
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceN-Hydroxyphthalimidyl diazoacetate (NHPI-DA): a modular methylene linchpin for the C–H alkylation of indoles†
Rajdip
Chowdhury
 and
Abraham
Mendoza
*
and
Abraham
Mendoza
*
Department of Organic Chemistry, Arrhenius laboratory, Stockholm University, 106 91 Stockholm, Sweden. E-mail: abraham.mendoza@su.se
First published on 29th March 2021
Abstract
Despite the extensive studies on the reactions between conventional diazocompounds and indoles, these are still limited by the independent synthesis of the carbene precursors, the specific catalysts, and the required multi-step manipulation of the products. In this work, we explore redox-active carbenes in the expedited and divergent synthesis of functionalized indoles. NHPI-DA displays unusual efficiency and selectivity to yield insertion products that can be swiftly elaborated into boron and carbon substituents that are particularly problematic in carbene-mediated reactions.
The systematic assembly of ubiquitous methylene units is currently limited by the different chemistries and reagents that are required to introduce various functions in the targeted aliphatic groups. This is the case for the conversion of heteroarenes (1) into important C–H alkylated derivatives 2 (R; Scheme 1A).1,2b,3 The highly electrophilic carbenes4 involved in these reactions are incompatible with most electron-rich or labile functions. In particular, boryl-,5a aryl-,5b alkyl-,5c,d and vinyl-5e methylidenes 3–6 would directly lead to valuable products, but are too unstable to participate in selective intermolecular C–H insertion. To overcome these limitations, our group envisaged a unified C–H alkylation strategy based on the early-stage C–H insertion of a versatile redox-active carbene 7,6 and subsequent late-stage diversification of the resulting intermediate 8 to obtain the modified heterocycles 2.2,7 This way, the initial C–H insertion would require only one carbene precursor 7 regardless of the final functionality desired in the products 2. Moreover, these moieties would even include those inherently incompatible with geminal carbene intermediates (i.e.3–6).
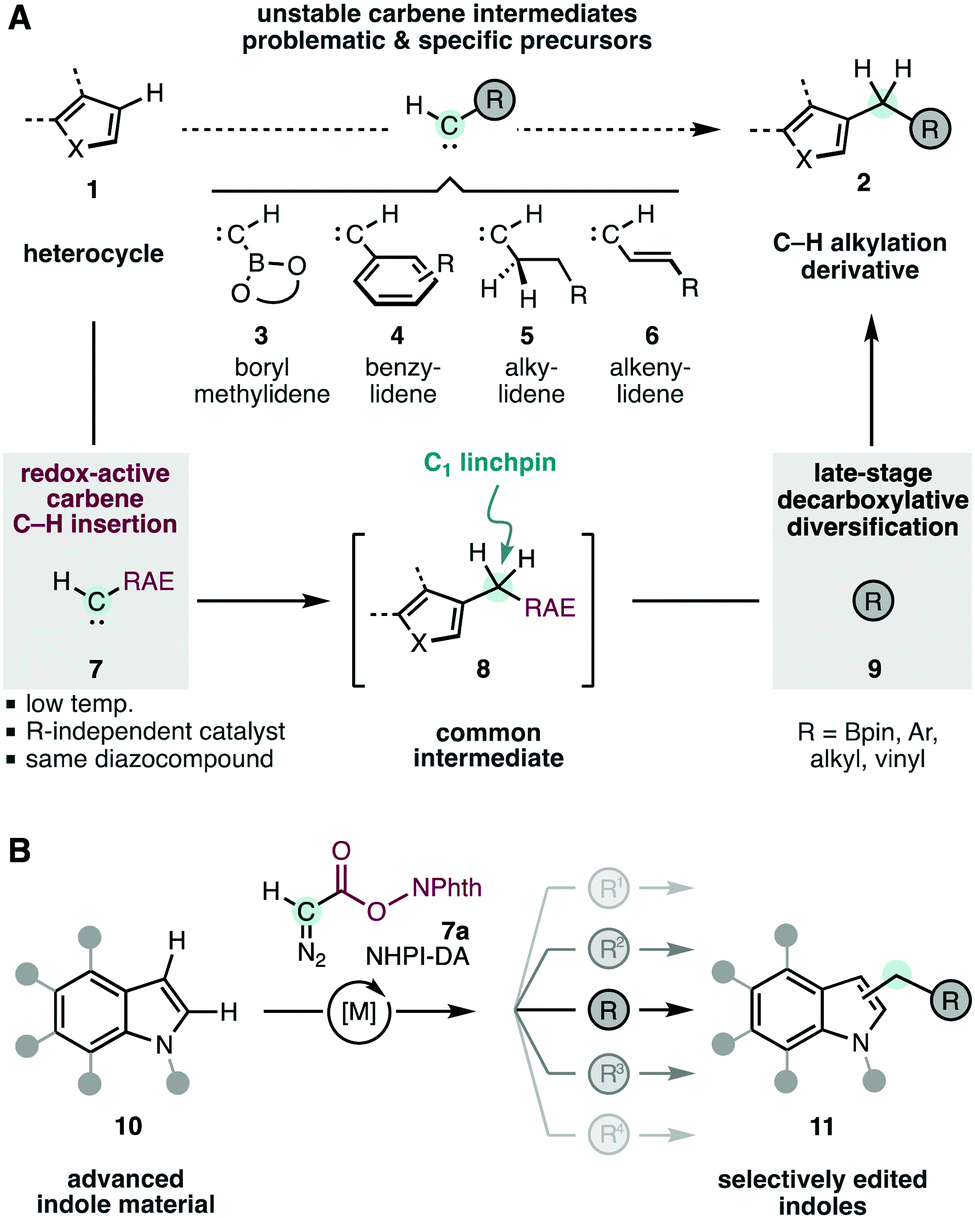 | ||
| Scheme 1 Heterocycle editing through C–H alkylation with redox-active carbenes. RAE, redox-active ester; PhthN, phthalimidyl. | ||
Recently, our group has discovered that N-hydroxyphtalimide (NHPI) esters can be orthogonal to geminal carbene intermediates.6 In particular, the diazocompound reagent NHPI-DA (7a) has displayed a surprisingly enhanced reactivity and selectivity in asymmetric cyclopropanation reactions.6 In this work we explore the utility of redox-active carbenes in C–H functionalization (Scheme 1B), which is unprecedented as far as we know. The transformation of indoles 10 into C3-alkyl derivatives 11 is particularly attractive for their importance in medicinal chemistry, natural products research, material science and chemical biology.8–13 The abundant literature on the reaction of metal carbenes with indoles4a,d,9–13 reveals that these reactions can produce mixtures of C3-8–11 or C2-insertion,12 and cyclopropanation of the C2,C3–π-bond.9c,11d,13 The mechanism of this reaction has been discussed in several studies.9b,c,10c,11b,d,13c These specific difficulties add to the more general challenge of taming the reactivity of carbenes bearing labile leaving groups directly connected to the highly reactive carbon.14
The reaction of the redox-active diazoacetate NHPI-DA (7a) in the model N-benzylindole (10a) was explored using a collection of the most successful catalysts that were used with conventional carbene precursors.9–13 It was found that the redox-active handle had a profound effect in the reactivity of the derived metal–carbene intermediate.6 The desired insertion product 12a is obtained only in low yields using rhodium, copper and iron catalysts that are successful with conventional diazoacetates (see ESI†). Extensive decomposition of NHPI-DA (7a) was observed but no other by-products were detected in the reaction crude by 1H-NMR. In line with our previous studies with chiral metallacyclic ruthenium catalysts,6b it was found that the achiral analogue 13 (Scheme 2)15 displayed optimal efficiency and selectivity. To the best of our knowledge, ruthenium metallacycles have not been reported to catalyze carbene transfer reactions on indoles. We found that the slow addition of the diazocompound 7a is optional, and similar results are obtained adding as little as 1.25 equiv. 7a in one shot (see ESI†). The performance of NHPI-DA (7a) contrasts with conventional diazocompounds that often require larger excess of the reagents and slow addition in similar reactions with indoles.8c,9a,b,11a,c,13a,d Moreover, the product is obtained with complete C3-selectivity, unlike some previous studies with C2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :C3 insertion selectivity issues.10 Using 13 as catalyst, conventional diazocompounds like ethyl diazoacetate are substantially less efficient (62%, see ESI†), and redox-active α-aryldiazoacetate reagents6a were unreactive. These observations illustrate the importance of the match between the catalyst and the carbene precursor for efficient insertion.
:C3 insertion selectivity issues.10 Using 13 as catalyst, conventional diazocompounds like ethyl diazoacetate are substantially less efficient (62%, see ESI†), and redox-active α-aryldiazoacetate reagents6a were unreactive. These observations illustrate the importance of the match between the catalyst and the carbene precursor for efficient insertion.
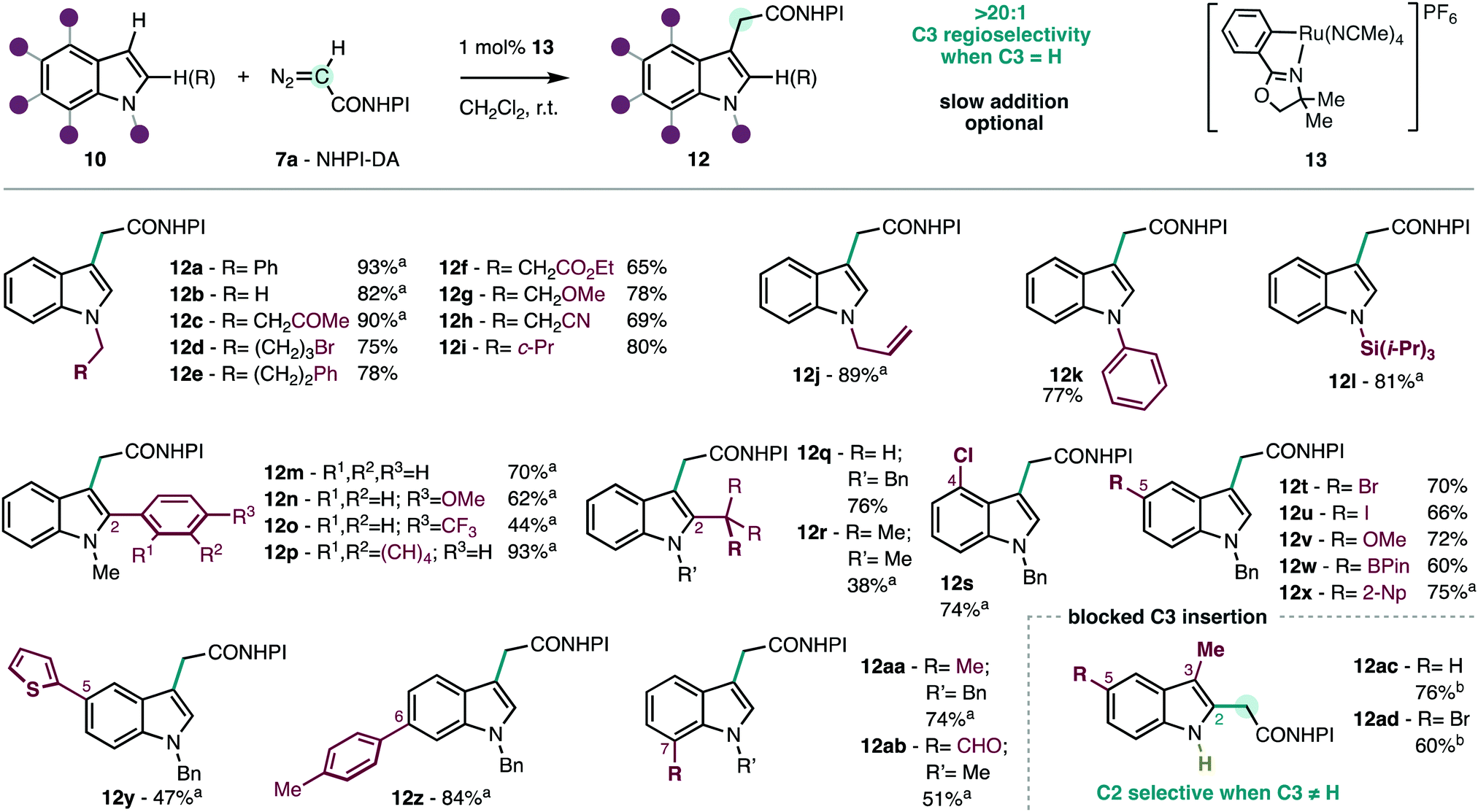 | ||
| Scheme 2 Scope of the C3-selective insertion of NHPI-DA (7a) into functionalized indoles. Conditions: 10 (0.2–0.3 mmol), 7a (1.25 equiv.), CH2Cl2 (0.1 M), 0.25–14 h, r.t. Isolated yields. aSlow addition (0.25–14 h; see ESI†). b7a (0.3 mmol), 10 (2.0 equiv.), [Ru(p-cymene)Cl2]2 (2 mol%). | ||
The performance and the selectivity of the insertion were explored in a series of indoles functionalized in all positions (Scheme 2). The substitution at the nitrogen atom is known to be crucial at defining the reactivity of the indole core.8–13 In our case, various types of aliphatic (12a–j,m–ac), aromatic (12k), and silicon (12l) substituents are suitable, including common functionality found in natural products (12b), and a cyclopropane drugs (12i). A wide range of functionalized aliphatics bearing ketone (12c),16 halide (12d), ester (12f), ether (12g) and nitrile (12h) functions are tolerated in this carbene transfer reaction, allowing for further diversification. Also, the unhindered olefin in 12j highlights the chemoselective insertion in the indole nucleus over the competing cyclopropanation that we have previously reported under similar conditions.6b This may be due to the higher nucleophilicity of the indole compared to the olefin, and the particular affinity of the metal–carbene intermediate for the former. Expectedly, indoles that are unfunctionalized at nitrogen (i.e. 1H-indole) are unsuitable due to competing N–H insertion, but conventional protecting groups like benzyl (12a), allyl (12j) and trialkylsilyl (12l) can provide access to the corresponding NH-indole derivatives, if desired. It was found that electron-withdrawing protecting groups at nitrogen deactivate the indole towards functionalization by electrophilic carbene intermediates. In the neighboring C2 position different aromatics (12m–p) are tolerated, including fluorinated groups (12o) and extended aromatics (12p). Aliphatic substituents are also tolerated (12q,r), despite the lower efficiency observed when a sterically hindered substituent is placed in the neighboring carbon (12r). The benzo-fused ring in the indole framework can also carry a wide range of functionality in C4 (12s), C5 (12t–y), C6 (12z) and C7 (12aa,ab), which include halides (12s–u), boronic ester (12w), aldehyde (12ab), and even an electron-rich heterocycle (12y). It is important to highlight that slow addition has been found to be optional in most cases (see Scheme 2 for details), and the products 12 can often be obtained in good yields adding NHPI-DA (7a) at once without a syringe pump. Despite the structural diversity of indoles 10a–ab used in this study, we have observed complete regioselectivity for the C3 insertion in all cases. This feature motivated further exploration of the insertion reaction at the C2 position of the indole nucleus with C3-functionalized substrates.10a,b,12 Given the complete C3 selectivity observed in C2-unfunctionalized substrates with the metallacyclic catalyst 13, its unsuitability for this purpose came as no surprise. After extensive experimentation, it was found that the simpler [Ru(p-cymene)Cl2]2 that had an intermediate performance with N-alkyl indoles (see ESI†), was an effective catalyst on NH-indole substrates to produce the derivatives 12ac,ad in good yields. The use of [Ru(p-cymene)Cl2]2 in the insertion of NH-indoles has been reported using α-aryldiazoacetates, which are significantly more stabilized than NHPI-DA (7a).12b Indeed, the selective C2-insertion into C3-substituted indoles with simple diazoacetates is rare9c,10c compared to stabilized carbene precursors bearing additional aryl or electron-withdrawing substituents.10a,b,17 Overall, the breadth of the scope, selectivity and efficiency demonstrated by NHPI-DA (7a) are remarkable in the context of related carbene transfer reactions to indoles,9c,10,11d and further demonstrates the unusual reactivity and selectivity of redox-active carbenes.6
With satisfactory conditions for the redox-active carbene transfer reaction, we studied the transformation of products 12 into methylborylated indoles 11a–m to illustrate the edge of this strategy at delivering the formal products of unavailable borylmethylidene5a (3) C–H insertion (Scheme 3). Among the various decarboxylative borylation protocols developed in recent years,2 we found that the conditions initially developed by Baran were most effective in this context.2a Moreover, we could telescope the borylation reaction performing a solvent switch, thus obtaining products 11a–m in a one-pot reaction from the functionalized indoles 10. Using this protocol, we could obtain methylborylated indoles with decorated aliphatics (11a–f), aromatic (11g) or silylated (11h) functions at nitrogen. Functionalization at other positions in the indole nucleus (11i–m) does not affect the performance of the borylation reaction. Due to the functional group tolerance of both the C–H insertion, and the decarboxylative borylation reactions the methylborylated indoles can be uneventfully decorated with ester (11c), nitrile (11d), ether (11e), silicon (11h) or halide (11k) functions. To put these results in perspective, boronic ester 11b has been synthesized through two different routes that require three steps from N-methylindole,2b,3 and can be now accomplished in a one-pot operation using the same material. The methylborylated indoles 11 have variable stability during chromatographic purification, which accounts for the lower isolated yields obtained in some cases.
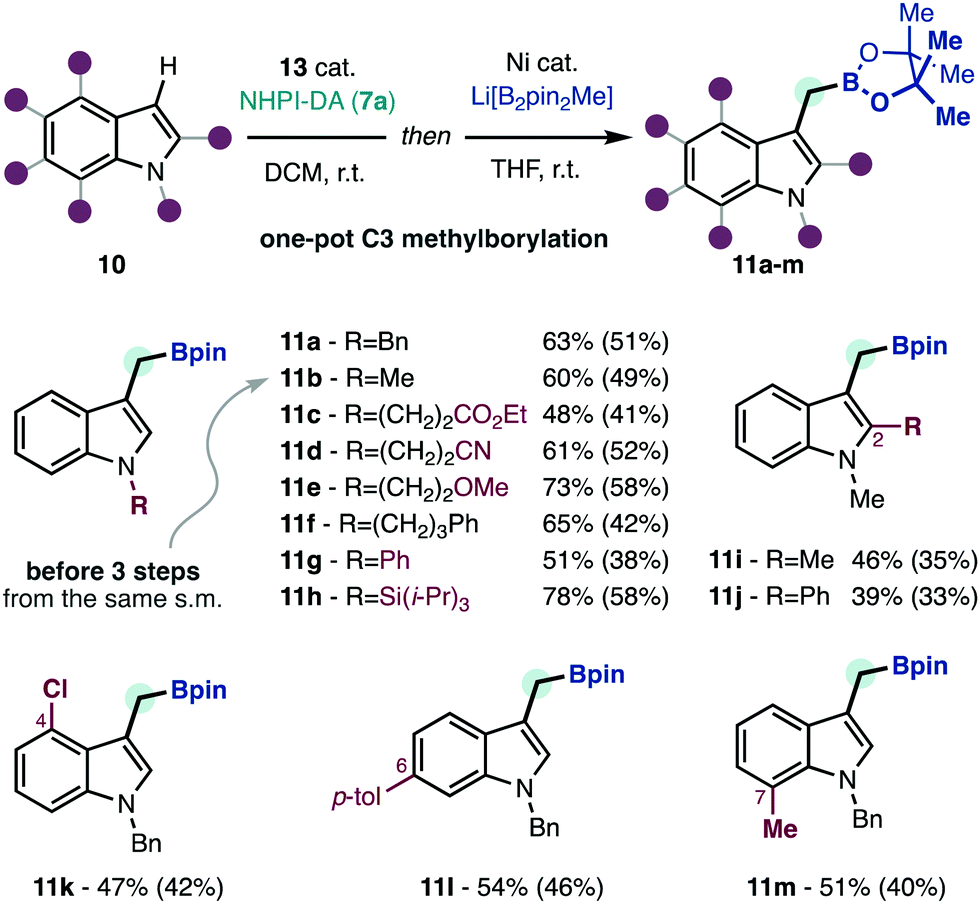 | ||
| Scheme 3 One-pot C3 methylborylation of functionalized indoles. Conditions: 10 (0.2–0.3 mmol), 7a (1.25 equiv.), CH2Cl2 (0.1 M), 0.25–14 h, r.t., then NiCl2·6H2O (0.10 equiv.), 4,4′-di-t-Bubipy (0.13 equiv.), MgBr2·OEt2 (1.5 equiv.), B2pin2 (3.0 equiv.), MeLi (3.3 equiv.), 0 °C to r.t., 2 h, Ar. Yields were determined by 1H-NMR using 1,1,2,2-tetrachloroethane as internal standard. Isolated yields in parenthesis. Pin, pinacol. | ||
The functionalized indole intermediates 12a,l are also well-suited for mild C–C coupling with different types of organometallics and Michael acceptors (Scheme 4).7 The corresponding products 11n–ab cannot be obtained through alternative carbene transfer reactions due to the particular instability of intermediates 4–6 (Scheme 1).5b–e After decarboxylative arylation (Scheme 4) the products 11n–p were obtained without the need to prepare and optimize the catalytic system for several aryl-diazomethane precursors. Vinyl carbenes are prone to oligomerize and isomerize but these substituents can be seamlessly incorporated to deliver the skipped unsaturated products 11q–s. In contrast with classic SN2′ allylations of indoles,18 the products 11q–s are obtained with high regioselectivity. Only minimal isomerization of the pendant alkene was observed in 11s. Functionalized alkyl groups (11t–ab), including protected alcohol (11t), heterocycle (11u), halide (11v), olefin (11w), or cyclopropane (11x) groups can also be included.7d Michael acceptors can also be utilized as alkyl group precursors in Giese-type reactions7b to deliver ketone (11z), nitrile (11aa), or ester (11ab) functions. Unlike conventional alkylation reactions that require the synthesis of the corresponding alkyl halides and high temperatures,1 these reactions occur at room temperature using a unified retrosynthetic approach. The strategic value of the linchpin approach presented herein is further illustrated by the product 11aa, which required 6 synthetic steps to be prepared from indole,19 and now can be obtained in half of those operations.
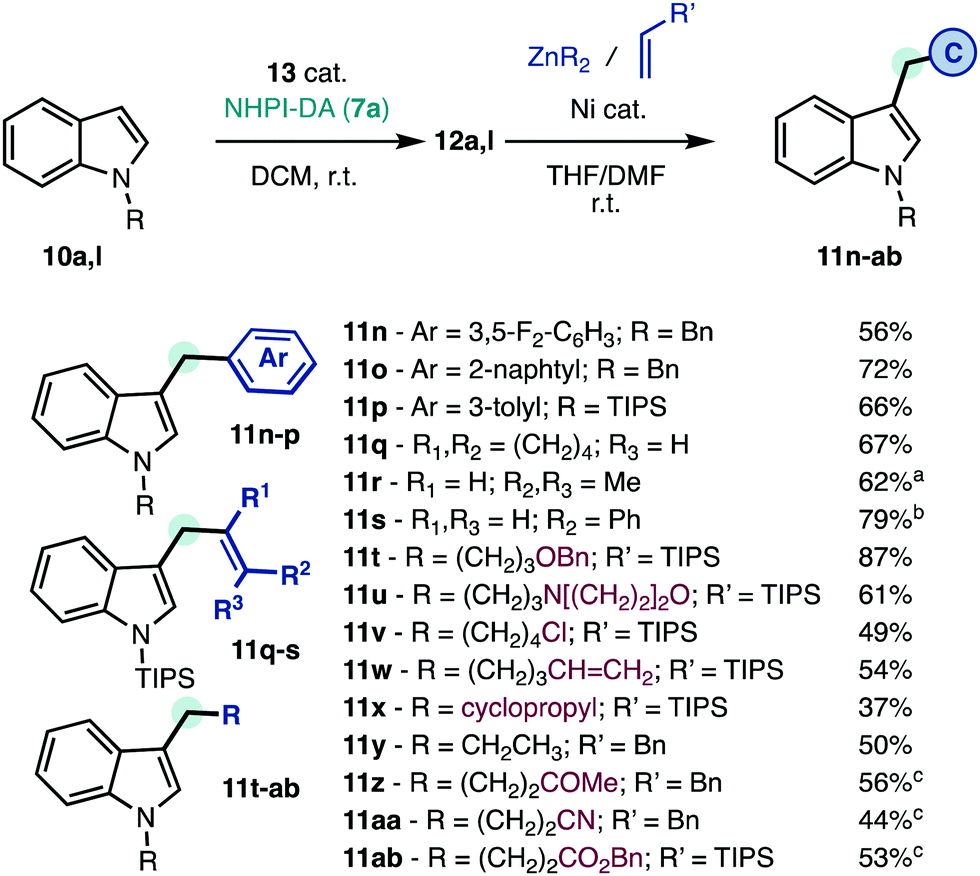 | ||
| Scheme 4 Unified alkylation of indoles at room temperature. See ESI† for details. Isolated yields. a>20:1 r.r. b>6:1 r.r. cGiese reaction with a Michael acceptor. r.r., regioisomeric ratio. | ||
In summary, a divergent C–H alkylation of indoles at room temperature is enabled by a redox-active single carbon linchpin. The exclusive regioselectivity and high efficiency of the insertion reaction with a redox-active carbene enable mild editing of the indole core. These results demonstrate the capacity of redox-active carbenes to enable formal C–H functionalization reactions in indoles by carbene intermediates that are problematic or inherently unstable like boryl-, aryl-, vinyl-, and alkyl-methylidenes.
Conflicts of interest
There are no conflicts to declare.Notes and references
- For representative alternative indole C3-alkylation methods, see:
(a) M. De Rosa and A. Soriente, Eur. J. Org. Chem., 2010, 1029 CrossRef CAS
; (b) H. E. Johnson and D. G. Crosby, J. Org. Chem., 1963, 28, 1246 CrossRef CAS
- For selected decarboxylative borylations, see:
(a) C. Li, J. Wang, L. M. Barton, S. Yu, M. Tian, D. S. Peters, M. Kumar, A. W. Yu, K. A. Johnson, A. K. Chatterjee, M. Yan and P. S. Baran, Science, 2017, 356, eaam7355 CrossRef PubMed
- H. Zhang, S. Hagihara and K. Itami, Chem. – Eur. J., 2015, 21, 16796 CrossRef CAS PubMed
- For selected reviews on α-diazo carbonyl compounds, see:
(a) H. M. Davies and S. J. Hedley, Chem. Soc. Rev., 2007, 36, 1109 RSC
-
(a) G. Benoit and A. B. Charette, J. Am. Chem. Soc., 2017, 139, 1364 CrossRef CAS PubMed
-
(a) Z. Yu and A. Mendoza, ACS Catal., 2019, 9, 7870 CrossRef CAS
- For selected decarboxylative C–C coupling reactions, see:
(a) J. Cornella, J. T. Edwards, T. Qin, S. Kawamura, J. Wang, C. M. Pan, R. Gianatassio, M. Schmidt, M. D. Eastgate and P. S. Baran, J. Am. Chem. Soc., 2016, 138, 2174 CrossRef CAS PubMed
- For selected seminal studies, see:
(a) G. M. Badger, B. J. Christie, H. J. Rodda and J. M. Pryke, J. Chem. Soc., 1958, 1958, 1179 RSC
- For selected C3-insertions with simple diazocompounds, see:
(a) K. J. Hock, A. Knorrscheidt, R. Hommelsheim, J. Ho, M. J. Weissenborn and R. M. Koenigs, Angew. Chem., Int. Ed., 2019, 58, 3630 CrossRef CAS PubMed
- The insertion of diazocompounds sometimes lead to mixtures of C2- and C3-insertion products. For examples using stabilized diazomalonates, see:
(a) M. B. Johansen and M. A. Kerr, Org. Lett., 2010, 12, 4956 CrossRef CAS PubMed
- For selected C3-insertions of α-functionalized diazoacetates, see:
(a) D. T. Boruta, O. Dmitrenko, G. P. A. Yap and J. M. Fox, Chem. Sci., 2012, 3, 1589 RSC
- Only stabilized α-aryldiazoacetates and diazomalonates undergo C2-selective insertion on C3-unfunctionalized indoles:
(a) J. Ghorai, M. Chaitanya and P. Anbarasan, Org. Biomol. Chem., 2018, 16, 7346 RSC
- For indole cyclopropanation, see:
(a) G. Ozuduru, T. Schubach and M. M. Boysen, Org. Lett., 2012, 14, 4990 CrossRef PubMed
-
(a) C. Schnaars, M. Hennum and T. Bonge-Hansen, J. Org. Chem., 2013, 78, 7488 CrossRef CAS PubMed
- S. Chanthamath, S. Thongjareun, K. Shibatomi and S. Iwasa, Tetrahedron Lett., 2012, 53, 4862 CrossRef CAS
- For our recent ketone synthesis, see: K. Colas, A. C. V. D. dos Santos and A. Mendoza, Org. Lett., 2019, 21, 7908 CrossRef CAS PubMed
- J. M. Antos, J. M. McFarland, A. T. Lavarone and M. B. Francis, J. Am. Chem. Soc., 2009, 131, 6301 CrossRef CAS PubMed
- M. Westermaier and H. Mayr, Org. Lett., 2006, 8, 4791 CrossRef CAS PubMed
-
(a) M. S. Pedras, Z. Minic, P. D. Thongbam, V. Bhaskar and S. Montaut, Phytochemistry, 2010, 71, 1952 CrossRef CAS PubMed
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1cc01026c; NMR raw data can be downloaded from Zenodo. See DOI: 10.5281/zenodo.4652793 |
| This journal is © The Royal Society of Chemistry 2021 |