
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
A dual-enzyme cleavable linker for antibody–drug conjugates†
Jonathan D.
Bargh
 a,
Stephen J.
Walsh
ab,
Nicola
Ashman
a,
Albert
Isidro-Llobet
c,
Jason S.
Carroll
b and
David R.
Spring
*a
a,
Stephen J.
Walsh
ab,
Nicola
Ashman
a,
Albert
Isidro-Llobet
c,
Jason S.
Carroll
b and
David R.
Spring
*a
aDepartment of Chemistry, University of Cambridge, Lensfield Road, Cambridge, CB2 1EW, UK. E-mail: spring@ch.cam.ac.uk
bCancer Research UK Cambridge Institute, University of Cambridge, Robinson Way, Cambridge, CB2 0RE, UK
cGSK, Gunnels Wood Road, Stevenage, SG1 2NY, UK
First published on 2nd March 2021
Abstract
A novel enzyme cleavable linker for antibody–drug conjugates is reported. The 3-O-sulfo-β-galactose linker is cleaved sequentially by two lysosomal enzymes – arylsulfatase A and β-galactosidase – to release the payload in targeted cells. An α-HER2 antibody–drug conjugate synthesised using this highly hydrophilic dual-cleavable linker exhibited excellent cytotoxicity and selectivity.
Antibody–drug conjugates (ADCs) now represent a major class of biotherapeutics, with nine approved by the Food and Drug Administration (FDA) and >60 more in clinical trials.1–3 By combining the tumour selectivity of monoclonal antibodies and the antitumour activity of small molecule payloads, a wide therapeutic window can be achieved.4,5 Key to the efficacy and safety of an ADC is a covalent linker between the two therapeutic components, which should help solubilise the typically hydrophobic payload and provide a stable linkage in circulation.6 Importantly, the linker usually also contains a cleavable trigger, which selectively releases the payload at target tumour cells.7 Enzyme-cleavable linkers represent a particularly attractive cleavable option, given their potential for high plasma stability and selective payload release in the lysosomes of target cells.
Thus far, the toolbox of enzyme-cleavable ADC linkers has included protease-,8–10 glycosidase-,11,12 pyrophosphatase-,13 acid phosphatase-14 and sulfatase-cleavable15,16 motifs. Of particular prevalence are cathepsin-cleavable dipeptides, now present in the majority of ADCs in clinical development. However, the mouse plasma instability and hydrophobicity of the dipeptide linkers can impede accurate preclinical evaluations and cause ADC aggregation.17–19 To address these shortcomings, we recently reported the arylsulfate motif as an effective sulfatase-cleavable group, capable of releasing drugs from ADCs with excellent cytotoxicity and cell-selectivity.15 The arylsulfate construct offered improved mouse plasma stability and hydrophilicity versus dipeptide linkers. However, the arylsulfate motif only represents a pseudo-substrate for lysosomal sulfatases, whose natural substrates are alkylsulfates.20
We looked to expand the toolbox of cleavable ADC linkers by mimicking sulfatide, the natural substrate of arylsulfatase A (ARSA).21 In its metabolic pathway, sulfatide is first hydrolysed by ARSA to reveal a β-galactosyl ceramide, which is then susceptible to β-galactosidase (β-gal)-mediated cleavage, producing the ceramide metabolite (Fig. 1A).22 We therefore designed a 3-O-sulfo-β-galactose linker, envisaged to hijack the natural dual enzymatic ARSA/β-gal metabolic cascade, thus releasing a payload in the target cell (Fig. 1B). This cleavable group was anticipated to exhibit particularly high hydrophilicity, due to the presence of both an anionic sulfate and a pyranose functionality. Furthermore, the dual-enzymatic process would afford excellent lysosome-selectivity to the conjugate. Given that efficient lysosomal cleavage of β-galactosyl ADC linkers has been previously reported, drug release from the linker was expected to be fast, provided that the first, ARSA-mediated cleavage was also efficient.11
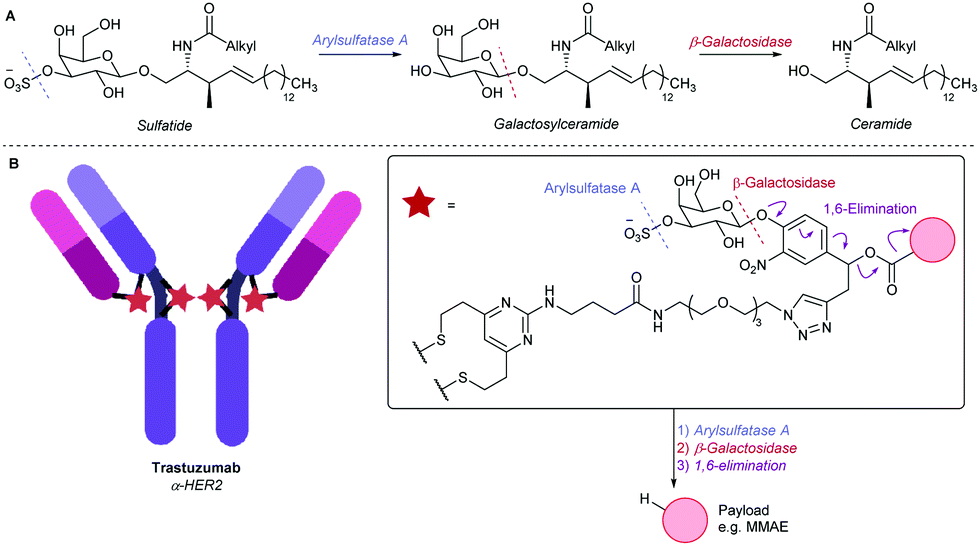 | ||
| Fig. 1 (A) The ARSA/β-gal-mediated metabolism of sulfatide. (B) An α-HER2 ADC employing the 3-O-sulfo-β-galactose linker motif. | ||
We first investigated linker-payload 1, bearing 7-amino-4-methyl coumarin (AMC) as a model payload, to measure real-time enzymolysis by fluorimetry. An analogous linker-payload 2 featuring the potent tubulin binder MMAE was also envisioned for in vitro ADC biological evaluation. This linker-MMAE construct therefore required antibody attachment, so a divinylpyrimidine (DVP) conjugation motif was also incorporated.23
Synthesis of both linker-payloads proceeded from acetyl-galactose 3 (Scheme 1).11 Carbamate 4a was synthesised by reaction with AMC-isocyanate 5 in the presence of a dibutyltin dilaurate catalyst. For the MMAE carbamate 4b, 3 was instead reacted with 4-chlorophenyl chloroformate to afford the 4-nitrophenyl carbonate 6, before displacement with MMAE. The acetyl-galactose-carbamates 4a and 4b were then hydrolysed to reveal tetrahydroxypyranoses 6a and 6b.11 In the key step of the synthesis, 6a and 6b were first reacted with dibutyltin oxide to form dibutylstannylene acetals, before reaction with SO3·NMe3.24,25 This procedure selectively sulfated at the equatorial 3-O-position, affording sulfates 1 and 7 in 67% and 55% yields, respectively. The sulfogalactose-MMAE 7 was further functionalised with a DVP conjugation motif, using copper-catalyzed azide–alkyne cycloaddition (CuAAC) chemistry, affording linker-payload 2. This route also facilitated the functionalisation of intermediate galactosyl 6b with DVP, affording galactosyl linker-payload 8, thus allowing a direct comparison between 3-O-sulfo-β-galactose and β-galactose ADC linkers.
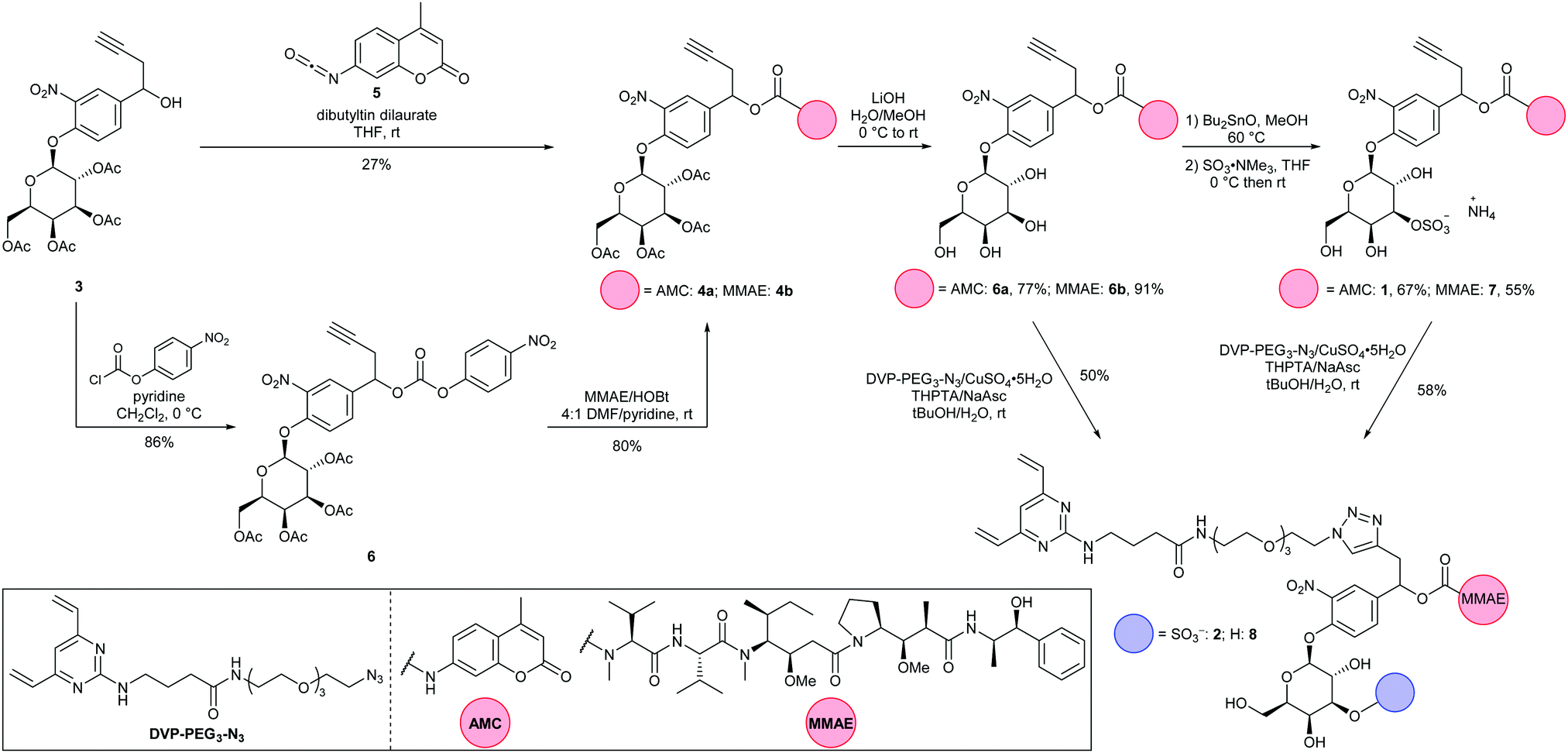 | ||
| Scheme 1 Synthesis of 3-O-sulfo-galactose-AMC 1 and linker-payloads 2 and 8. | ||
To validate its enzyme sensitivity, sulfogalactose-AMC 1 was incubated with isolated ARSA and β-gal enzymes and the fluorescence monitored over time (Fig. 2). As expected, no significant increase in fluorescence was observed upon incubation with either ARSA or β-gal alone. In contrast, incubation with both enzymes resulted in a dramatic increase in fluorescence intensity, suggesting that dual-enzyme catalysis is required for the hydrolysis of 1. Moreover, the extracellular stability of 1 was demonstrated, with no observable AMC release observed over 20 hours in human or mouse plasma (Fig. S3, ESI†). Together, these results suggest the 3-O-sulfo-β-galactose motif will facilitate tumour cell targeting by discriminating between lysosomal and circulatory conditions.
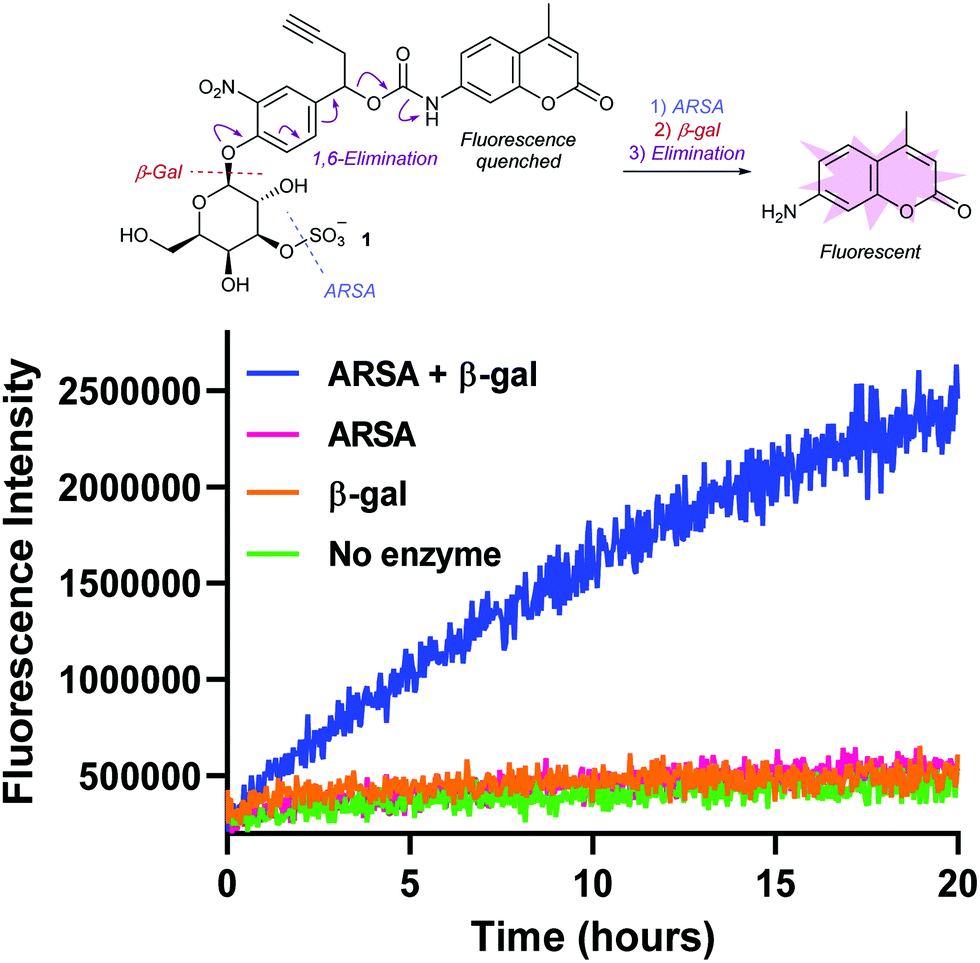 | ||
| Fig. 2 Fluorometric measurement of AMC release from probe 1 by incubation with ARSA and β-gal enzymes. λex = 350 nm, λem = 460 nm. | ||
To evaluate the biological properties of the 3-O-sulfo-β-galactose linker, linker-payloads 2 and 8 were conjugated to α-HER2 antibody trastuzumab (Fig. 3). As with previous IgG conjugations employing the DVP motif,15,23,26,27 the four interchain disulfides of trastuzumab were first reduced by treatment with tris(2-carboxyethyl)phosphine hydrochloride (TCEP). Upon subsequent addition of 2 or 8, the reduced disulfides were rebridged via the bis-reactive DVP, affording conjugates ADC 1 and ADC 2 with four drugs/antibody. The excellent aqueous solubility of sulfo-galactose 2 was exemplified by its complete solubility in water at 20 mM, thereby facilitating protein bioconjugation at 0% organic co-solvent. Conversely, preparation of a 20 mM stock of galactose 8 required DMSO for dissolution, thus the ensuing bioconjugation was not 100% aqueous. Although the small amount of co-solvent (3% DMSO) was not deleterious to protein stability in this case, such differences in linker-payload solubilities suggest that a 3-O-sulfo-β-galactose linker may facilitate bioconjugation with a more lipophilic payload.
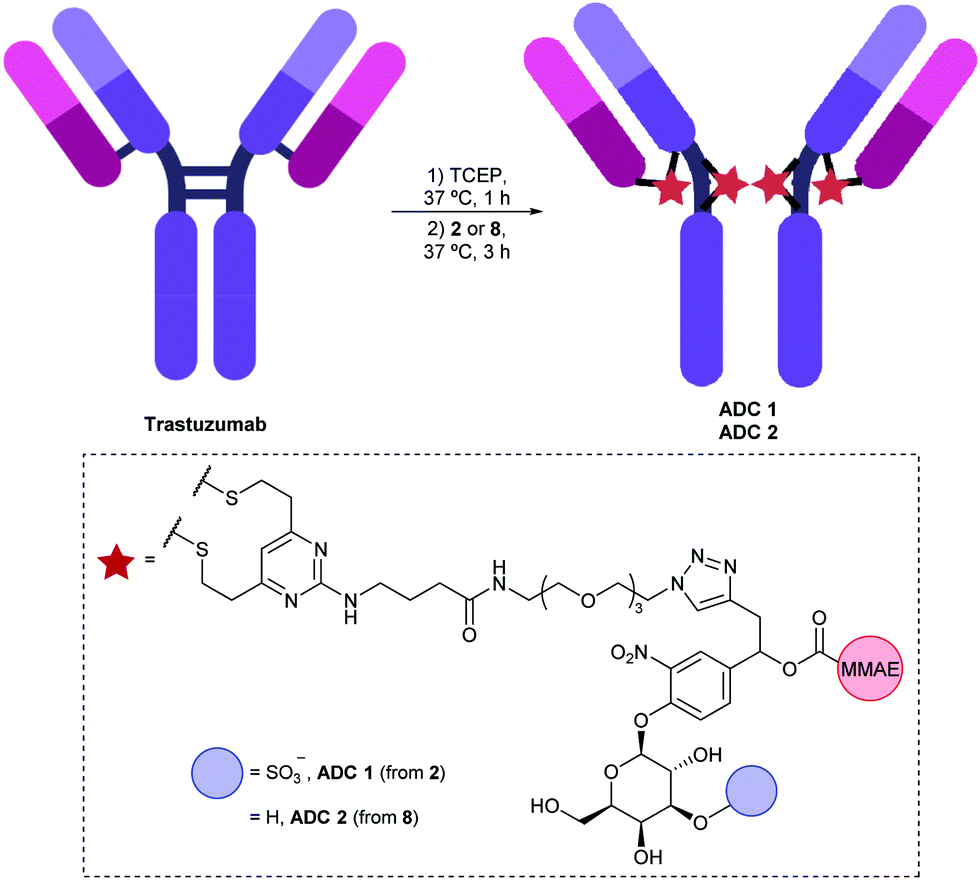 | ||
| Fig. 3 Bioconjugation of linker-payloads 2 and 8 to trastuzumab to afford ADC 1 and ADC 2 respectively. | ||
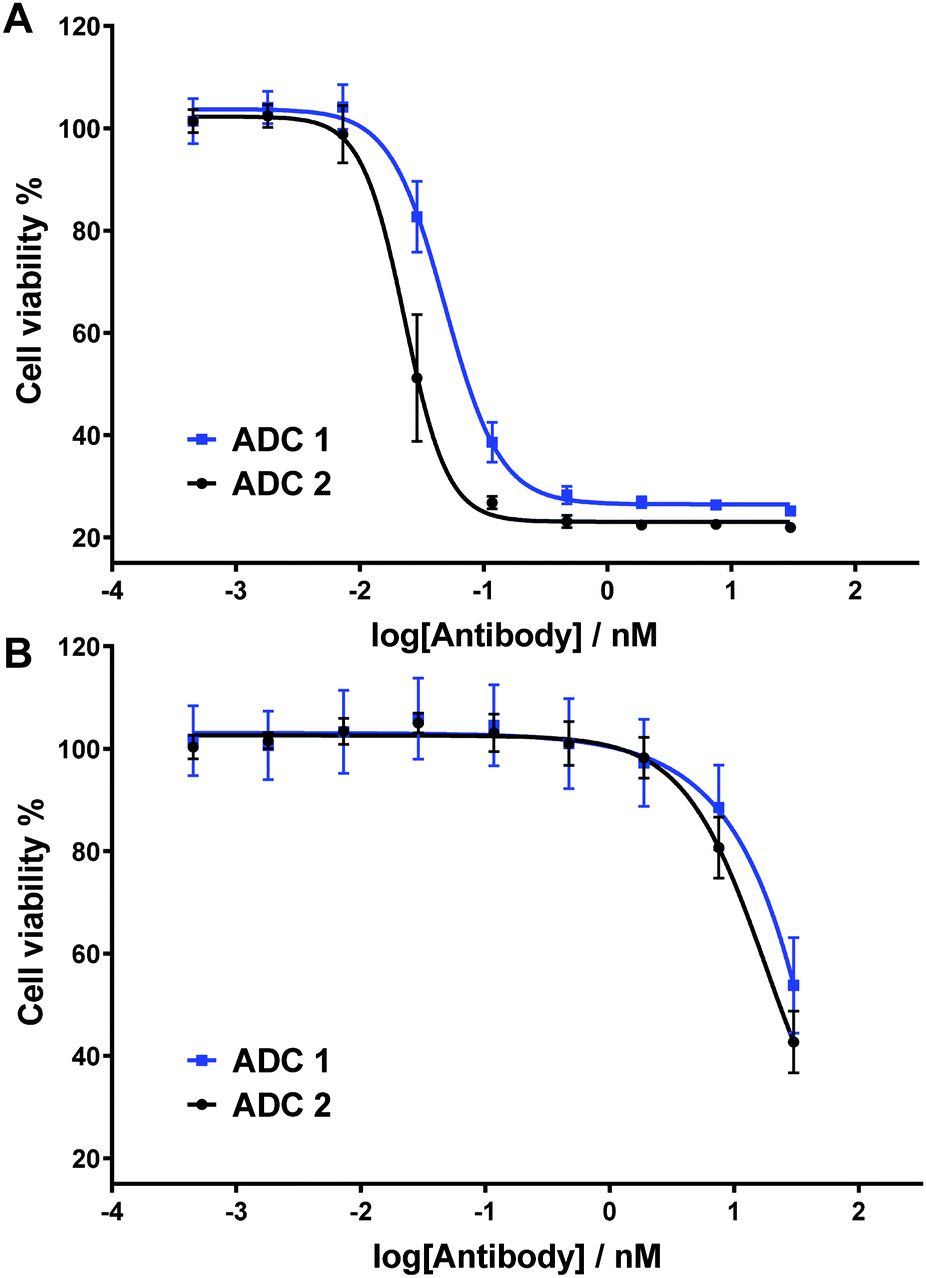 | ||
| Fig. 4 In vitro biological evaluation of ADC 1 and ADC 2 in (A) HER2-positive SKBR3 cells and (B) HER2-negative MCF7 cells. Viability data shows the mean of three independent experiments and error bars represent standard error of the mean. | ||
Next, the two ADCs were tested against HER2-positive SKBR3 cells to determine their dose-dependent cytotoxicity (Fig. 4A). We have previously demonstrated that under the same assay conditions and with analogous non-cleavable or poorly-cleavable ADCs, ADC potency is dramatically reduced versus rapidly cleavable ADCs, thus confirming the relationship between linker cleavage efficiency and cytotoxicity.15 Gratifyingly, sulfogalactose ADC 1 was extremely potent, with IC50 = 49 pM. This cytotoxicity is comparable to our most potent analogous arylsulfate ADC, (IC50 [SKBR3] = 40 pM), as well as the cathepsin-cleavable Val-Ala-PABC-MMAE ADC (IC50 [SKBR3] = 41 pM).15 It is therefore probable that the ARSA/β-gal dual-enzymatic cascade occurs in lysosomes at a comparable rate to the enzymolysis of arylsulfate linkers and the Val-Ala dipeptide linkers. Although the non-sulfo ADC 2 was slightly more cytotoxic (IC50 = 23 pM) (Fig. 4A), we postulate that the hydrophilicity gained upon sulfation of the galactosyl motif may deem the 3-O-sulfo-β-galactose motif more appropriate for certain hydrophobic linker-payloads. The ADCs were also tested against HER2-negative MCF7 cells (Fig. 4B), with both exhibiting relatively low potency and thus indicating their linkers’ selectivity for intracellular cleavage.
In conclusion, the 3-O-sulfo-β-galactose motif is a highly hydrophilic and cleavable ADC linker motif. The ARSA/β-gal dual-enzymatic cleavage was confirmed by virtue of fluorescence probe 1, signifying the linker's strict requirement for cleavage by both lysosomal enzymes for drug release. The motif was successfully attached to trastuzumab in a completely aqueous bioconjugation reaction and the resulting ADC 1 was highly potent and selective. It is anticipated that the motif will be widely applicable to a range of hydrophobic payloads and antibodies, given its favourable properties.
J. D. B. acknowledges an iCASE studentship from GlaxoSmithKline/EPSRC. N. A. acknowledges an iCASE studentship from AstraZeneca. D. R. S. acknowledges support from the Engineering and Physical Sciences Research Council (EP/P020291/1) and Royal Society (Wolfson Research Merit Award). The Spring lab acknowledges general lab support from the EPSRC, BBSRC, MRC and Royal Society.
Conflicts of interest
There are no conflicts to declare.References
- L. Gauzy-Lazo, I. Sassoon and M.-P. P. Brun, SLAS Discovery, 2020, 25, 1–26 Search PubMed.
- M. Nejadmoghaddam, A. Minai-tehrani and R. Ghahremanzadeh, Avicenna J. Med. Biotechnol., 2019, 11, 3–23 Search PubMed.
- S. J. Walsh, J. D. Bargh, F. M. Dannheim, A. R. Hanby, H. Seki, A. J. Counsell, X. Ou, E. Fowler, N. Ashman, Y. Takada, A. Isidro-Llobet, J. S. Parker, J. S. Carroll and D. R. Spring, Chem. Soc. Rev., 2021, 50, 1305–1353 RSC.
- A. Beck, L. Goetsch, C. Dumontet and N. Corvaïa, Nat. Rev. Drug Discovery, 2017, 16, 315–337 CrossRef CAS PubMed.
- V. Chudasama, Drug Discovery Today: Technol., 2018, 30, 1–2 CrossRef PubMed.
- B. Nolting, Methods Mol. Biol., 2013, 1045, 71–100 CrossRef PubMed.
- J. D. Bargh, A. Isidro-Llobet, J. S. Parker and D. R. Spring, Chem. Soc. Rev., 2019, 48, 4361–4374 RSC.
- S. O. Doronina, B. E. Toki, M. Y. Torgov, B. A. Mendelsohn, C. G. Cerveny, D. F. Chace, R. L. DeBlanc, R. P. Gearing, T. D. Bovee, C. B. Siegall, J. A. Francisco, A. F. Wahl, D. L. Meyer and P. D. Senter, Nat. Biotechnol., 2003, 21, 778–784 CrossRef CAS PubMed.
- Y. Shiose, Y. Ochi, H. Kuga, F. Yamashita and M. Hashida, Biol. Pharm. Bull., 2007, 30, 2365–2370 CrossRef CAS PubMed.
- B. Wei, J. Gunzner-Toste, H. Yao, T. Wang, J. Wang, Z. Xu, J. Chen, J. Wai, J. Nonomiya, S. P. Tsai, J. Chuh, K. R. Kozak, Y. Liu, S. F. Yu, J. Lau, G. Li, G. D. Phillips, D. Leipold, A. Kamath, D. Su, K. Xu, C. Eigenbrot, S. Steinbacher, R. Ohri, H. Raab, L. R. Staben, G. Zhao, J. A. Flygare, T. H. Pillow, V. Verma, L. A. Masterson, P. W. Howard and B. Safina, J. Med. Chem., 2018, 61, 989–1000 CrossRef CAS PubMed.
- S. Kolodych, C. Michel, S. Delacroix, O. Koniev, A. Ehkirch, J. Eberova, S. Cianférani, B. Renoux, W. Krezel, P. Poinot, C. D. Muller, S. Papot and A. Wagner, Eur. J. Med. Chem., 2017, 142, 376–382 CrossRef CAS PubMed.
- S. C. Jeffrey, J. B. Andreyka, S. X. Bernhardt, K. M. Kissler, T. Kline, J. S. Lenox, R. F. Moser, M. T. Nguyen, N. M. Okeley, I. J. Stone, X. Zhang and P. D. Senter, Bioconjugate Chem., 2006, 17, 831–840 CrossRef CAS PubMed.
- J. C. Kern, M. Cancilla, D. Dooney, K. Kwasnjuk, R. Zhang, M. Beaumont, I. Figueroa, S. C. Hsieh, L. Liang, D. Tomazela, J. Zhang, P. E. Brandish, A. Palmieri, P. Stivers, M. Cheng, G. Feng, P. Geda, S. Shah, A. Beck, D. Bresson, J. Firdos, D. Gately, N. Knudsen, A. Manibusan, P. G. Schultz, Y. Sun and R. M. Garbaccio, J. Am. Chem. Soc., 2016, 138, 1430–1445 CrossRef CAS PubMed.
- J. C. Kern, D. Dooney, R. Zhang, L. Liang, P. E. Brandish, M. Cheng, G. Feng, A. Beck, D. Bresson, J. Firdos, D. Gately, N. Knudsen, A. Manibusan, Y. Sun and R. M. Garbaccio, Bioconjugate Chem., 2016, 27, 2081–2088 CrossRef CAS PubMed.
- J. D. Bargh, S. J. Walsh, A. Isidro-Llobet, S. Omarjee, J. S. Carroll and D. R. Spring, Chem. Sci., 2020, 11, 2375–2380 RSC.
- D. Rabuka, J. Lui and S. Chuprakov, World Patent, WO2020096775, 2020 Search PubMed.
- M. Dorywalska, R. Dushin, L. Moine, S. E. Farias, D. Zhou, T. Navaratnam, V. Lui, A. Hasa-Moreno, M. G. Casas, T.-T. Tran, K. Delaria, S.-H. Liu, D. Foletti, C. J. O’Donnell, J. Pons, D. L. Shelton, A. Rajpal and P. Strop, Mol. Cancer Ther., 2016, 15, 958–970 CrossRef CAS PubMed.
- P. J. Burke, P. D. Senter, D. W. Meyer, J. B. Miyamoto, M. Anderson, B. E. Toki, G. Manikumar, M. C. Wani, D. J. Kroll and S. C. Jeffrey, Bioconjugate Chem., 2009, 20, 1242–1250 CrossRef CAS PubMed.
- S. C. Jeffrey, M. T. Nguyen, R. F. Moser, D. L. Meyer, J. B. Miyamoto and P. D. Senter, Bioorg. Med. Chem. Lett., 2007, 17, 2278–2280 CrossRef CAS PubMed.
- S. R. Hanson, M. D. Best and C. H. Wong, Angew. Chem., Int. Ed., 2004, 43, 5736–5763 CrossRef CAS PubMed.
- G. Lukatela, N. Krauss, K. Theis, T. Selmer, V. Gieseimann, K. Von Figura and W. Saenger, Biochemistry, 1998, 37, 3654–3664 CrossRef CAS PubMed.
- Y. Yang, Y. Feng, X. Zhang, T. Nakajima, N. Tanaka, E. Sugiyama, Y. Kamijo and T. Aoyama, Tohoku J. Exp. Med., 2016, 240, 113–122 CrossRef CAS PubMed.
- S. J. Walsh, S. Omarjee, W. R. J. D. Galloway, T. T. L. Kwan, H. F. Sore, J. S. Parker, M. Hyvönen, J. S. Carroll and D. R. Spring, Chem. Sci., 2019, 10, 694–700 RSC.
- B. Guilbert, N. J. Davis, M. Pearce, R. T. Aplin and S. L. Flitsch, Tetrahedron: Asymmetry, 1994, 5, 2163–2178 CrossRef CAS.
- M. Jäger and A. J. Minnaard, Chem. Commun., 2016, 52, 656–664 RSC.
- S. J. Walsh, J. Iegre, H. Seki, J. D. Bargh, H. F. Sore, J. S. Parker, J. S. Carroll and D. R. Spring, Org. Biomol. Chem., 2020, 18, 4224–4230 RSC.
- J. Charoenpattarapreeda, S. J. Walsh, J. S. Carroll and D. R. Spring, Angew. Chem., Int. Ed., 2020, 59, 23045–23050 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1cc00957e |
| This journal is © The Royal Society of Chemistry 2021 |