynamic stability of (Z)-1q is manifested in greater reactivity toward nucleophilic attack, and/or the steric requirements of the reaction are better accommodated by (Z)-1q. Moreover, the major enantiomer of 2y is identical for both reactions (see the ESI† for tentative stereochemical models). These results contrast with several other examples of enantioselective 1,4-additions of carbon nucleophiles to electron-deficient alkenes where E- and Z-isomers of the substrates give opposite enantiomers of the products.11 However, reactions where E- and Z-isomers give the same major enantiomers of 1,4-addition products are also known.1j,11b
:1 mixture of inseparable E- and Z-isomers, respectively. Evidently, the steric hindrance imparted by the t-butyl group had a negative effect on the efficiency of arylative cyclization. Interestingly, however, the analogous reaction with the stereoisomeric substrate (Z)-1q gave 2y in 90% yield and >99% ee (eqn (2)). The markedly different propensity of (E)-1q and (Z)-1q to undergo the desired reaction is reminiscent of our prior work in enantioselective nickel-catalyzed intramolecular allylic alkenylations, where Z-allylic phosphates gave arylative cyclization products but the corresponding E-isomers did not.2b The reasons for the differing results obtained from (E)-1q and (Z)-1q are not clear, but perhaps the lower thermodynamic stability of (Z)-1q is manifested in greater reactivity toward nucleophilic attack, and/or the steric requirements of the reaction are better accommodated by (Z)-1q. Moreover, the major enantiomer of 2y is identical for both reactions (see the ESI† for tentative stereochemical models). These results contrast with several other examples of enantioselective 1,4-additions of carbon nucleophiles to electron-deficient alkenes where E- and Z-isomers of the substrates give opposite enantiomers of the products.11 However, reactions where E- and Z-isomers give the same major enantiomers of 1,4-addition products are also known.1j,11b
 | (1) |
 | (2) |
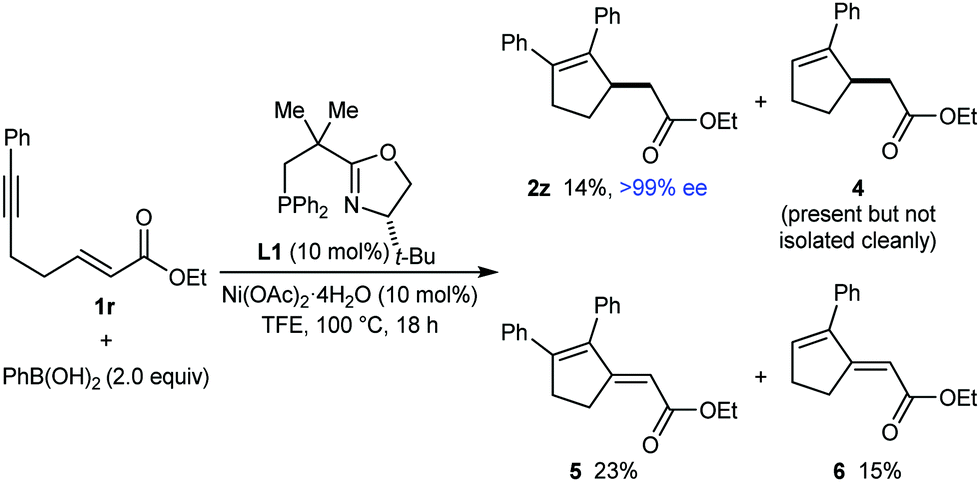
Thus far, only enones or nitroalkenes had been used as electrophiles. Interestingly, use of an α,β-unsatured ester gave other types of products (eqn (3)). Substrate 1r reacted with PhB(OH)2 (2.0 equiv.) in the presence of 10 mol% each of Ni(OAc)2·4H2O and (S)-t-Bu-NeoPHOX (L1) to give the arylative cyclization product 2z (14%, >99% ee), conjugated dienes 5 (23% yield) and 6 (15% yield) resulting from Heck-type cyclizations,12,13 and what appeared to be the reductive cyclization product 4, which could not be isolated cleanly. These results can be explained by considering the mechanism of nickel-catalyzed anti-carbometallative cyclizations that we have proposed previously (Scheme 2).2,4c Reaction of 1r and PhB(OH)2 would, after arylnickelation and reversible E/Z isomerization,2,4c lead to alkenylnickel species 7. A syn-stereospecific migratory insertion of the alkene2b would then give the C-bound nickel enolate 8. Protodenickelation of 8 by TFE gives the arylative cyclization product 2z. However, the low yield of 2z suggests that this step is slow compared with substrates containing ketones or nitro groups (Tables 1 and 2).14 In competition with protodenickelation of 8, bond rotation to give 8′ and stereospecific syn-β-hydride elimination gives diene 5 and a nickel hydride species 9. The nickel hydride 9 can then enter analogous reaction pathways with substrate 1r but via alkyne hydronickelation to give 10 and eventually, the reductive cyclization product 4 and diene 6.
 | (3) |
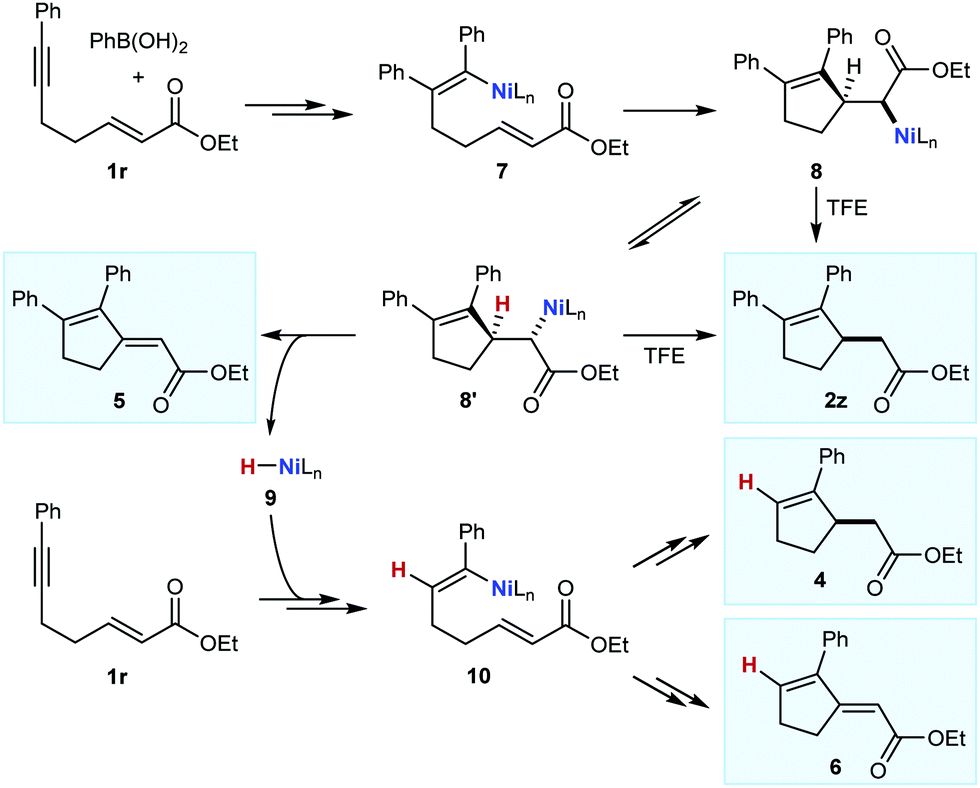 | ||
| Scheme 2 Mechanistic rationale of the formation of 2z and 4–6. | ||
Conjugate dienes were also obtained from substrates containing an α,β-unsaturated nitrile (eqn (4) and (5)). The reaction of PhB(OH)2 with (E)-1s gave an inseparable 13:1 mixture of dienes (E)-11 (18% yield) and 12 (1.4% yield), and the remainder of the material was a mixture of unidentified products (eqn (4)). None of the desired product 2aa was detected. In contrast, the stereoisomeric substrate (Z)-1s gave 2aa in 60% yield and 99% ee, along with diene (Z)-11 in 25% yield (eqn (5)). The observation that the Z-isomer of the substrate is more effective in providing the product 2aa is similar to the results shown in eqn (1) and (2). For a mechanistic rationale of the production of different stereoisomers of dienes (E)-11 and (Z)-11 from (E)-1s and (Z)-1s, respectively, see the ESI.†
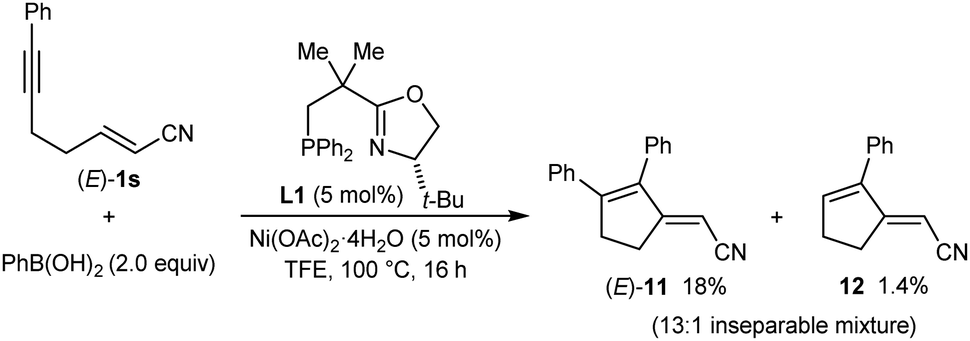 | (4) |
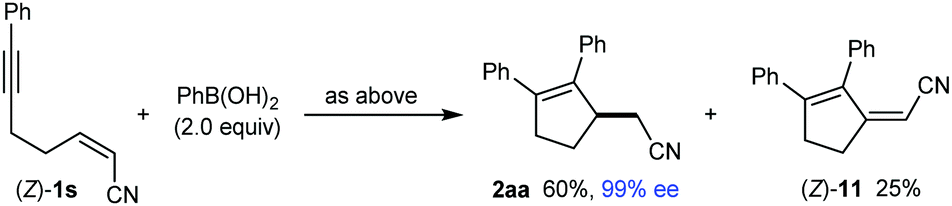 | (5) |
Next, the formation of six-membered rings was attempted. However, reaction of 13 (a higher homologue of substrate 1h that successfully gave product 2h (see Table 1)) with PhB(OH)2 (2.0 equiv.) in the presence of 10 mol% each of Ni(OAc)2·4H2O and (S)-t-Bu-NeoPHOX (L1) failed to provide the desired six-membered arylative cyclization product. Instead, a 2.3:1 mixture of inseparable stereoisomeric alkyne hydroarylation products (E)-14 and (Z)-14, respectively, was obtained in 57% yield (Scheme 3). Replacing (S)-t-Bu-NeoPHOX (L1) with other chiral phosphine–oxazoline ligands did not lead to any improvement.8 However, use of the achiral ligand pyphos (L2) gave racemic 15 in 47% yield (Scheme 3).15
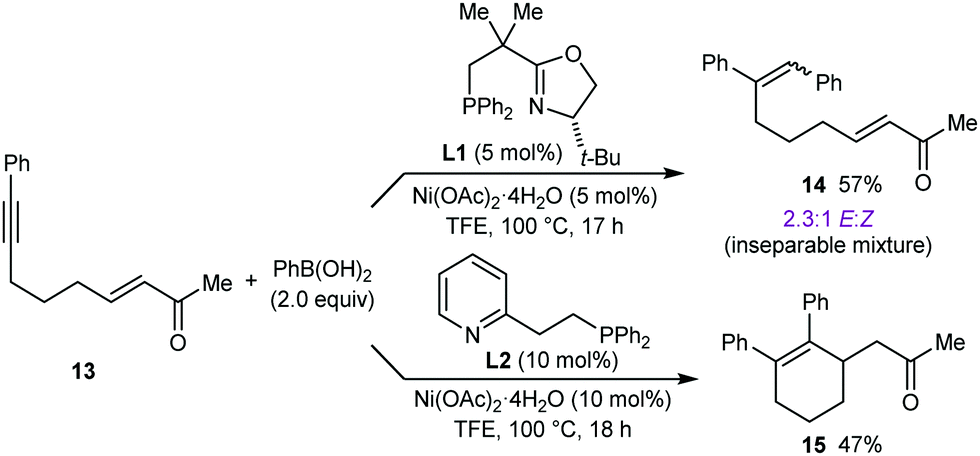 | ||
| Scheme 3 | ||
The reaction of PhB(OH)2 with substrate 16a, which contains a para-toluenesulfonamide group, gave only a complex mixture of unidentified products (Scheme 4). As with 13, improved results were not obtained with other chiral phosphine–oxazoline ligands8 but use of pyphos (L2) gave the racemic arylative cyclization product 17 in 52% yield.
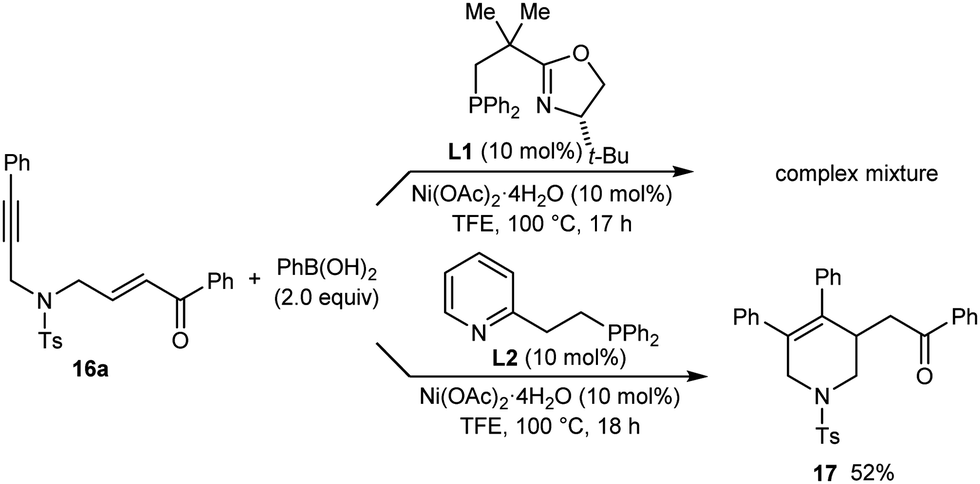 | ||
| Scheme 4 | ||
Given the results shown in Schemes 3 and 4, it was not surprising that substrate 16b (see eqn (6)), which contains an α,β-unsaturated ester rather than an α,β-unsaturated ketone, did not provide the desired arylative cyclization product when it was reacted with PhB(OH)2 using L1 as the chiral ligand. However, unlike for substrates 13 and 16a, it was interesting to observe that (S)-t-Bu-PHOX (L3) was an effective chiral ligand in the arylative cyclization of 16b, which reacted smoothly with PhB(OH)2 (2.0 equiv.) in the presence of 10 mol% each of Ni(OAc)2·4H2O and L3 to give tetrahydropyridine 18 in 59% yield and 89% ee (eqn (6)).
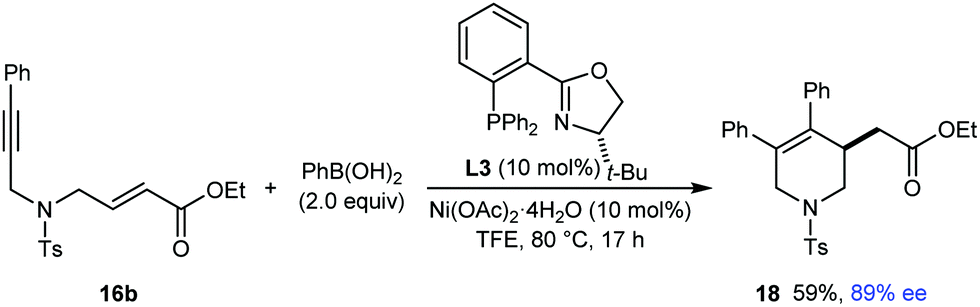 | (6) |
In summary, we have reported enantioselective nickel-catalyzed anti-carbometallative cyclizations of (hetero)arylboronic acids and alkenylboronic acids with acyclic substrates containing an alkyne tethered to an enone, nitroalkene, α,β-unsaturated ester, or α,β-unsaturated nitrile. The products are various non-fused chiral carbo- and heterocycles, and the enantioselectivities are excellent in most cases (often ≥99% ee). These results represent a substantial increase in the scope over our previous work.2 Interesting findings comparing the efficiencies of E/Z stereoisomers of certain substrates, and the isolation of products resulting from β-hydride eliminations and reductive cyclizations have also been described (eqn (1)–(5)).16
This work was supported by the Engineering and Physical Sciences Research Council and AstraZeneca [Industrial CASE Studentship, grant number EP/S513854/1]; the University of Nottingham; and GlaxoSmithKline.
Conflicts of interest
There are no conflicts to declare.References
- For representative examples, see: (a) R. Shintani, K. Okamoto, Y. Otomaru, K. Ueyama and T. Hayashi, J. Am. Chem. Soc., 2005, 127, 54–55 CrossRef CAS PubMed; (b) R. Shintani, A. Tsurusaki, K. Okamoto and T. Hayashi, Angew. Chem., Int. Ed., 2005, 44, 3909–3912 CrossRef CAS PubMed; (c) J. Song, Q. Shen, F. Xu and X. Lu, Org. Lett., 2007, 9, 2947–2950 CrossRef CAS PubMed; (d) X. Han and X. Lu, Org. Lett., 2010, 12, 108–111 CrossRef CAS PubMed; (e) Z.-T. He, B. Tian, Y. Fukui, X. Tong, P. Tian and G.-Q. Lin, Angew. Chem., Int. Ed., 2013, 52, 5314–5318 CrossRef CAS PubMed; (f) J. Keilitz, S. G. Newman and M. Lautens, Org. Lett., 2013, 15, 1148–1151 CrossRef CAS PubMed; (g) Y. Li and M.-H. Xu, Org. Lett., 2014, 16, 2712–2715 CrossRef CAS PubMed; (h) F. Serpier, B. Flamme, J.-L. Brayer, B. Folléas and S. Darses, Org. Lett., 2015, 17, 1720–1723 CrossRef CAS PubMed; (i) A. Selmani and S. Darses, Org. Lett., 2019, 21, 8122–8126 CrossRef CAS PubMed; (j) A. Selmani and S. Darses, Org. Chem. Front, 2019, 6, 3978–3982 RSC; (k) A. Groves, J. Sun, H. R. I. Parke, M. Callingham, S. P. Argent, L. J. Taylor and H. W. Lam, Chem. Sci., 2020, 11, 2759–2764 RSC; (l) A. Selmani and S. Darses, Org. Lett., 2020, 22, 2681–2686 CrossRef CAS PubMed.
- (a) C. Clarke, C. A. Incerti-Pradillos and H. W. Lam, J. Am. Chem. Soc., 2016, 138, 8068–8071 CrossRef CAS PubMed; (b) C. Yap, G. M. J. Lenagh-Snow, S. N. Karad, W. Lewis, L. J. Diorazio and H. W. Lam, Angew. Chem., Int. Ed., 2017, 56, 8216–8220 CrossRef CAS PubMed; (c) S. N. Karad, H. Panchal, C. Clarke, W. Lewis and H. W. Lam, Angew. Chem., Int. Ed., 2018, 57, 9122–9125 CrossRef CAS PubMed.
- Z. Lu, X.-D. Hu, H. Zhang, X.-W. Zhang, J. Cai, M. Usman, H. Cong and W.-B. Liu, J. Am. Chem. Soc., 2020, 142, 7328–7333 CrossRef CAS PubMed.
- (a) X. Zhang, X. Xie and Y. Liu, Chem. Sci., 2016, 7, 5815–5820 RSC; (b) G. R. Kumar, R. Kumar, M. Rajesh and M. S. Reddy, Chem. Commun., 2018, 54, 759–762 RSC; (c) S. M. Gillbard, C.-H. Chung, S. N. Karad, H. Panchal, W. Lewis and H. W. Lam, Chem. Commun., 2018, 54, 11769–11772 RSC.
- For reviews on nickel-catalyzed difunctionalization of alkynes, see: (a) S. E. Bottcher, L. E. Hutchinson and D. J. Wilger, Synthesis, 2020, 52, 2807–2820 CrossRef CAS; (b) W. Liu and W. Kong, Org. Chem. Front, 2020, 7, 3941–3955 RSC.
- (a) M. Hari Babu, G. Ranjith Kumar, R. Kant and M. Sridhar Reddy, Chem. Commun., 2017, 53, 3894–3897 RSC; (b) M. Rajesh, M. K. R. Singam, S. Puri, S. Balasubramanian and M. Sridhar Reddy, J. Org. Chem., 2018, 83, 15361–15371 CrossRef CAS PubMed; (c) N. Iqbal, N. Iqbal, D. Maiti and E. J. Cho, Angew. Chem., Int. Ed., 2019, 58, 15808–15812 CrossRef CAS PubMed; (d) M. K. R. Singam, A. Nagireddy, M. Rajesh, V. Ganesh and M. S. Reddy, Org. Chem. Front, 2020, 7, 30–34 RSC; (e) J. Chen, Y. Wang, Z. Ding and W. Kong, Nat. Commun., 2020, 11, 1882 CrossRef CAS PubMed; (f) Z. Zhou, W. Liu and W. Kong, Org. Lett., 2020, 22, 6982–6987 CrossRef CAS PubMed; (g) Z. Zhou, J. Chen, H. Chen and W. Kong, Chem. Sci, 2020, 11, 10204–10211 RSC.
- T. Igarashi, S. Arai and A. Nishida, J. Org. Chem., 2013, 78, 4366–4372 CrossRef CAS PubMed.
- Other phosphine − oxazoline ligands evaluated included (R)-Ph-PHOX, (S)-i-Pr-PHOX, and (S)-t-BuPHOX (L3).
- M. G. Schrems and A. Pfaltz, Chem. Commun., 2009, 6210–6212 RSC.
- The absolute configurations of products 2a, 2r, 2s, and 2y were determined by X-ray crystallography, and those of the remaining products were assigned by analogy.
- For examples, see: (a) T. Hayashi, T. Senda, Y. Takaya and M. Ogasawara, J. Am. Chem. Soc., 1999, 121, 11591–11592 CrossRef CAS; (b) S. R. Harutyunyan, F. López, W. R. Browne, A. Correa, D. Peña, R. Badorrey, A. Meetsma, A. J. Minnaard and B. L. Feringa, J. Am. Chem. Soc., 2006, 128, 9103–9118 CrossRef CAS PubMed; (c) S.-Y. Wang, S.-J. Ji and T.-P. Loh, J. Am. Chem. Soc., 2007, 129, 276–277 CrossRef CAS PubMed; (d) P. Mauleón, I. Alonso, M. R. Rivero and J. C. Carretero, J. Org. Chem., 2007, 72, 9924–9935 CrossRef PubMed; (e) R. Shintani and T. Hayashi, Org. Lett., 2011, 13, 350–352 CrossRef CAS PubMed.
- H. Yokoyama, T. Satoh, T. Furuhata, M. Miyazawa and Y. Hirai, Synlett, 2006, 2649–2651 CAS.
- (a) J.-I. I. Kim, B. A. Patel and R. F. Heck, J. Org. Chem., 1981, 46, 1067–1073 CrossRef CAS; (b) P. M. Wovkulich, K. Shankaran, J. Kiegiel and M. R. Uskokovic, J. Org. Chem., 1993, 58, 832–839 CrossRef CAS; (c) O. Dirat, C. Kouklovsky and Y. Langlois, J. Org. Chem., 1998, 63, 6634–6642 CrossRef CAS; (d) K. S. Yoo, C. H. Yoon and K. W. Jung, J. Am. Chem. Soc., 2006, 128, 16384–16393 CrossRef CAS PubMed; (e) A. N. Cuzzupe, C. A. Hutton, M. J. Lilly, R. K. Mann, K. J. McRae, S. C. Zammit and M. A. Rizzacasa, J. Org. Chem., 2001, 66, 2382–2393 CrossRef CAS PubMed; (f) R. Manoharan, R. Logeswaran and M. Jeganmohan, J. Org. Chem., 2019, 84, 14830–14843 CrossRef CAS PubMed.
- A possible reason is that protodenickelation proceeds faster via the O-bound, rather than the C-bound nickel enolate, and ketone-derived enolates are more likely to exist as the O-bound form compared with ester-derived enolates. Similarly, protodenickelation of nickel nitronates is likely to be more rapid than ester-derived nickel enolates because of a higher ratio of O- vs. C-bound forms.
- Product 15 contained an inseparable impurity and therefore the yield was calculated by 1H NMR analysis using an internal standard.
- The research data associated with this publication can be found at: http://dx.doi.org/10.17639/nott.7109 .
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental procedures, full spectroscopic data for new compounds, and crystallographic data for 2a, 2r, 2s, and 2y. CCDC 2040010–2040013. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d1cc01166a |
| ‡ To whom enquires regarding X-ray crystallography should be addressed. |
| This journal is © The Royal Society of Chemistry 2021 |