
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceNucleophilic transformations of azido-containing carbonyl compounds via protection of the azido group†
Takahiro
Aimi
a,
Tomohiro
Meguro
a,
Akihiro
Kobayashi
ab,
Takamitsu
Hosoya
 *a and
Suguru
Yoshida
*ab
*a and
Suguru
Yoshida
*ab
aLaboratory of Chemical Bioscience, Institute of Biomaterials and Bioengineering, Tokyo Medical and Dental University (TMDU), 2-3-10 Kanda-Surugadai, Chiyoda-ku, Tokyo 101-0062, Japan. E-mail: s-yoshida@rs.tus.ac.jp; thosoya.cb@tmd.ac.jp
bDepartment of Biological Science and Technology, Faculty of Advanced Engineering, Tokyo University of Science, 6-3-1 Niijuku, Katsushika-ku, Tokyo 125-8585, Japan
First published on 18th May 2021
Abstract
Nucleophilic transformations of azido-containing carbonyl compounds are discussed. The phosphazide formation from azides and di(tert-butyl)(4-(dimethylamino)phenyl)phosphine (Amphos) enabled transformations of carbonyl groups with nucleophiles such as lithium aluminum hydride and organometallic reagents. The good stability of the phosphazide moiety allowed us to perform consecutive transformations of a diazide through triazole formation and the Grignard reaction.
Azides are of great significance in a broad range of research fields including synthetic organic chemistry, chemical biology, and materials chemistry.1 Diverse reliable transformations of azides such as copper-catalyzed azide–alkyne cycloaddition (CuAAC),2 strain-promoted azide–alkyne cycloaddition (SPAAC),3 the Staudinger reaction,4 and reduction into amines5 have been used in the synthesis of various organonitrogen compounds (Fig. 1A). One drawback of azide chemistry is that an azido group is easily damaged with various nucleophiles due to the high electrophilicity.1,6,7 We herein disclose selective nucleophilic transformations of a carbonyl group through the protection of the azido group (Fig. 1B and C).
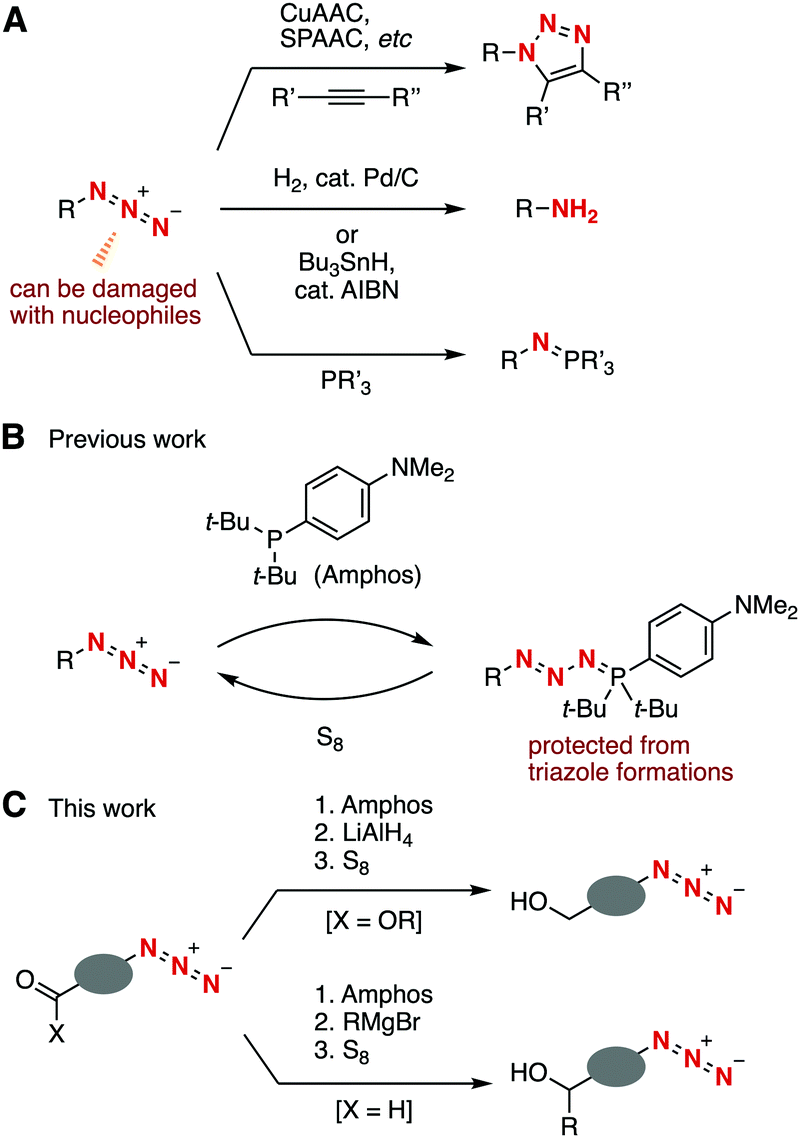 | ||
| Fig. 1 Backgrounds and plan of this study. (A) Conventional transformations of azides. (B) Our previous work. (C) This work. | ||
In the course of our studies on azide4d,e,8,9 and organophosphorus chemistry,9,10 we recently found a protection method for the azido group from CuAAC and SPAAC reactions by phosphazide formation with di(tert-butyl)(4-(dimethylamino)phenyl)phosphine (Amphos) (Fig. 1B).9 The phosphazide formation took place smoothly without forming azaylides through a denitrogenation process. The deprotection was easily realized by treating with elemental sulfur to afford azides in high yields. Considering the significance of azides in synthetic organic chemistry, we focused on the stability of phosphazides under nucleophilic or radical conditions for transformations of azides, which were still unclear due to the possible equilibrium liberation of azides from phosphazides under various conditions.9
We firstly examined denitrogenative reductions of azide 1a and phosphazide 4 (Fig. 2). As a result, aniline 2 was obtained by hydrogenation of azide 1a catalyzed by Pd/C under a hydrogen atmosphere (Fig. 2, a-1) or radical-mediated reduction with tributylstannane and 2,2′-azodiisobutyronitrile (AIBN) (Fig. 2, a-2). The Staudinger reaction of azide 1a with triphenylphosphine furnished azaylide 3 in high yield (Fig. 2, a-3). In contrast, we found that phosphazide 4 prepared from azide 1a with Amphos was stable under the reductive conditions using Pd/C and H2 (Fig. 2, b-1), tributylstannane and AIBN (Fig. 2, b-2), or triphenylphosphine (Fig. 2, b-3). These results obviously indicated that azides can be protected by phosphazide formation under various reduction conditions, although azides can form in equilibrium from phosphazides.9
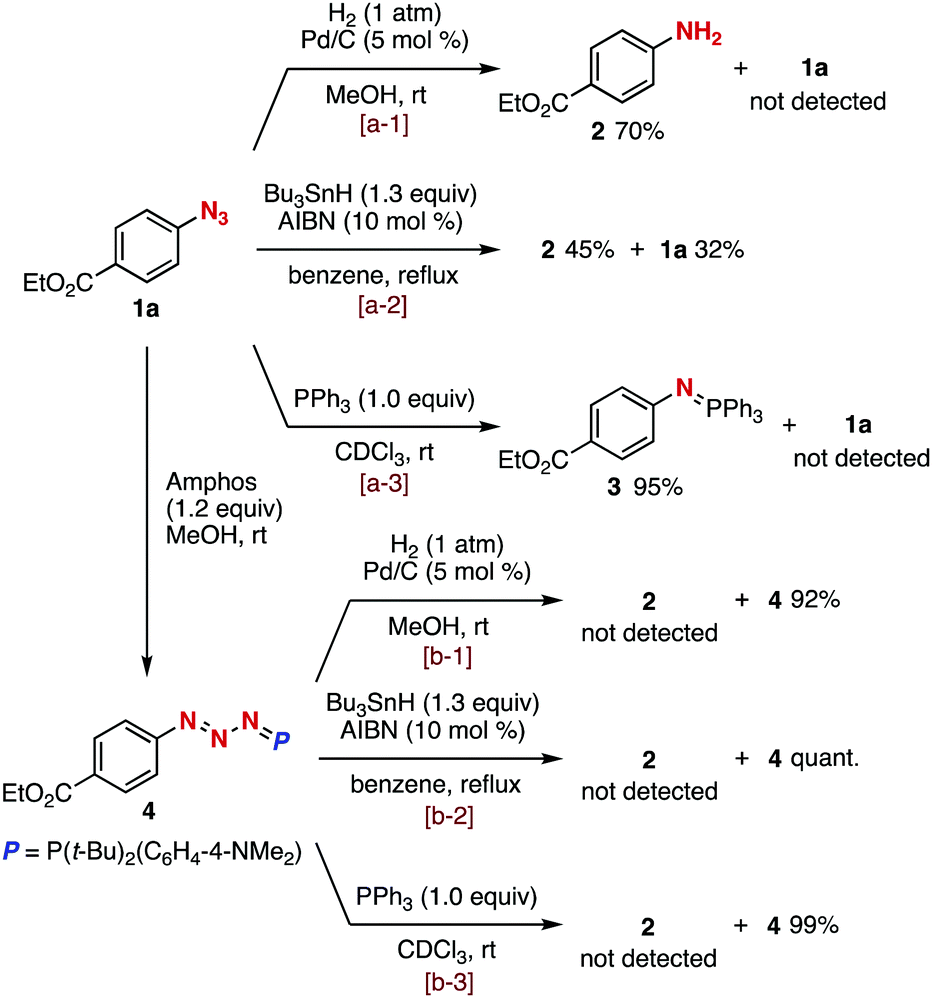 | ||
| Fig. 2 Transformations of azide 1a and phosphazide 4 under reductive conditions. | ||
Ester-selective lithium aluminum hydride (LAH) reduction was achieved through the phosphazide formation (Fig. 3).6 While the treatment of azide 1a with LAH in tetrahydrofuran (THF) at −20 °C provided 4-aminobenzyl alcohol (5) and 4-(ethoxycarbonyl)aniline (2) via reduction of the azido group (Fig. 3A, upper), pretreatment of azide 1a with Amphos at room temperature followed by the addition of LAH at −20 °C and subsequent deprotection using elemental sulfur successfully afforded 4-azidobenzyl alcohol (6a) by selective reduction of the ethoxycarbonyl group leaving the azido group untouched (Fig. 3A, lower).11 It is worth noting that the phosphazide moiety tolerated highly nucleophilic LAH. We also succeeded in the ester-selective LAH reduction of a broad range of azido-substituted esters 1via the formation of phosphazides (Fig. 3B). Indeed, phosphazide formation, LAH reduction, and deprotection with elemental sulfur took place efficiently using esters 1 to afford the corresponding alcohols 6b–6f in moderate to good yields via the protection of the heteroaromatic and primary, secondary, and tertiary alkyl azido groups. These results clearly show that a wide range of azido groups were protected using Amphos even in the presence of highly nucleophilic reagents such as LAH.
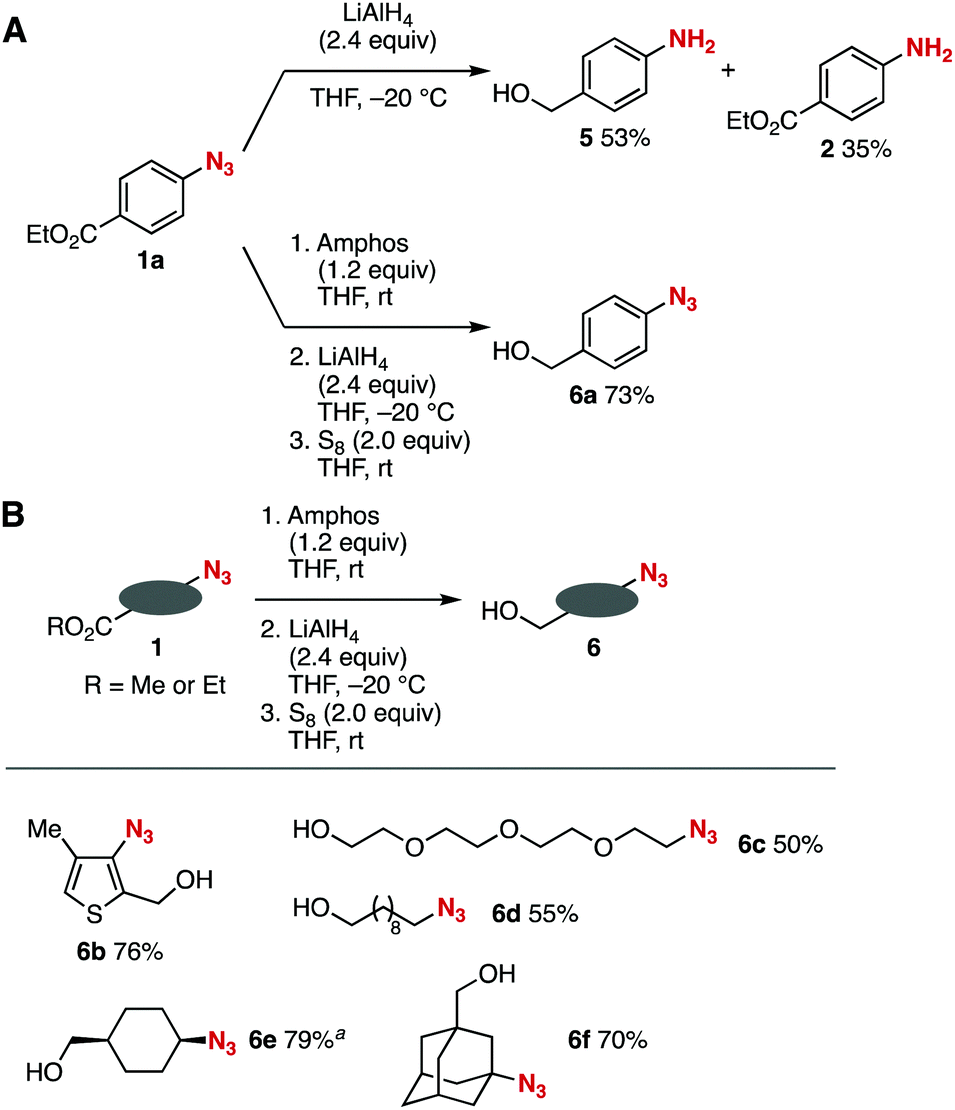 | ||
| Fig. 3 Reduction of azido-substituted esters 1 with LiAlH4. (A) Reductions with or without the protection of azide 1a. (B) Scope of synthesized azido-substituted alcohols 6. See ESI† for the structures of 1. | ||
The good stability of phosphazides toward nucleophiles allowed for alcohol synthesis from 4-azidobenzaldehyde (7a) with organomagnesium or organolithium reagents (Fig. 4).7,12 The reaction between aldehyde 7a and ethylmagnesium bromide furnished aniline 9 in moderate yield along with a trace amount of alcohol 8a due to the denitrogenation reduction of the azido group (Fig. 4A, upper). In sharp contrast, we accomplished an efficient synthesis of alcohol 8a from aldehyde 7a and ethylmagnesium bromide without damaging the highly electrophilic aromatic azido group (Fig. 4A, lower). Indeed, the phosphazide formation of azide 7a with Amphos proceeded smoothly leaving the formyl group. Then, the addition of ethylmagnesium bromide to the resulting mixture followed by the deprotection with elemental sulfur furnished alcohol 8a in high yield keeping the azido group unreacted. The reaction performed at a 1 mmol scale also proceeded efficiently to afford alcohol 8a in good yield. Thus, this simple single-purification procedure enabled the efficient Grignard reaction of the formyl group without damaging the azido group.
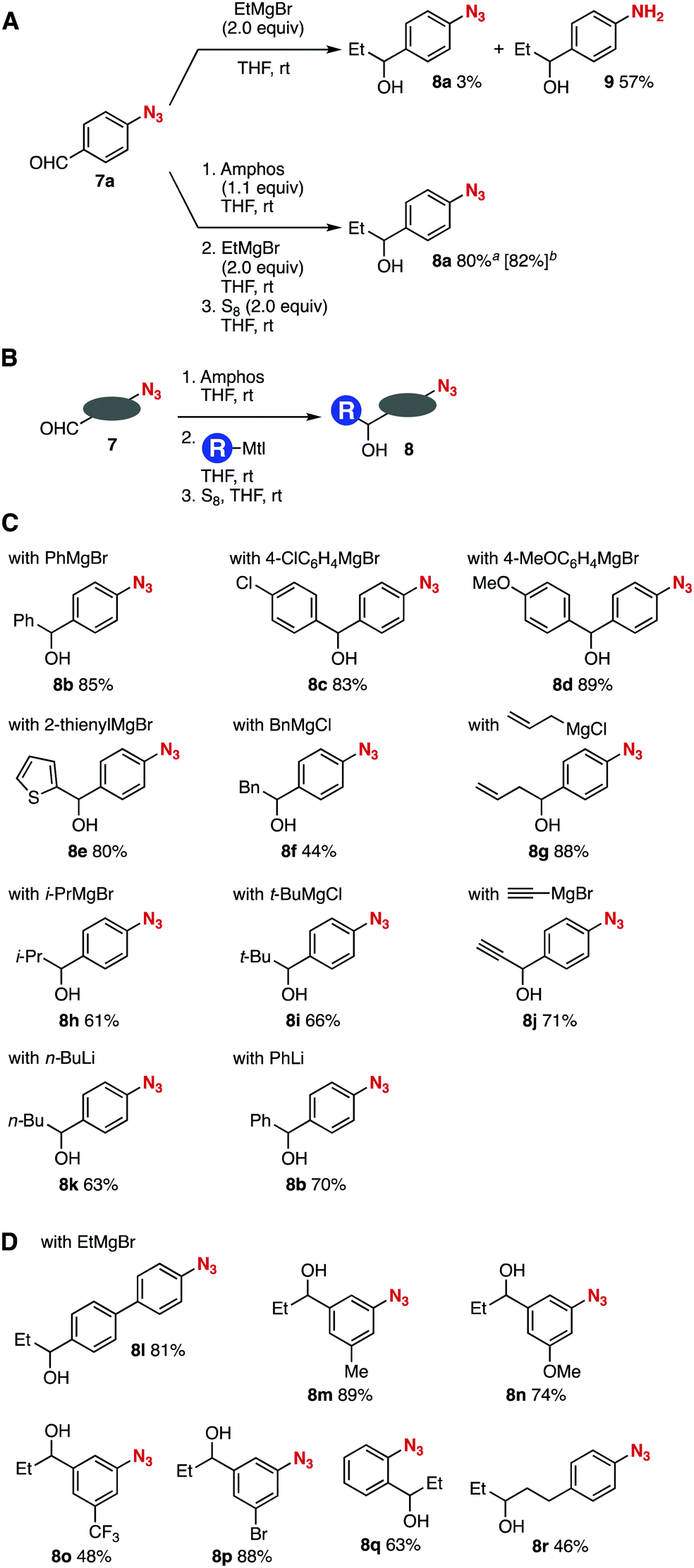 | ||
| Fig. 4 Azido-substituted alcohol synthesis with organomagnesium or organolithium reagents. (A) Grignard reaction with or without the protection of azide 7a. (B) General scheme. (C) Scope of synthesized azido-substituted alcohols 8 from 7a. (D) Scope of synthesized azido-substituted alcohols 8 from various azido-substituted aldehydes 7. See ESI† for the structures of 7. aThe isolated yield for the reaction conducted on a 0.2 mmol scale. bThe isolated yield for the reaction conducted on a 1 mmol scale. | ||
A wide variety of carbanions successfully participated in the azido-substituted alcohol synthesis from aldehydes 7 (Fig. 4B and C). For example, phenylation, 4-chloro- and 4-methoxyphenylation, and 2-thienylation of aldehyde 7a efficiently proceeded to provide alcohols 8b–8e possessing an azido group in good yields. Benzylation and allylation of aldehyde 7a also took place smoothly to afford azido-substituted alcohols 8f and 8g in moderate to high yields. Bulky alkyl Grignard reagents such as isopropylmagnesium bromide and tert-butylmagnesium chloride reacted with aldehyde 7a avoiding the reaction at the azido group. Of note, we achieved the introduction of a terminal alkyne moiety by the reaction using ethynylmagnesium bromide, furnishing azido-substituted propargyl alcohol 8j in good yield, which will serve in click chemistry. Also, organolithium reagents including n-butyllithium and phenyllithium were applicable to the addition reaction without damaging the phosphazide moiety.
The Grignard reactions of various aldehydes 7 bearing an azido group were accomplished to provide alcohols 8l–8r in moderate to good yields (Fig. 4B and D). A broad range of aldehydes 7 efficiently reacted with ethylmagnesium bromide at the formyl group without damaging the azido group via phosphazide formation to yield alcohols 8l–8p leaving methoxy, trifluoromethyl, and bromo groups intact. The azide protection followed by the Grignard reaction and deprotection uneventfully took place when using o-azidobenzaldehyde to provide 8q in good yield. Synthesis of secondary alcohol 8r was also achieved from an aliphatic aldehyde (3-(4-azidophenyl)propanal) and ethylmagnesium bromide via the azide protection with Amphos. These results clearly show that nucleophilic additions of formyl-substituted azides with a broad range of organomagnesium and organolithium reagents through the robust phosphazides realized the preparation of diverse alcohols having an azido group.
A synthetic utility of the Grignard reaction of aldehydes having azido groups was showcased by the consecutive reactions between diazide 10, cycloalkyne 12, and 4-methoxyphenylmagnesium bromide (Fig. 5). Firstly, the treatment of diazide 10 with an equimolar amount of Amphos resulted in the selective protection of the aromatic azido group by virtue of the stabilization by the benzene ring without phosphazide formation at the benzyl azide moiety. Secondly, the SPAAC reaction of phosphazide 11 with cycloalkyne 12 at the remaining benzylic azido group proceeded smoothly to afford cycloadduct 13. Thirdly, the Grignard reaction of aldehyde 13 with 4-methoxyphenylmagnesium bromide followed by the deprotection with elemental sulfur provided azide 14 in high yield. Since azides are of great importance in not only synthetic chemistry but also chemical biology, this four-step two-pot method will allow us to construct a vast chemical library of a wide variety of triazoles having an aromatic azide moiety from formyl-substituted diazide 10, alkynes, and organometallic reagents through the phosphazide formation.
 | ||
| Fig. 5 Synthesis of azide 14 from diazide 10. | ||
In summary, we have accomplished nucleophilic transformations of diverse azides through phosphazide formation. Good stability of the phosphazide intermediates realized reduction of esters by LAH and addition reaction of aldehydes with organometallic reagents to provide a wide variety of azido-substituted alcohols. Further studies to examine detailed properties of phosphazides, especially equilibrium liberation of azides, and to expand the applicability of the azide protection method are underway in our group.
The authors thank Dr Takashi Niwa (RIKEN, Center for Biosystems Dynamics Research (BDR)) and Dr Yuki Sakata (Tokyo Medical and Dental University) for HRMS analyses and Assoc. Prof. Dr Hiroki Tanimoto for helpful discussions. This work was supported by JSPS KAKENHI Grant Numbers JP19K05451 (C; S. Y.) and JP18H02104 (B; T. H.); the Naito Foundation (S. Y.); the Japan Agency for Medical Research and Development (AMED) under Grant Number JP20am0101098 (Platform Project for Supporting Drug Discovery and Life Science Research, BINDS); and the Cooperative Research Project of Research Center for Biomedical Engineering.
Conflicts of interest
There are no conflicts to declare.Notes and references
- (a) S. Bräse, C. Gil, K. Knepper and V. Zimmermann, Angew. Chem., Int. Ed., 2005, 44, 5188 CrossRef PubMed; (b) S. Bräse and K. Banert, Organic Azides: Syntheses and Applications, John Wiley & Sons, Ltd, Chichester, 2010 Search PubMed; (c) H. Tanimoto and K. Kakiuchi, Nat. Prod. Commun., 2013, 8, 1021 CrossRef CAS PubMed; (d) K. Banert, Synthesis, 2016, 48, 2361 CrossRef CAS; (e) D. Huang and G. Yan, Adv. Synth. Catal., 2017, 359, 1600 CrossRef CAS; (f) S. Yoshida, Org. Biomol. Chem., 2020, 18, 1550 RSC; (g) Z.-K. Liu, Q.-Q. Zhao, Y. Gao, Y.-X. Hou and X.-Q. Hu, Adv. Synth. Catal., 2020, 363, 411 CrossRef.
- (a) C. W. Tornøe, C. Christensen and M. Meldal, J. Org. Chem., 2002, 67, 3057 CrossRef PubMed; (b) V. V. Rostovtsev, L. G. Green, V. V. Fokin and K. B. Sharpless, Angew. Chem., Int. Ed., 2002, 41, 2596 CrossRef CAS; (c) M. Meldal and C. W. Tornøe, Chem. Rev., 2008, 108, 2952 CrossRef CAS PubMed; (d) A. Mandoli, Molecules, 2016, 21, 1174 CrossRef PubMed.
- (a) N. J. Agard, J. A. Prescher and C. R. Bertozzi, J. Am. Chem. Soc., 2004, 126, 15046 CrossRef CAS PubMed; (b) J. A. Codelli, J. M. Baskin, N. J. Agard and C. R. Bertozzi, J. Am. Chem. Soc., 2008, 130, 11486 CrossRef CAS PubMed; (c) X. Ning, J. Guo, M. A. Wolfert and G.-J. Boons, Angew. Chem., Int. Ed., 2008, 47, 2253 CrossRef CAS PubMed; (d) A. A. Poloukhtine, N. E. Mbua, M. A. Wolfert, G.-J. Boons and V. V. Popik, J. Am. Chem. Soc., 2009, 131, 15769 CrossRef CAS PubMed; (e) J. Dommerholt, S. Schmidt, R. Temming, L. J. A. Hendriks, F. P. J. T. Rutjes, J. C. M. van Hest, D. J. Lefeber, P. Friedl and F. L. van Delft, Angew. Chem., Int. Ed., 2010, 49, 9422 CrossRef CAS PubMed; (f) J. C. Jewett, E. M. Sletten and C. R. Bertozzi, J. Am. Chem. Soc., 2010, 132, 3688 CrossRef CAS PubMed; (g) S. Yoshida, Y. Hatakeyama, K. Johmoto, H. Uekusa and T. Hosoya, J. Am. Chem. Soc., 2014, 136, 13590 CrossRef CAS PubMed; (h) R. Ni, N. Mitsuda, T. Kashiwagi, K. Igawa and K. Tomooka, Angew. Chem., Int. Ed., 2014, 54, 1190 CrossRef PubMed; (i) R. R. Ramsubhag and G. B. Dudley, Org. Biomol. Chem., 2016, 14, 5028 RSC; (j) K. Kaneda, R. Naruse and S. Yamamoto, Org. Lett., 2017, 19, 1096 CrossRef CAS PubMed; (k) E. G. Burke, B. Gold, T. T. Hoang, R. T. Raines and J. M. Schomaker, J. Am. Chem. Soc., 2017, 139, 8029 CrossRef CAS PubMed; (l) C. Lis, S. Rubner, C. Gröst, R. Hoffmann, D. Knappe and T. Berg, Chem. – Eur. J, 2018, 24, 13762 CrossRef CAS PubMed; (m) S. Yoshida, T. Kuribara, H. Ito, T. Meguro, Y. Nishiyama, F. Karaki, Y. Hatakeyama, Y. Koike, I. Kii and T. Hosoya, Chem. Commun., 2019, 55, 3556 RSC; (n) K. Adachi, T. Meguro, Y. Sakata, K. Igawa, K. Tomooka, T. Hosoya and S. Yoshida, Chem. Commun., 2020, 56, 9823 RSC; (o) N. Makio, T. Kuribara, K. Adachi, Y. Hatakeyama, T. Meguro, Y. Sakata, K. Igawa, K. Tomooka, T. Hosoya and S. Yoshida, Chem. Commun., 2020, 56, 11449 RSC.
- (a) H. Staudinger and J. Meyer, Helv. Chim. Acta, 1919, 2, 635 CrossRef CAS; (b) E. Saxon and C. R. Bertozzi, Science, 2000, 287, 2007 CrossRef CAS PubMed; (c) S. S. van Berkel, M. B. van Eldijk and J. C. M. van Hest, Angew. Chem., Int. Ed., 2011, 50, 8806 CrossRef CAS PubMed; (d) T. Meguro, S. Yoshida and T. Hosoya, Chem. Lett., 2017, 46, 473 CrossRef CAS; (e) T. Meguro, N. Terashima, H. Ito, Y. Koike, I. Kii, S. Yoshida and T. Hosoya, Chem. Commun., 2018, 54, 7904 RSC; (f) C. Bednarek, I. Wehl, N. Jung, U. Schepers and S. Bräse, Chem. Rev., 2020, 120, 4301 CrossRef CAS PubMed.
- (a) F. Rolla, J. Org. Chem., 1982, 47, 4327 CrossRef CAS; (b) S. N. Maiti, P. Spevak and A. V. N. Reddy, Synth. Commun., 1988, 18, 1201 CrossRef CAS; (c) G. V. Reddy, G. V. Rao and D. S. Iyengar, Tetrahedron Lett., 1999, 40, 3937 CrossRef CAS; (d) A. M. Salunkhe, P. V. Ramachandran and H. C. Brown, Tetrahedron, 2002, 58, 10059 CrossRef CAS; (e) A. Kamal, N. Shankaraiah, N. Markandeya and C. S. Reddy, Synlett, 2008, 1297 CrossRef CAS; (f) A. Kamal, N. Markandeya, N. Shankaraiah, C. R. Reddy, S. Prabhakar, C. S. Reddy, M. N. Eberlin and L. S. Santos, Chem. – Eur. J., 2009, 15, 7215 CrossRef CAS PubMed; (g) T. Maegawa, T. Takahashi, M. Yoshimura, H. Suzuka, Y. Monguchi and H. Sajiki, Adv. Synth. Catal., 2009, 351, 2091 CrossRef CAS.
- J. H. Boyer, J. Am. Chem. Soc., 1951, 73, 5865 CrossRef CAS.
- For reactions of azides with organometallic reagents, see: (a) P. A. S. Smith, C. D. Rowe and L. B. Bruner, J. Org. Chem., 1969, 34, 3430 CrossRef CAS; (b) D. H. Sieh, D. J. Wilbur and C. J. Michejda, J. Am. Chem. Soc., 1980, 102, 3883 CrossRef CAS; (c) B. M. Trost and W. H. Pearson, J. Am. Chem. Soc., 1981, 103, 2483 CrossRef CAS; (d) B. M. Trost and W. H. Pearson, J. Am. Chem. Soc., 1983, 105, 1054 CrossRef CAS; (e) K. Nishiyama and N. Tanaka, J. Chem. Soc., Chem. Commun., 1983, 1322 RSC; (f) R. H. Smith, Jr. and C. J. Michejda, Synthesis, 1983, 476 CrossRef; (g) G. W. Kabalka and G. Li, Tetrahedron Lett., 1997, 38, 5777 CrossRef CAS; (h) A. Nakhai, B. Stensland, P. H. Svensson and J. Bergman, Eur. J. Org. Chem., 2010, 6588 CrossRef CAS; (i) A. A. Suleymanov, R. Scopelliti, F. F. Tirani and K. Severin, Org. Lett., 2018, 20, 3323 CrossRef CAS PubMed; (j) S. Graßl, J. Singer and P. Knochel, Angew. Chem., Int. Ed., 2020, 59, 335 CrossRef PubMed.
- For our recent azide chemistry, see: (a) S. Yoshida, S. Goto, Y. Nishiyama, Y. Hazama, M. Kondo, T. Matsushita and T. Hosoya, Chem. Lett., 2019, 48, 1038 CrossRef CAS; (b) H. Takemura, S. Goto, T. Hosoya and S. Yoshida, Chem. Commun., 2020, 56, 15541 RSC; (c) T. Meguro, Y. Sakata, T. Morita, T. Hosoya and S. Yoshida, Chem. Commun., 2020, 56, 4720 RSC; (d) N. Terashima, Y. Sakata, T. Meguro, T. Hosoya and S. Yoshida, Chem. Commun., 2020, 56, 14003 RSC; (e) S. Yoshida, Y. Sakata, Y. Misawa, T. Morita, T. Kuribara, H. Ito, Y. Koike, I. Kii and T. Hosoya, Chem. Commun., 2021, 57, 899 RSC.
- T. Meguro, S. Yoshida, K. Igawa, K. Tomooka and T. Hosoya, Org. Lett., 2018, 20, 4126 CrossRef CAS PubMed.
- For our organophosphorus chemistry, see: (a) S. Yoshida, K. Igawa and K. Tomooka, J. Am. Chem. Soc., 2012, 134, 19358 CrossRef CAS PubMed; (b) S. Yoshida and T. Hosoya, Chem. Lett., 2013, 42, 583 CrossRef CAS; (c) Y. Nishiyama, Y. Hazama, S. Yoshida and T. Hosoya, Org. Lett., 2017, 19, 3899 CrossRef CAS PubMed; (d) Y. Nishiyama, S. Kamada, S. Yoshida and T. Hosoya, Chem. Lett., 2018, 47, 1216 CrossRef CAS; (e) Y. Nishiyama, T. Hosoya and S. Yoshida, Chem. Commun., 2020, 56, 5771 RSC.
- Alcohol 6a was not obtained when triphenylphosphine was used instead of Amphos. Treatment of azide 1a with tri(t-butyl)phosphinonium tetrafluoroborate and triethylamine followed by LiAlH4 reduction and subsequent deprotection with elemental sulfur furnished alcohol 6a in 61% yield.
- For transformations of carbonyl group without damaging azido group, see: (a) S. Hanessian, R. R. Vakiti, S. Dorich, S. Banerjee, F. Lecomte, J. R. DelValle, J. Zhang and B. Deschênes-Simard, Angew. Chem., Int. Ed., 2011, 50, 3497 CrossRef CAS PubMed; (b) S. Hanessian, R. R. Vakiti, S. Dorich, S. Banerjee and B. Deschênes-Simard, J. Org. Chem., 2012, 77, 9458 CrossRef CAS PubMed; (c) K. Morihiro, N. Ankenbruck, B. Lukasak and A. Deiters, J. Am. Chem. Soc., 2017, 139, 13909 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures, and characterization of new compounds including NMR spectra. See DOI: 10.1039/d1cc01143j |
| This journal is © The Royal Society of Chemistry 2021 |