
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAromaticity in catalysis: metal ligand cooperation via ligand dearomatization and rearomatization†
Théo P.
Gonçalves‡
,
Indranil
Dutta‡
 and
Kuo-Wei
Huang
*
and
Kuo-Wei
Huang
*
KAUST Catalysis Center and Division of Physical Sciences and Engineering, King Abdullah University of Science and Technology, Thuwal 23955-6900, Saudi Arabia. E-mail: hkw@kaust.edu.sa
First published on 9th February 2021
Abstract
Unlike the conventional model of transition metal catalysis, ligands in metal–ligand cooperative (or bifunctional) catalysis are involved in the substrate activations. Such processes have offered unique mechanistic understandings and led to new concepts for the catalyst design. In particular, unprecedented activities were discovered when the ligand could undergo dearomatization–rearomatization reactions during the catalytic cycle. Aromatization can provide an extra driving force to thermodynamics; consequently, it brings a new perspective to ligand platform design for catalysis. While numerous applications were demonstrated, the influences of changing ligand aromatic properties were often overlooked. In this article, representative ligand systems will be highlighted and a comparison between the Milstein and the Huang pincer systems will be discussed to provide theoretical and conceptual insights.
 Théo P. Gonçalves | Théo Piechota Gonçalves was born in Recife, Brazil. He received his BSc for the University Federal of Pernambuco (UFPE, Brazil) in 2009, MSc in total synthesis and PhD in computational chemistry at the Southampton University, UK in 2014. He then moved to KAUST to work in the catalysis field as a Post-Doc with Prof. Kuo-Wei Huang. In 2018, he was promoted to the research scientist position at the same institution. His research interests focus on applying computational tools to study of organic, organometallic reactions and properties. He is also interested in the relationship between aromaticity, chemical bonding, and chemical reactivity. |
 Indranil Dutta | Indranil Dutta received his MSc at the University of Hyderabad, India in Inorganic Chemistry. He pursued his PhD degree on metal–metal and metal–ligand cooperation strategies for organic transformation at Indian Institute of Technology, Kanpur, India. Afterwards he moved to Osaka University, Japan as a Post-Doc before moving to KAUST. Since 2019, he has been a Post-Doc working with Prof. Kuo-Wei Huang. His broad research interests include the design of various cooperative catalysts and their reactivity towards small molecule activation and in particular for dehydrogenation chemistry. His present work focuses on formic acid dehydrogenation and CO2 utilization. |
 Kuo-Wei Huang | Kuo-Wei Huang is a Professor of Chemical Science at King Abdullah University of Science and Technology. He received his BS from National Taiwan University as a Yuan T. Lee Fellow and PhD from Stanford University as a Regina Casper Fellow. Before joining KAUST, he was an Assistant Professor at the National University of Singapore and a Goldhaber Distinguished Fellow at Brookhaven National Laboratory. The research interests of his group include CO2 utilization, hydrogen storage, and small molecule activation by the PN3(P) platform his group has developed and pioneered. He was recently highlighted in the “Pioneers and Influencers in Organometallic Chemistry” series in Organometallics 2020. |
1. Introduction
In the conventional model of transition-metal catalysis, the reactivities of the catalyst can be manipulated by modifying the steric and electronic properties of coordinating ligands and the transformations occur at the metal center.1,2 For example, in an oxidative addition process, a given X–Y bond is split with the formation of corresponding M–X and M–Y bonds (M = metal), and the oxidation state and the coordination number of the metal are both increased by 2. In a reductive elimination reaction (the reverse of oxidative addition and vice versa), M–X and M–Y bonds are cleaved to generate X–Y, and the oxidation state and the coordination number of the metal are both decreased by 2 (Fig. 1). In contrast, when metal–ligand cooperation (MLC) takes place, the cooperating ligand participates in the substrate activation and in general, the oxidation state of the metal center remains intact (Fig. 1).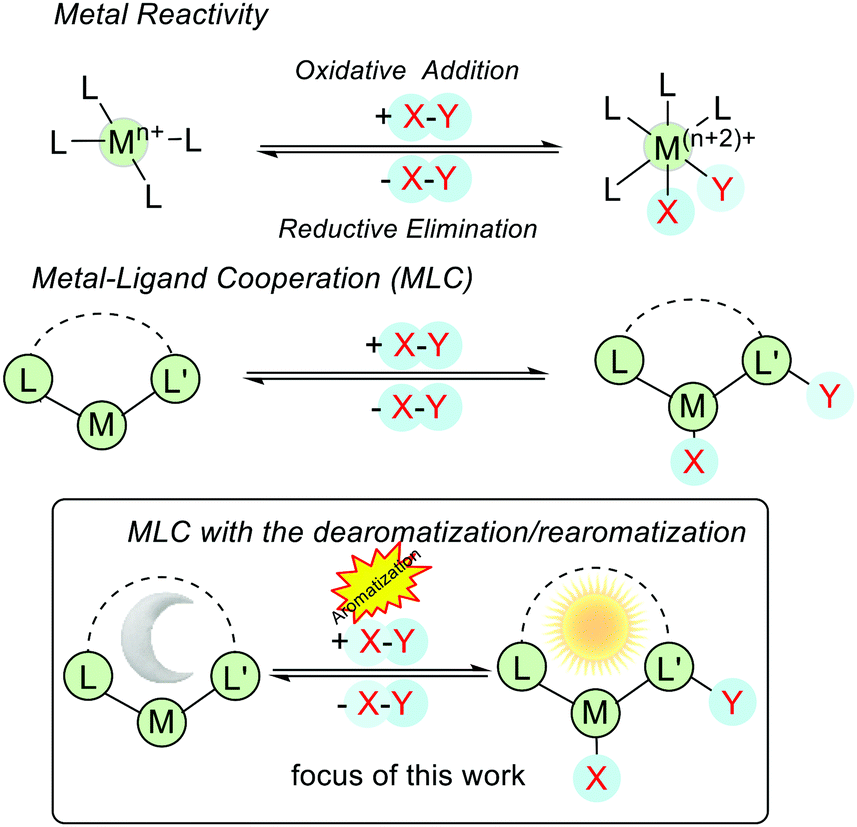 | ||
| Fig. 1 Different modes of substrate activations in conventional and metal–ligand cooperative catalysis. | ||
MLC has been identified in several hydrogenative and dehydrogenative processes catalyzed by naturally occurring metalloenzymes (alcohol dehydrogenase, hydrogenase catalase, hydrolase, etc.).3–5 Following the pioneering works by Shvo6–9 and Noyori10–14 in the mid-1980s and 1990s, respectively, this concept has gained increasing attention and has been incorporated into the design of synthetic catalysts. The synergistic interplay between metal and ligand(s) accounts for the improved reactivity and selectivity that would not have been achieved if the metal had acted alone, opening up a new dimension for bond activation and catalysis.15–19
The Noyori-type reactivity has been well studied and developed.15,16 In a typical catalytic cycle (Fig. 2),20 the coordinatively unsaturated active intermediate (NB) is generated via a base mediated conversion of the coordinatively saturated precursor (NA). NB subsequently facilitates the heterolytic cleavage of molecular hydrogen (H2) (or using isopropanol as the H2 source) across the Ru–amide bond to generate an amino Ru–hydride species (NC). The carbonyl substrate then binds to NC in the second coordination sphere and two hydrogens of antagonistic polarities are transferred to the substrate in a concerted manner via an outer-sphere mechanism with the regeneration of the active catalyst (NB). The term, bifunctionality, was coined to underline the roles of the Lewis basic ligand(s) and the Lewis acidic metal center to polarize a neutral H2 molecule,21–24 and analogous MLC reactivities were observed in many recent examples.25
 | ||
| Fig. 2 Ketone hydrogenation and interconversion of amido/amine forms in Noyori's catalyst (Ru: ruthenium, Ar = aryl, and R = alkyl). | ||
Unlike the Noyori's model, a non-coordinating atom in the ligand partakes in the Shvo MLC system (Fig. 3). The cyclopentadienone ligand in SA is converted to an aromatic hydroxycyclopentadienyl moiety in SB upon treatment with a H2 donor (e.g. isopropanol) along with the change of the formal oxidation state of Ru from 0 to +2. However, the roles of the ligand aromatic properties did not receive much attention until another groundbreaking discovery was made on MLC involving aromatization–dearomatization processes of PNP and PNN pyridine-based pincer complexes by Milstein and co-workers (Fig. 3).26–29 Finally, inspired by the distinct kinetic and thermodynamic properties in our PN3(P) pincer ligand platform by replacing the CH/CH2 spacers in the Milstein system to N/NH arms (Fig. 3),30–32 the influences of aromatic stabilization energies (ASE) on the thermodynamics of MLC were elucidated.33 In this distinctive subclass, MLC via aromatization–dearomatization, the relation between metal and ligand is even more rigorous. In this article, representative systems where ligands undergo aromatization–dearomatization during MLC will be highlighted. A comprehensive comparison between the Milstein and the Huang pincer systems will be provided. After introducing the known MLC system via aromatization–dearomatization, and a short introduction to the theoretical aspect of the aromaticity, a comprehensive comparison between the Milstein and the Huang pincer systems will be provided. Our aim is to familiarize the readers with the recent conceptual understanding of aromaticity and its contribution to the thermodynamics of the catalytic reactions.
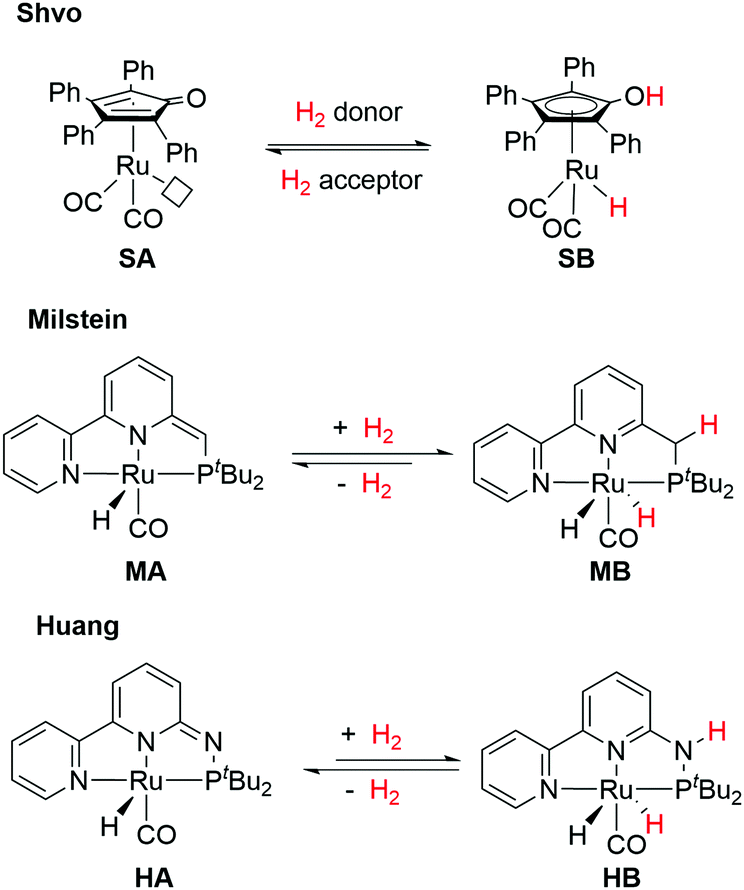 | ||
| Fig. 3 The Shvo, Milstein, and Huang metal–ligand cooperative systems. | ||
2. Ligand aromatization–dearomatization
2.1 Shvo and analogous systems
The very first example of ligand assisted bifunctional substrate activation was introduced by the Shvo group.6–9 Reaction of Ru3(CO)12 with diphenyl acetylene under CO atmosphere followed by water treatment yielded a dinuclear species known as Shvo's complex 1 which was identified as an efficient catalyst for transfer hydrogenation of ketones. The Shvo's complex was further explored for other transformations, such as transfer hydrogenation of imines, hydrogenation of ketones, aldehydes, alkenes, alkynes and alcohol dehydrogenation.34–36 These reactivities are attributed to the thermal dissociation of complex 1 into the two catalytically active monomeric species: a proposed reactive complex, SA (16e, dehydrogenation catalyst), and SB (18e, hydrogenation catalyst) (Fig. 4). SB has a η5-bound chemically non-innocent hydroxycyclopentadienyl ligand, which can act as a proton donor with the Ru–H center as a hydride donor. SA is coordinatively unsaturated with a η4-cyclopentadienone ligand, which serves as a proton acceptor with the Ru center as a hydride acceptor.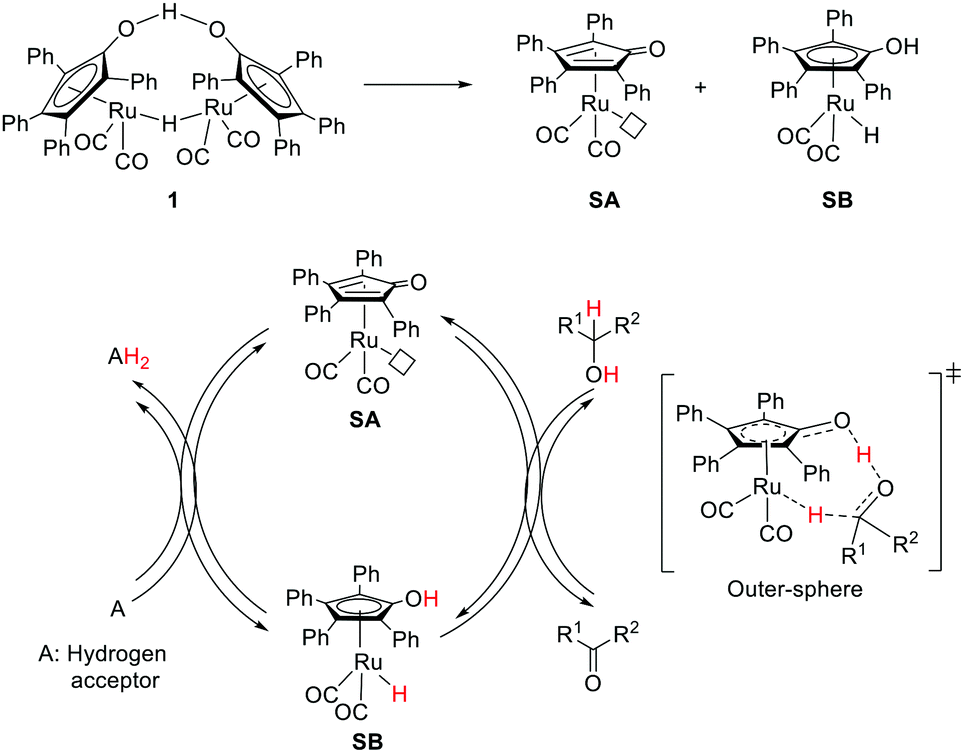 | ||
| Fig. 4 Shvo complexes and the proposed mechanistic pathway for transfer hydrogenation. | ||
An outer-sphere mechanism was proposed for the transfer hydrogenation of ketones. Extensive mechanistic studies including kinetic isotope effects, deuterium labelling experiments and theoretical calculations, suggested a concerted proton (–OH group at the hydroxycyclopentadienyl ligand) and hydride (Ru–H) transfer from the 18e complex (SB) to the carbonyl group with the concurrent formation of SA (Fig. 4).37,38 This simultaneous proton/hydride transfer step requires only one single coordination site around the metal and hence may proceed without any ring slippage. The outer-sphere route was also supported by the kinetic isotope effects in the alcohol dehydrogenation, where SA served as an active catalyst.39 Overall, the Shvo MLC is realized by the reversible interconversion between the aromatic η5-hydroxycyclopentadienyl ligand and the η4-cyclopentadienone moiety along with the 2e redox activity of the Ru center to facilitate bond activation events.
The Shvo-type MLC reactivities appear to be general. Analogous iron hydride hydroxycyclopentadienyl systems were investigated by Casey and Guan.40 The Knölker complex (2)41 was found to be efficient towards hydrogenation of carbonyl compounds and aldimines, and alcohol oxidation.40,42 Similar to the Shvo catalyst, interconvertible iron species are accounted for by the bifunctional substrate activation. The iron hydride species 2 is active for hydrogenation whereas the resulting coordinatively unsaturated 3 is active for dehydrogenation (oxidation). Extensive kinetic studies, stoichiometric experiments, and computational analysis support a concerted proton/hydride transfer process in an outer-sphere mechanism where hydrogen transfer from 2 is the rate-limiting step (Fig. 5).40,43
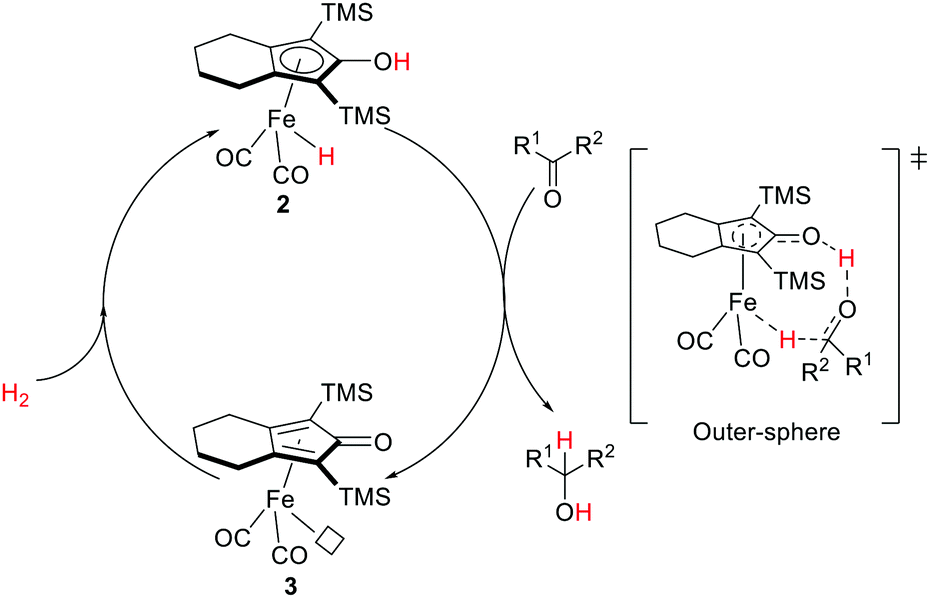 | ||
| Fig. 5 Hydrogenation of ketone catalyzed by the Knölker complex. | ||
Nozaki and co-workers have designed a number of iridium (Ir) and rhodium complexes bearing a hydroxycyclopentadienyl ligand and tested their catalytic activities towards acceptor-less dehydrogenation of C–C single bonds.44 The Ir complex having an OH group in the Cp ring (4) displayed the best activity compared to those of other silyl-protected ones and the rhodium analog (Fig. 6). The design principle was guided by MLC and the –OH group in alkyl hydride complex 7 (18e) was hypothesized to drive the release of dihydrogen via heterolytic bond formation with concurrent generation of unsaturated 16e alkyl complex 8, followed by a successive β-hydride elimination to afford the alkene product. H/D exchange studies and computational analysis suggest that this β-hydride elimination is the rate limiting step. The regenerated complex 5 may then undergo an intramolecular proton transfer to give intermediate 6, which has two open coordination sites suitable for oxidative addition of a C–H bond. Very interestingly, complex 4 is also active for catalyzing the hydrogenolysis of C–O bonds of arenols and aryl methyl ethers (Fig. 7).45 The unique reactivity and selectivity was proposed to proceed via a similar concerted transition state to that of ketone hydrogenation by the Shvo catalyst.
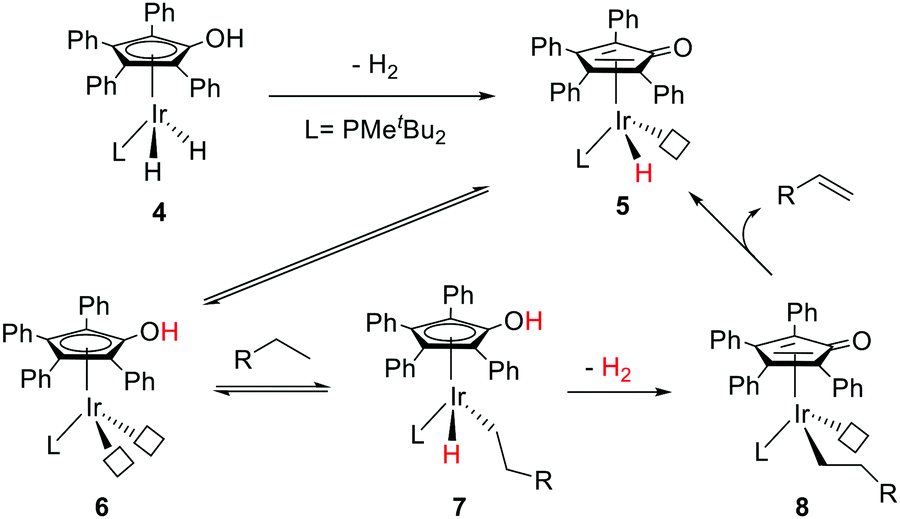 | ||
| Fig. 6 MLC assisted dehydrogenation of C–C single bonds catalyzed by the Ir analog (4) of the Knölker complex. | ||
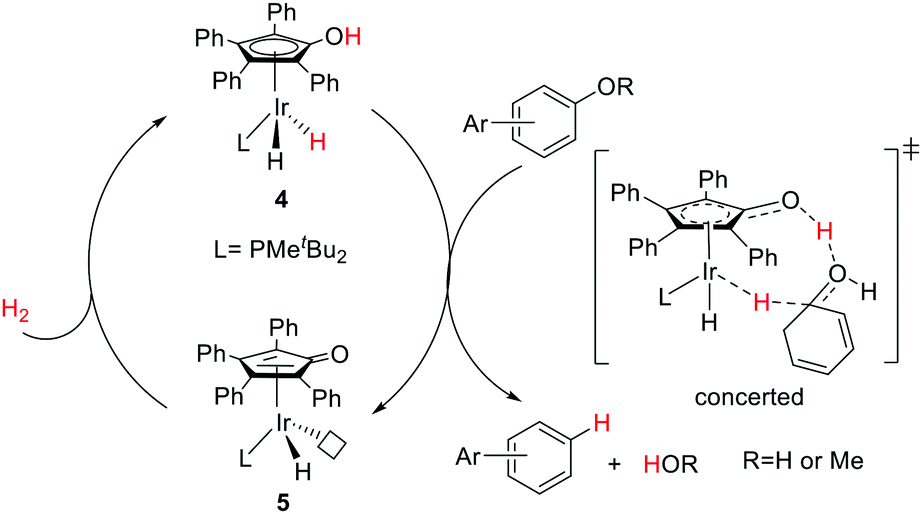 | ||
| Fig. 7 Proposed mechanism for the hydrogenolysis of arenols and aryl methyl ethers by 4. | ||
2.2 Bipyridine and related systems
Interconversion between hydroxypyridine derivatives and their pyridone tautomers can be mediated by the presence of acids or bases,46 and such changes can considerably alter the π-electron donating effects when serving as ligands in transition metal complexes.47 Himeda and co-workers first demonstrated the crucial roles of the –OH groups functionality in the 4,7-dihydroxy-1,10-phenanthroline moiety during catalysis. The superior catalytic aptitude was attributed to the formation of oxyanion species having strong electron donating ability and enhanced water solubility (Fig. 8a).48 The catalytic activities of a range of half-sandwich complexes bearing 6,6′-dihydroxy-2,2′-bipyridine or 4,4′-dihydroxy-2,2′-bipyridine backbone towards CO2 and bicarbonate hydrogenation were examined, and under basic conditions, the generated oxyanionic forms dramatically enhanced the catalytic efficiency.48–50 Detailed mechanistic studies revealed that the ortho –OH groups (6,6′-dihydroxy-2,2′-bipyridine) could act as an internal base to promote H2 activation via MLC (Fig. 8b).51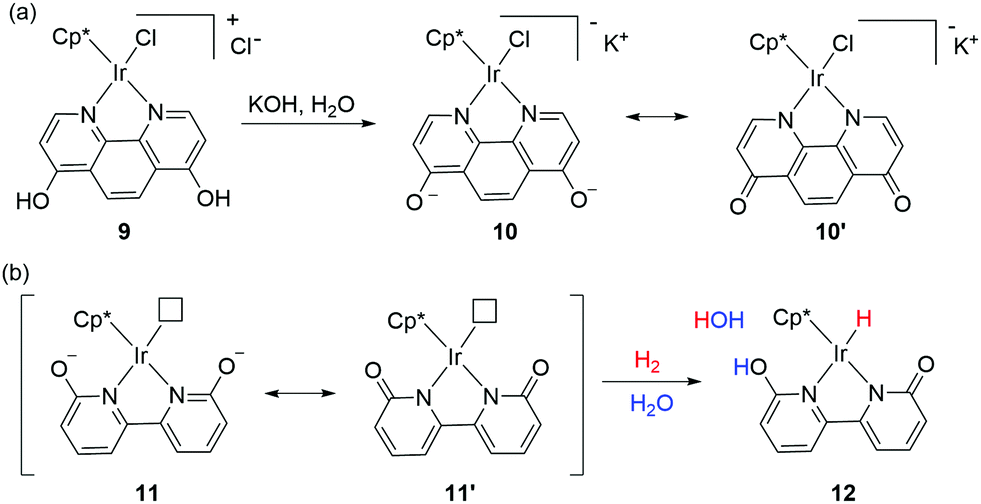 | ||
| Fig. 8 (a) Formation of oxyanionic species of the Ir complex with 4,7-dihydroxy-1,10-phenanthroline under basic conditions. (b) MLC and H2 activation by an Ir complex bearing a dihydroxy-2,2′-bipyridine ligand (Cp*: 1,2,3,4,5-pentamethylcyclopentadienyl). | ||
Pioneering work from Yamaguchi, Fujita, and co-workers also established the decisive role of the –OH group(s) during the dehydrogenative alcohol oxidation catalyzed by an Ir complex tethered to 6,6′-dihydroxy-2,2′-bipyridine.52,53 The pivotal role of ligand assisted alcohol activation involving aromatization–dearomatization was realized. In the proposed mechanism (Fig. 9), H2O is removed from the dicationic precatalyst (13) to generate a coordinatively unsaturated monocationic active catalytic species (14) followed by the alcohol activation to produce an alkoxo Ir intermediate (15). Subsequent elimination reaction results in the formation of the carbonyl product and a hydrido Ir intermediate (16) that releases H2 to complete the catalytic cycle.
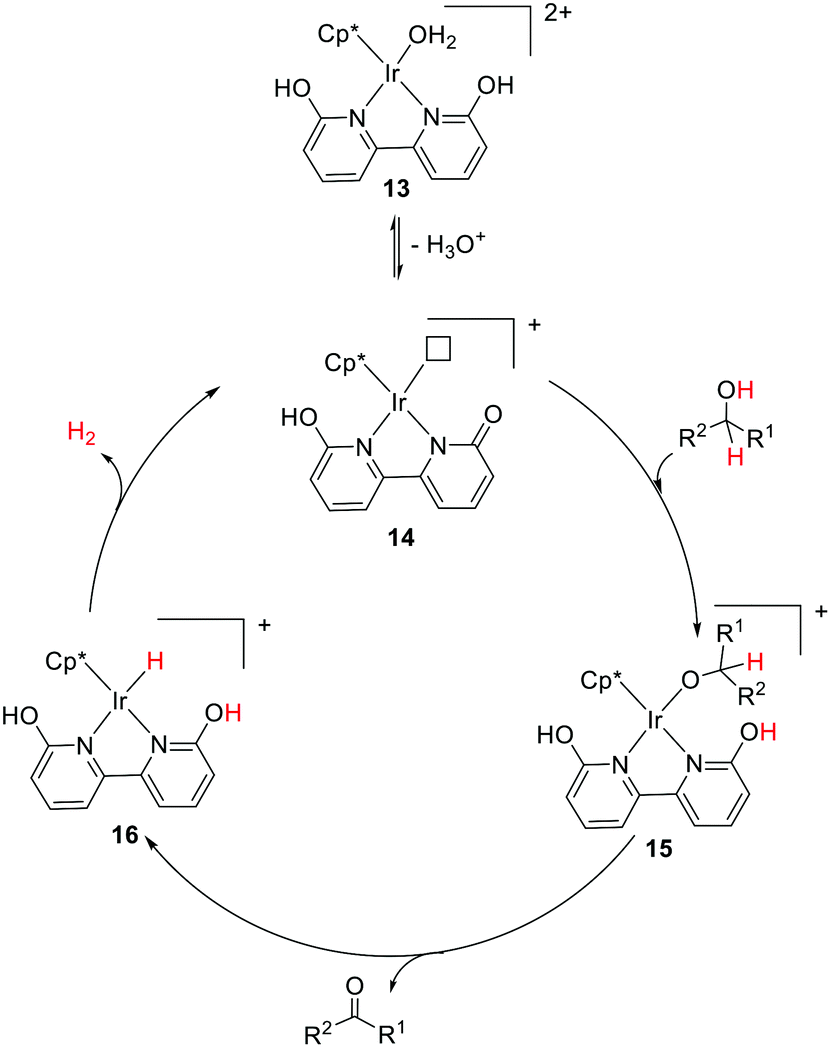 | ||
| Fig. 9 Proposed mechanistic pathway for alcohol dehydrogenation using the Cp*Ir complex bearing dihydroxy-2,2′-bipyridine. | ||
Another unique example of MLC was observed in Ru complex 17 possessing a 4,5-diazafluorenide ligand towards heterolytic H2 activation.54 The proton migrates to the 9th position of the central ring of 17 leading to dearomatization, and the hydride goes to the Ru center to generate 18 (Fig. 10). Controlled studies using D2 resulted in D incorporation into the 9th position of the ring and the formation of a Ru–D bond. Interestingly, D incorporation was also observed at the ortho-C–H of the PPh3 ligands, which was attributed to the concomitant reversible cyclometalation. Reaction of partially deuterated 17 with H2 led to protium re-incorporation in the ring, Ru and ortho-C of PPh3, further confirming the reversibility of the process.
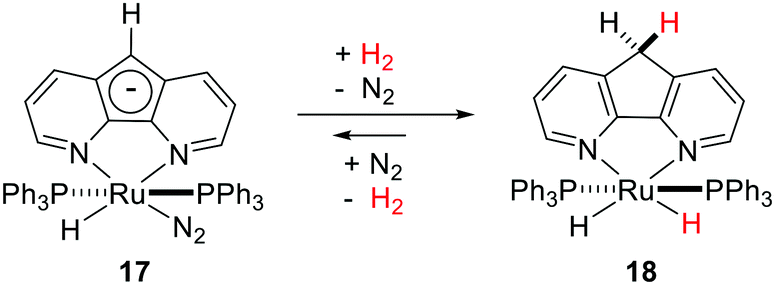 | ||
| Fig. 10 MLC and H2 activation by 4,5-diazafluorenide complex 17. | ||
The Milstein group reported a tetradentate phenanthroline-based bisphosphine ligand and the corresponding Ru complex (19).55 Upon treatment with a base, the resulting dearomatized species 20 could undergo unusual stepwise MLC aromatization/deconjugation processes in the presence of H2 to give complex 22 with trans-dihydride complex 21 as a possible intermediate (Fig. 11). Further reaction of 22 with H2 was not observed. Notably, hydrogenation of the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C double bond of the ligand backbone is reversible as complex 20 can be formed by heating 22 in an open system. Both 20 and 22 catalyze the dehydrogenative coupling of primary alcohols to esters efficiently, however, they show limited activity in the hydrogenation of esters.
C double bond of the ligand backbone is reversible as complex 20 can be formed by heating 22 in an open system. Both 20 and 22 catalyze the dehydrogenative coupling of primary alcohols to esters efficiently, however, they show limited activity in the hydrogenation of esters.
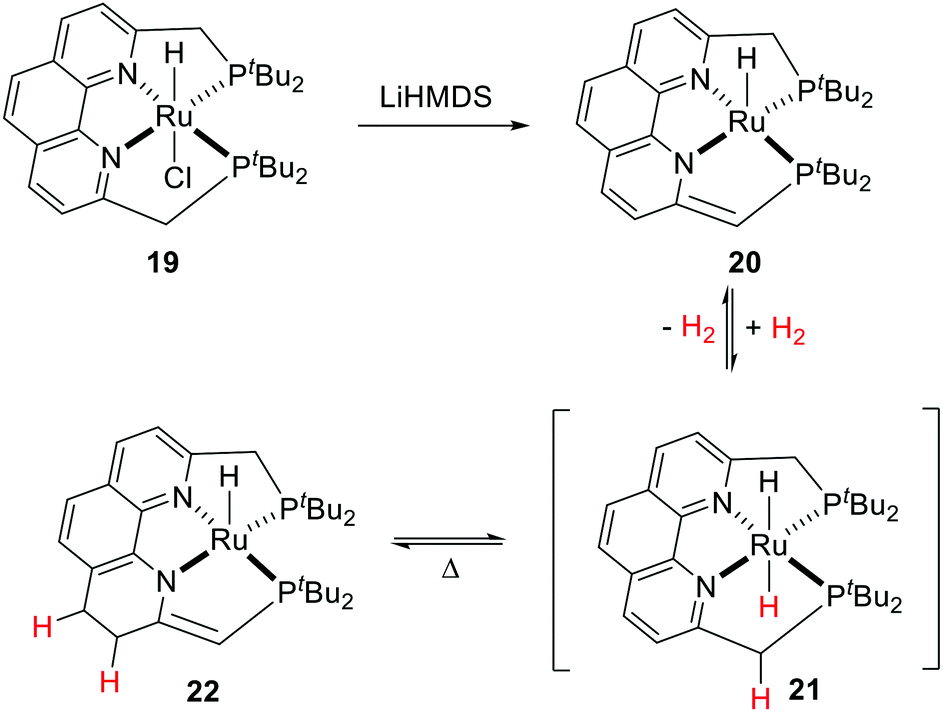 | ||
| Fig. 11 Reversible H2 activation by a Ru phenanthroline–bisphosphine complex (LiHMDS: lithium bis(trimethylsilyl)amide). | ||
2.3 Pyridine-based pincer systems
Pincer-type metal complexes have been extensively investigated since the pioneering reports on the coordination chemistry in the 1970s by the Nelson and Shaw groups.56,57 While the ligand reactivity of the pyridine-based pincer complex was first reported by Sacco in 1988,58 it was not until 2005 when Milstein utilized MLC with the dearomatization–rearomatization of the central pyridine to mediate a series of dehydrogenative coupling and hydrogenation reactions.26–28,59–63 The plausible mechanism was later supported and revised by Wang based on DFT (density functional theory) calculations (Fig. 12).64,65 The activation of the H–H σ bond by complex 23 was proposed to proceed via formal [2+2+2] addition (transition state 24) to the sp2 carbon of the CH arm and the Ru center, involving two π electrons of the CC bond, the electron lone pair of the dearomatized pyridine's nitrogen, and two σ electrons of H2. The aromaticity from the involvement of six electrons may stabilize the transition states to form the rearomatized trans-dihydride 25. Hydride transfer from 25 to a carbonyl substrate, such as methyl formate, can then occur to yield intermediate 26, which undergoes proton transfer from the CH2 moiety to the bound methoxymethoxide. The significant discoveries by the Milstein group encourage the development of synthetic applications of a series of pyridine-based transition metal pincer catalysts.25,27,66
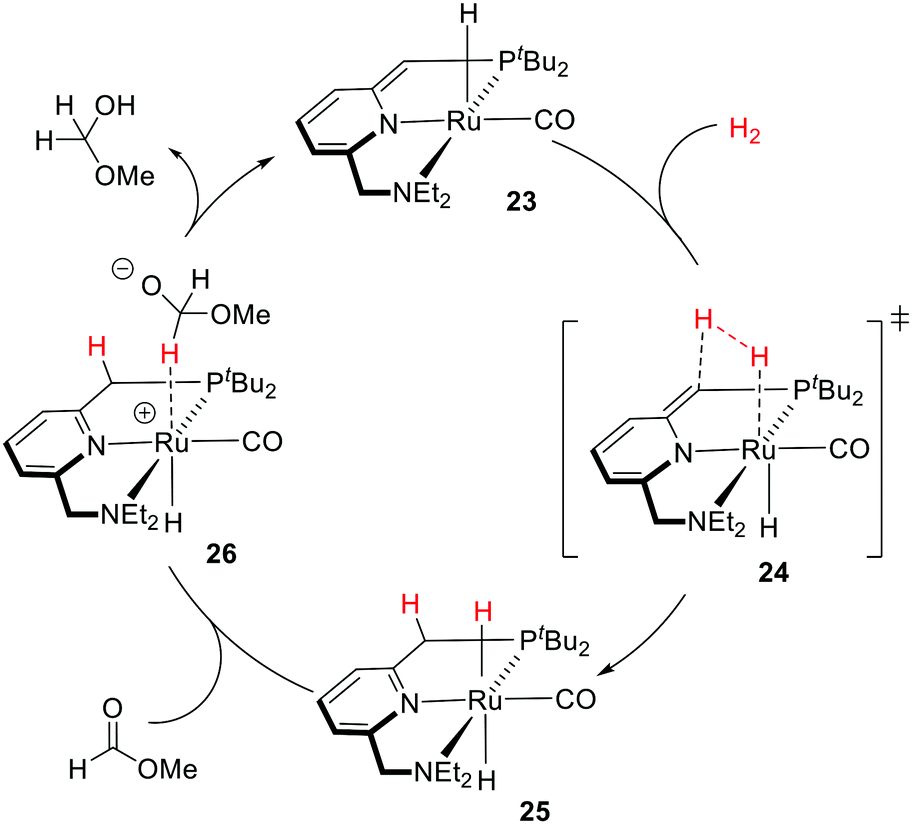 | ||
| Fig. 12 Computed plausible mechanism by Wang for the first step of methyl formate hydrogenation catalyzed by Milstein's catalyst 23. | ||
Motivated by Milstein's work and other studies on coordination chemistry,67–69 we were prompted to examine the effects by replacing the CH2 arm(s) with one or two NH groups in the conventional PNN and PNP systems because of the different acidity and bond strengths of the C–H and N–H bonds.31,32,70,71 Remarkably, in our PN3(P) complexes, distinct thermodynamic and kinetic properties were observed compared to those of CH2 analogs.30,72–74 For example, in the hydrogenation reaction, the MLC rearomatization from MA to MB or 23 to 25 is exergonic, but that of HA to HB is endergonic. Furthermore, proton transfer shuttles (e.g. water or alcohol) play a crucial role in mediating the H2 activation and transfer processes in the Huang system. A two-water shuttle is needed to convert HA and H2 to HB, through transition state 27, while the hydrogen transfer steps from HB to carbonyl substrates (transition state 28) only require one-water shuttle (Fig. 13). Indeed, diverse catalytic activities were identified across different transition metal complexes on the Milstein-type 2,6-lutidine-based PNN/PCN and Huang's PN3(P) platforms.70 All these observations suggest that a closer look at the effects of aromaticity to understand energetics between various reaction pathways is necessary.
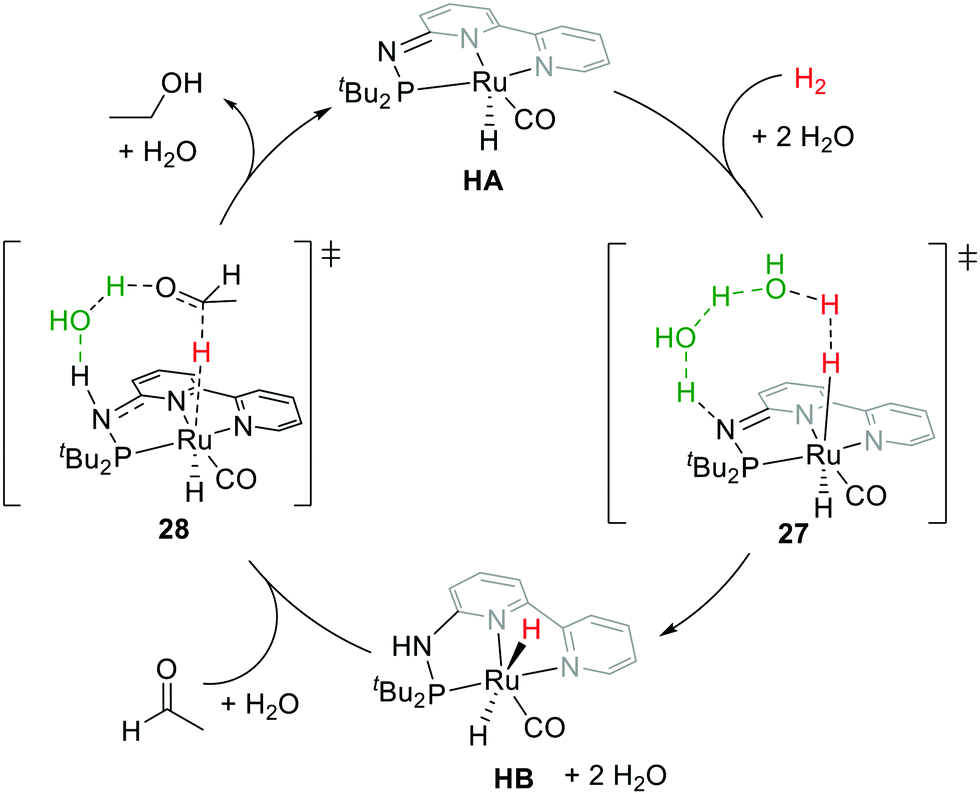 | ||
| Fig. 13 Computed plausible mechanism of acetaldehyde hydrogenation catalyzed by Huang's catalyst HA. | ||
3. Aromaticity
3.1 Historical foundation of aromaticity and computational methods
Our modern understanding of bonding has a long history, and through evolution of models many concepts have risen.75 Before insolation of benzene by Faraday in 1825,76 some compounds were already described to have an![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) “aromatic”smell. Kekulé in 1865 suggested the cyclohexatrienebenzenestructure, which is considered to be the originof the structuralbasisof aromaticity.77 One year later, Erlenmeyer noticed that in aromatic structures substitution was more favorable than addition, providing evidence of the connection between aromaticity and reactivity for the first time.78 Remarkably, Hückel theoretically explained unusual aromatic stability by the theoryofcyclic(4n + 2)πelectronsystems,79,80 and finally Pauling associated resonance and valencebonds with aromaticity.81 The experimental proof of additional aromatic stability was later provided by Kistiakowski to support Pauling's statement.82 Aromaticity was soon recognized as one of the most fundamental concepts in organic chemistry.83–89 As it is not the intention of this article to provide an in-depth review on aromaticity, interested readers are referred to the relevant literature.
“aromatic”smell. Kekulé in 1865 suggested the cyclohexatrienebenzenestructure, which is considered to be the originof the structuralbasisof aromaticity.77 One year later, Erlenmeyer noticed that in aromatic structures substitution was more favorable than addition, providing evidence of the connection between aromaticity and reactivity for the first time.78 Remarkably, Hückel theoretically explained unusual aromatic stability by the theoryofcyclic(4n + 2)πelectronsystems,79,80 and finally Pauling associated resonance and valencebonds with aromaticity.81 The experimental proof of additional aromatic stability was later provided by Kistiakowski to support Pauling's statement.82 Aromaticity was soon recognized as one of the most fundamental concepts in organic chemistry.83–89 As it is not the intention of this article to provide an in-depth review on aromaticity, interested readers are referred to the relevant literature.
While a precise definition has not yet been materialized, experimental scientists and theoreticians continue to develop and explore various tools in an effort to characterize and quantify aromaticity. Although numerous descriptors have been suggested,85 unfortunately, there is no perfect option that can be used as the general quantitative measure.90,91 As aromaticity is connected to induced ring currents, magnetic properties are important for its evaluation and detection.85 The Harmonic Oscillator Model of Aromaticity (HOMA) is a simple and successful tool for aromaticity measuring only based on the geometry,92 but Nucleus Independent Chemical Shifts (NICS) is one of the most widely used for investigating aromaticity based on the magnetic criteria.85,93,94 The negative NICS value of a ring designates the presence of the induced diatropic ring current, which is linked with aromaticity. The positive value indicates the paratropic ring current which is comprehended as antiaromaticity.95 While it is well known that sometimes NICS suffers from inferior results, it is normally accepted despite doubtful views.88 Compared with other indices of aromaticity probes localized in the geometrical center of the ring, (NICS(0)iso) may misalign the results.95 Placing the NICS probe 1 Å above/below the ring, designated as NICS(1)iso, was found to be a better indicator of the ring current.96,97 Further improvement was observed when out-of-plane zz components of the NICS tensor was employed, designated as NICS(1)zz, showing excellent correlation with the other aromaticity indices.94,97 It is also convenient to describe aromaticity with the Aromatic Stabilization Energy (ASE)98 that can be calculated as a difference of the strength of (hyper)conjugation between the cyclic molecule and the acyclic reference compound via an energy decomposition analysis (EDA) approach.99,100 Additionally, the Valence Bond Self-Consistent Field (VBSCF) method can also be useful in such calculations.101
3.2 Aromaticity and reactivities
In nature, the most remarkable reaction involving dearomatization–rearomatization is the NADH/NAD+ redox pair. It operates highly effectively reducing biologically important carbonyls compounds (Fig. 14A).102 In such a system, the 4H-pyridine moiety releases a hydride (H+ and 2e−) to gain aromaticity. Due to the thermodynamics of the biological system, the process is reversible and a dearomatized state can be restored. To illustrate the role of aromaticity in the NAD+/NADH redox system, Mucsi et al.102 proposed an analogy to the lift elevators (Fig. 14B). The resonance enthalpy is stored in the ring, it acts as a “counterweight” and has the ability to balance changes in bonding or other properties. With the aid of a counterweight, elevator lift motors achieve work with less effort, similar to aromatization which facilitates the release of the hydride in NADH. Due to the stabilization energy of the delocalized electrons in the aromatic ring, aromatic compounds are in general stable, thus remain more chemically resistant in comparison to their aliphatic or olefinic counterparts. However, in some particular cases with electron-rich aromatic compounds, the dearomatization process can be facilitated.103–105,132 It was recently demonstrated that the enantioselective dearomatization of the indole ring through a phosphinecatalyzed [3+2] annulation reaction was significantly influenced by the aromatic character of the indole.106 The computational studies have shown that the aromaticity of the indole ring was modified by the substituents on the pyrrole ring. Notably, the aromaticity index of the indole ring, including NICS(1)zz and corresponding ASE obtained via Schleyer's protocol,94 correlated with the yields of the reaction very well. Thus, using less aromatic pyrrole rings gave a higher reaction output.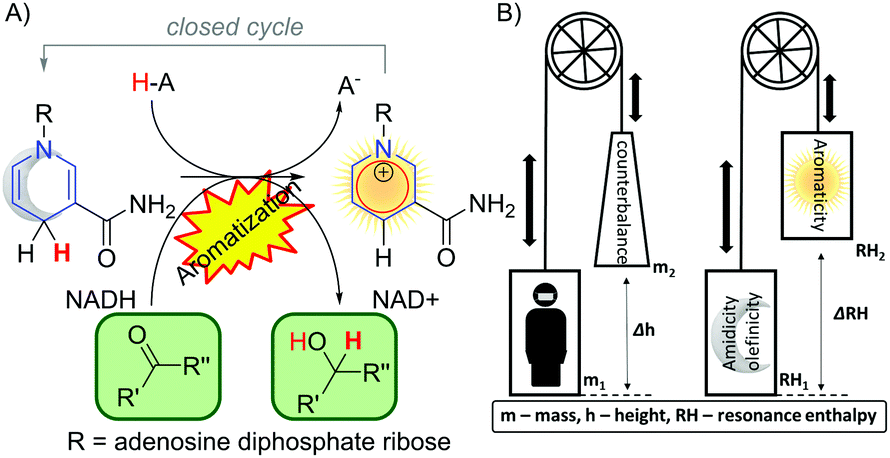 | ||
| Fig. 14 (A) Nicotinamide Adenine Dinucleotide (NAD) as an example of the dearomatization–rearomatization reaction in biological systems. (B) Pictorial demonstration of energy balancing and storage for a lift which associates with energy storage achieved by aromaticity as proposed by Mucsi et al. | ||
The aromaticity study by the Wu group demonstrated the influences of the hydrogen bonds on the enhancement and reduction of the aromaticity in pyridine based rings.107–109 Those hydrogen-bonded systems are important in biologically relevant processes and aromaticity was found to be one of the driving forces for the selectivity in base-pairing interactions.110 The aromaticity on small dimers with hydrogen bonding interactions increases the cyclic 4n + 2 π-electron delocalization (Fig. 15A) for (pseudo)dearomatized rings. However, hydrogen bonding interactions reduce aromaticity for aromatic sextets.
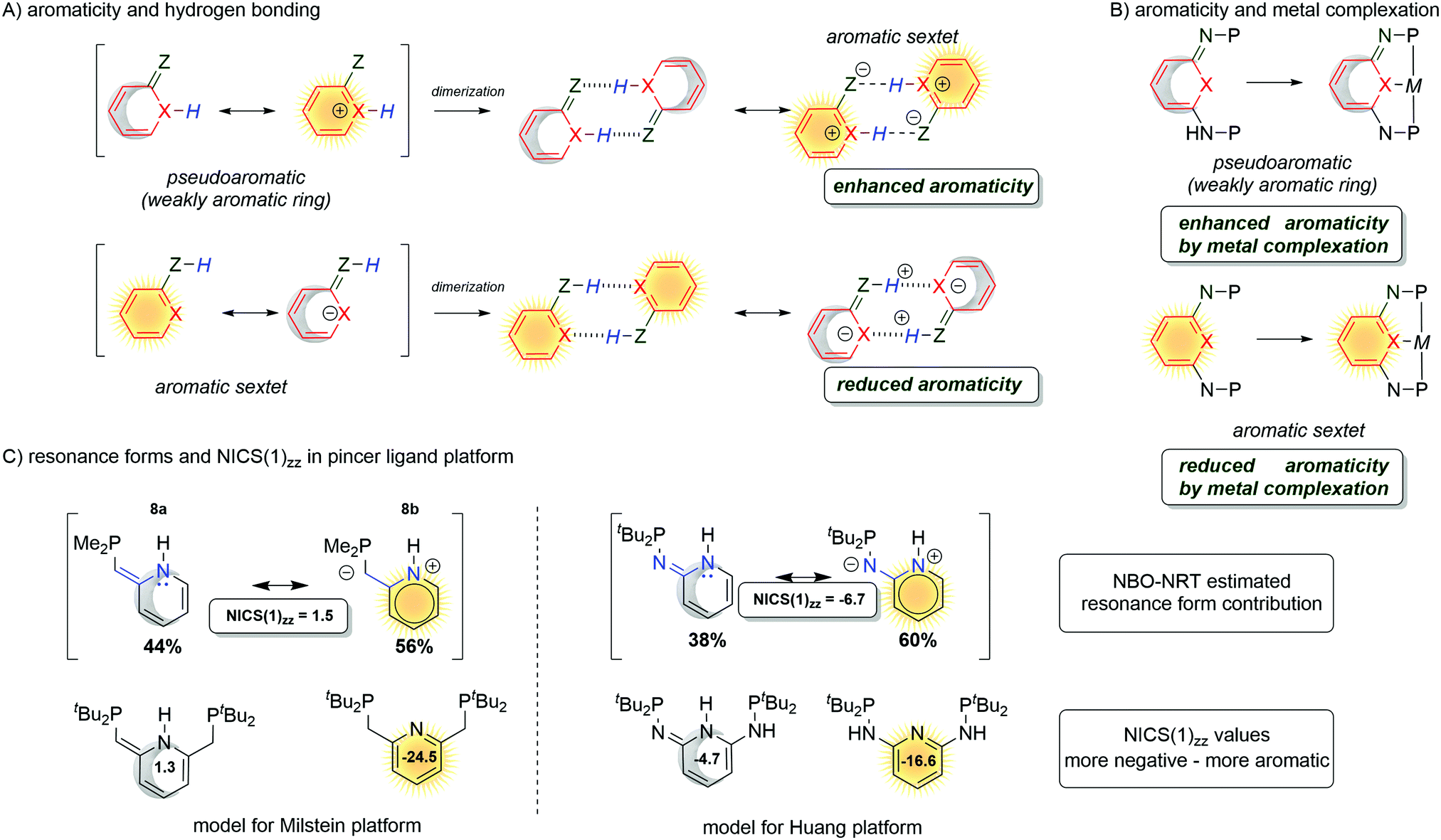 | ||
| Fig. 15 Aromaticity change in quinoidal/pyridine moiety influenced by hydrogen bonding (A), metal complexation (B) and CH/NH arms pyridine (C). | ||
3.3 The roles of aromaticity in metal ligand cooperation
The above-mentioned exemplary system by Wu and co-workers can serve as a prototypic model for understanding key interactions between the metal and ligands in pincer complexes reacting via aromatization/dearomatization.111 The enhancement and decrease of the aromaticity are also observed when a metal is coordinated to the pyridine-based ligand. Consequently, the resulting complex is expected to enhance the aromaticity when the (dearomatized) pyridine ring is non-aromatic ((pseudo)dearomatized), and decrease the aromaticity when the pyridine ring is aromatic (Fig. 15B).33 The unique character of this system origins in double bond conjugation which allows the presence of resonance with aromatic zwitterionic forms. Although being always present in aromatic mesomeric structures, their intensity depends on the structure. Remarkably, the presence of nitrogen linkers in the Huang platform shows NICS(1)zz values more negative (more aromatic) for (pseudo)dearomatized forms than for the Milstein platform model (Fig. 15C).33As we just showed, the pyridine base ligands provide access to many catalytic hydrogenation reactions, and the aromatization/dearomatization catalyst design concepts can be readily practiced on this platform. As discovered by Milstein in 2005,26 a plausible mechanism states that the catalyst becomes aromatic as a result of accepting H2 from desaturation of the substrate (Fig. 16). In the next step, the release of H2 regenerates the initial dearomatized form of the catalyst, therefore closing the cycle. The cycle can follow opposite directions if the right conditions are used.112 Thus, the overall reaction can be seen as the composition of the events related to the bond breaking/forming, aromatization, and hydrogen evolution steps. In the key step of the catalytic cycle, the activation of the substrate (C–H and X–H) is dictated by the energetic balance of bonds being formed/broken (M–H and Z–H) and the gain of the aromaticity. Here, it is worth mentioning that in many dearomatized pyridine structures an aromatic resonance form is present.113,114 Thus the (pseudo)dearomatized concept applies for those molecules with weak aromatic stabilization energy. For historic reasons, herein, we chose the symbol of the sun (sunflower)85 to represent aromatic rings and the moon as the counterpart to illustrate (pseudo)dearomatized (nonaromatic) rings. Prior to our work, there were limited known applications utilizing and discussing aromaticity descriptors in the field of catalysis. The dearomatization–rearomatization process is a key factor to understand the MLC reactivities (Fig. 17). Even though many factors influence the reactivity of the Shvo, Milstein, and Huang systems, the ring nature is the key to understand the thermodynamics differences. Using H2 activation as a model reaction, the Shvo and PNx(P) platforms become aromatized while accepting H2. Simultaneously, the double bond migrates forming an aromatic ring and the interaction between the metal and ligand becomes weaker. Although reactive sides are not part of the pyridine or cyclopentadienyl moiety, aromatization of them contributes to the reaction energy, providing an additional driving force to either promote or inhibit the reactivity. Consequently, the extra electronic stabilization energy arising from the ASE of the ligand has potential in modifying or tuning the reaction energetics.
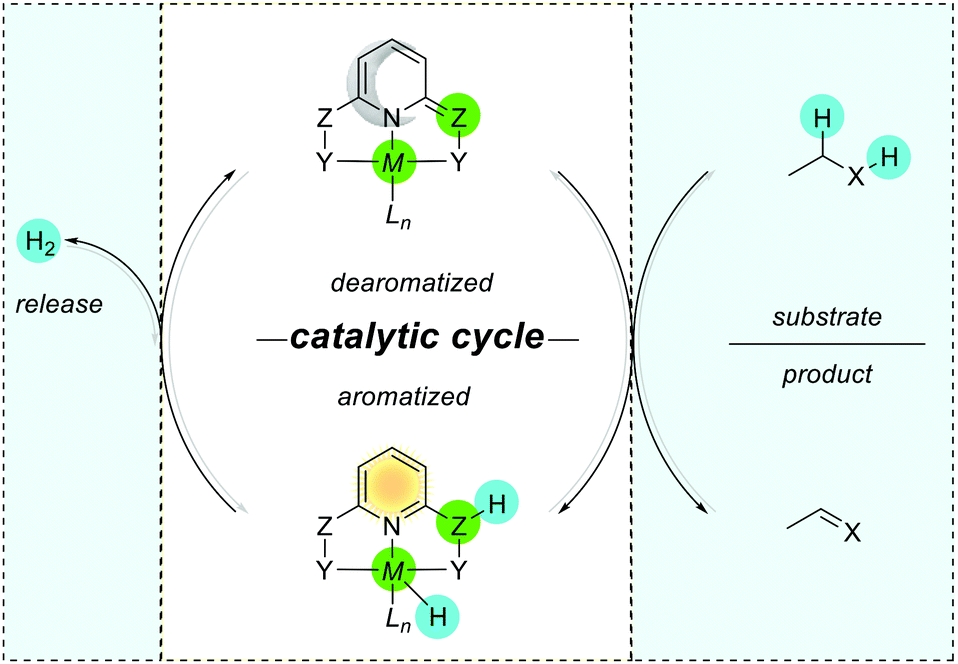 | ||
| Fig. 16 Schematic representation of the MLC aromatization/dearomatization catalytic cycle in pincer complexes. Z = C for Milstein and Z = C for the Huang system (Y = PR2; ex. tBu, iPr or any alkyl/aryl group, Z = C for Milstein and Z = C for the Huang system). | ||
 | ||
| Fig. 17 Reactivity of the Shvo, Milstein, and Huang catalyst systems via the aromatization/dearomatization process. | ||
Hence, tuning bond energies and aromatization may influence the chemical output. The aromaticity criteria, as described in the previous paragraph, are still based on the qualitative interpretation. For the study on the ligand aromaticity with small structural changes, the NICS(1)zz descriptor was found to be appropriate for such estimation. Similarly, the bond dissociation energies can be estimated or calculated with reasonable precision with experiments or computational calculations. For example the H2N–H bond is stronger than H3C–H115 by 3.3/1.8 kcal mol−1 (experiments/calculated) and the ArNH–H bond is stronger than ArCH2–H by 2.3/1.6 kcal mol−1 (experiments/calculated).115,116 As strictly different kinetic and thermodynamic properties were observed on the PN3(P) pincer complexes compared to those of their CH2 PNN/PNP counterparts by Milstein, based on our previous studies on aromaticity,117–120 we were prompted to evaluate the potential roles of aromaticity in the MLC processes. Since the N–H bond is stronger than C–H, one might envision hydrogenation should be more thermodynamically favored for the C-arm rather than the N-arm (Fig. 17, Z = C or N). Surprisingly, the trend for the pincer platform is opposite and the “rearomatization” process for the Huang system is endergonic whereas that for the Milstein system is exergonic with around 10 kcal of the spacer effect (Fig. 18A).
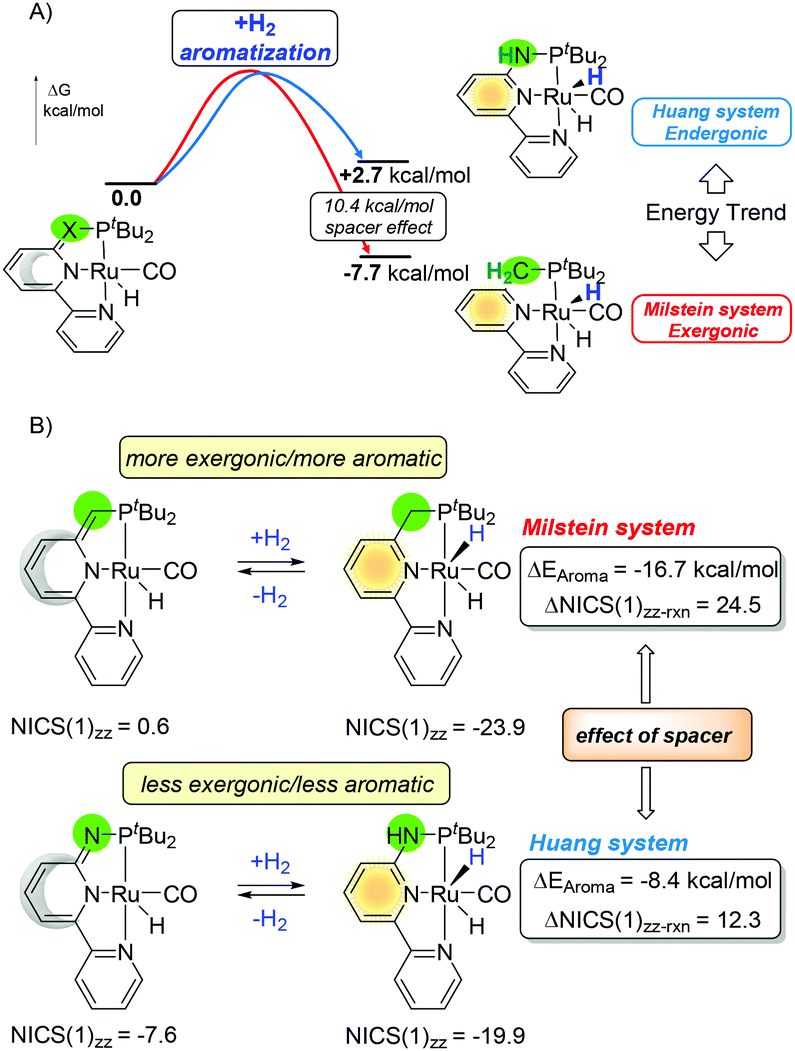 | ||
| Fig. 18 Effects of the arms on the (A) thermodynamics of the reaction and (B) NICS(1)zz values and estimated aromatic stabilization energies ΔEaroma for the hydrogenation reaction. | ||
In an effort to elucidate the origin of the distinct thermodynamic trends of the hydrogenation for the Milstein and the Huang systems, the difference in ASE (ΔEAroma) was estimated by the correlation with ΔNICS(1)zz results.33 Although the formation of new Ru–H and C/N–H bonds thermodynamically favors the H2 activation, the energy from aromatization of the central pyridine ring changes the outcome. While the formation of the stronger N–H bond delivers a larger reaction driving force than that of the C–H moiety, the reaction thermodynamics follows an opposite trend than expected and cannot be fully comprehended by only considering those chemical bonds. The main reason lies behind the aromatization of the central pyridine ring (Fig. 18B). The NICS(1)zz values of 0.6 for the Milstein and −7.6 for the Huang complexes, respectively, strongly suggest that the Huang platform has a much higher aromatic character than that of Milstein in the “dearomatized” form. Consequently, the ASE gained from the hydrogenation is larger in the Milstein system than that in the Huang platform. In the PNX(P) model, the hydrogenation thermodynamics follows the formation of a more aromatic ring rather than the generation of stronger chemical bonds.
In comparison to the Shvo system, the PNX(P) pincer platforms have greater room for modifications (Fig. 17 and 19) as they enable diversified designs for various donor atoms (Y and Y′), spacers (Z), other spectator ligands (L), etc. to fine-tune the steric and electronic properties of the resulting complexes. More importantly, the Huang system shows a considerable degree of aromaticity, in a “(pseudo)dearomatized” form. In such a flexible (pseudo)dearomatized frame, the metal center can act as a Lewis acid side and the unsaturated arm as a basic side. Thus as a result of resonance, the system expresses a highly intriguing ligand centered reactivity (Fig. 19, LCR), and can be triggered by the non-innocent character of the ligand and/or enabled by the zwitterionic resonance characters.121,122
 | ||
| Fig. 19 Two activation modes in catalysts influenced by the aromatization. Catalytic reduction of CO2 to methanol and methylamines via Ligand Centred Reactivity (LCR). | ||
For instance, the presented LCR leads to activation of Si–H and catalytic reduction of CO2 to methanol122 and methylamines,123 rather than insertion to the Ni–H bond observed in other pincer platforms (Fig. 19B).123
The N spacer of Huang's PN3P ligand platform possesses unique σ electron-donating properties and such ligand-specific reactivity is triggered by the aromaticity and metal ability to stabilize the positive charge. As we already discussed before, the aromatic zwitterionic structure plays a role in the (pseudo)dearomatized ring, and the σ electron-donating properties can be understood by mesomeric effects. Both zwitterionic aromatic forms and electron-donating abilities were investigated by Natural Bond Order analysis and Tolman electronic effects (Fig. 20A).124 The σ-donation of the nitrogen arm in the Ni–H complex was calculated to be stronger than those of nitrogen bases, such as DBU (1,8-diazabicyclo[5.4.0]undec-7-ene), DHIP (2,3-dihydroimidazo[1,2-a]pyridine), and TBD (1,5,7-triazabicyclo[4.4.0]dec-5-ene). The presence of the (pseudo)dearomatized ring increases the zwitterionic contribution from 8.2% → 21.5% in DBU to DHIP and 15.7% → 39.9% in TBD to PN3P*Ni–H which reflects charge distribution within the molecules. A similar trend is observed when the Tolman electronic effect is taken into account. In this approach, the A1 C–O vibrational mode from the ligand–Ni(CO)3 complex is employed as a relative measure of the electron donating or withdrawing ability of a ligand.124 Considering such vibration as the σ electron-donating quality, DHIP is a better electron donor than pyridine, DABCO and PN3P*Ni–H is the strongest, having the lowest frequency of 2060.8 cm−1 (Fig. 20B), clearly indicative of the strong donation ability of the iminic nitrogen. That theoretical findings explain the formation of the molecular structures with distinguishing nitrogen metal interaction (Fig. 20C)121,125 and formation of which can be rationalized by the charge distribution due to the mesomeric form ((pseudo)dearomatized).
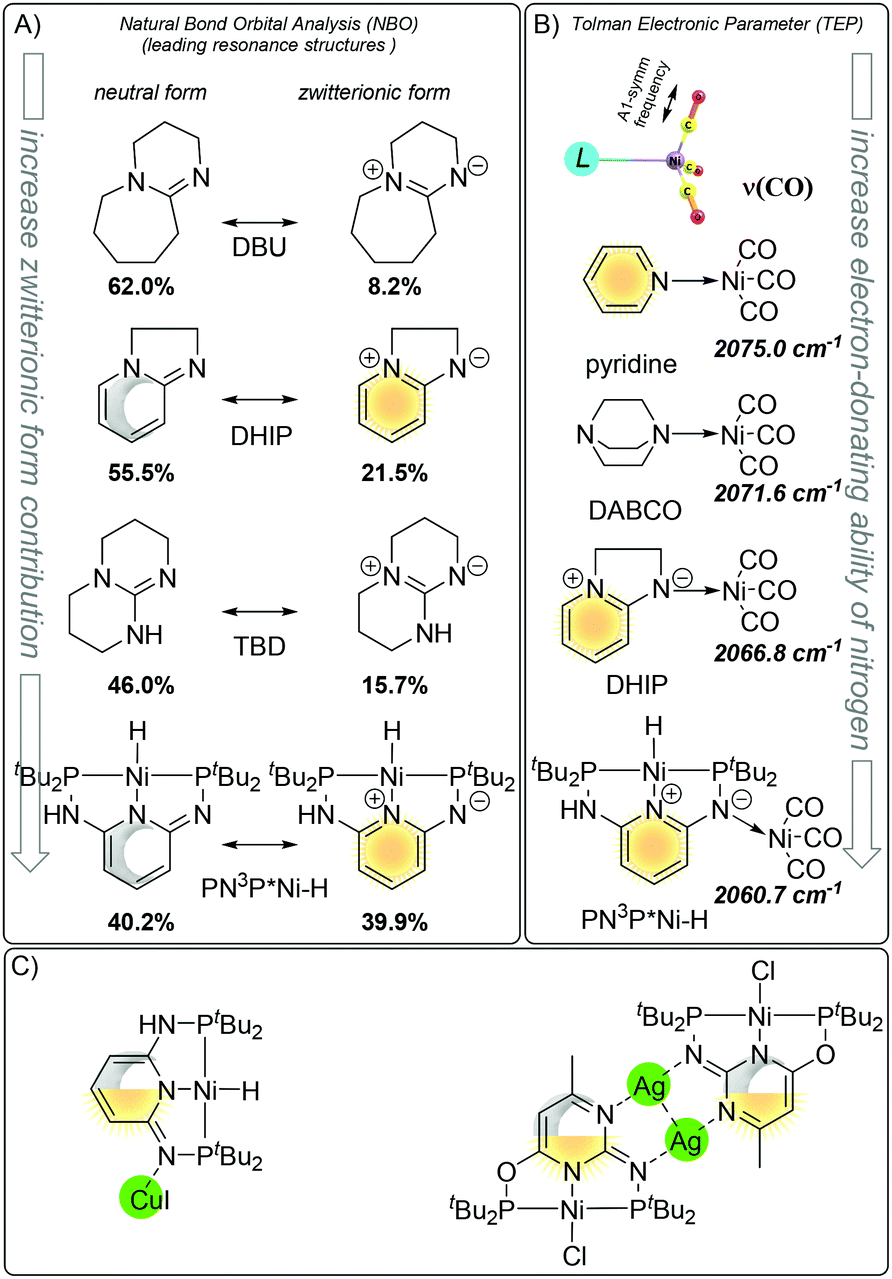 | ||
| Fig. 20 Nitrogen σ donation ability via resonances forms. (A) Aromatic zwitterionic resonance form contribution in nitrogen bases. (B) Tolman electronic parameter. (C) Pincer characterized by X-ray. | ||
The experimental126 and computational127 studies on triazine-based PN3PMn complexes also demonstrated the influence of the aromaticity on the thermodynamics (Fig. 21A). Along with deprotonation of the arm which impacts aromaticity of the central ring (NICS(1)zz values 0.17 → −8.53 and 1.08 → −4.43), the reaction energy for the dehydrogenation became less exergonic (−10.8 → −4.8 kcal mol−1). Similar to the PNxP system in Fig. 18, the reaction energies correlate with NICS(1)zz values, although the changes are not as significant (ΔNICS(1)zz values −8.70 and −5.51). Thus, the deprotonation of the arm leads to lower value aromatization (ΔNICS(1)zz) and increases the reaction energy. The experimental measured bond dissociation energy (BDE) of the Mn–H is stronger (60.9 ± 1.2 kcal mol−1) than Ru–H (53.3 ± 3.5 kcal mol−1).128 Hence, in contrast to the Milstein and Huang platforms, the hydrogenation in triazine-based PN3P-pincer complexes gains more energy from bond formation and less from the aromatization. If iridium metal is used instead, the hydrogenation become endergonic (Fig. 20B).74
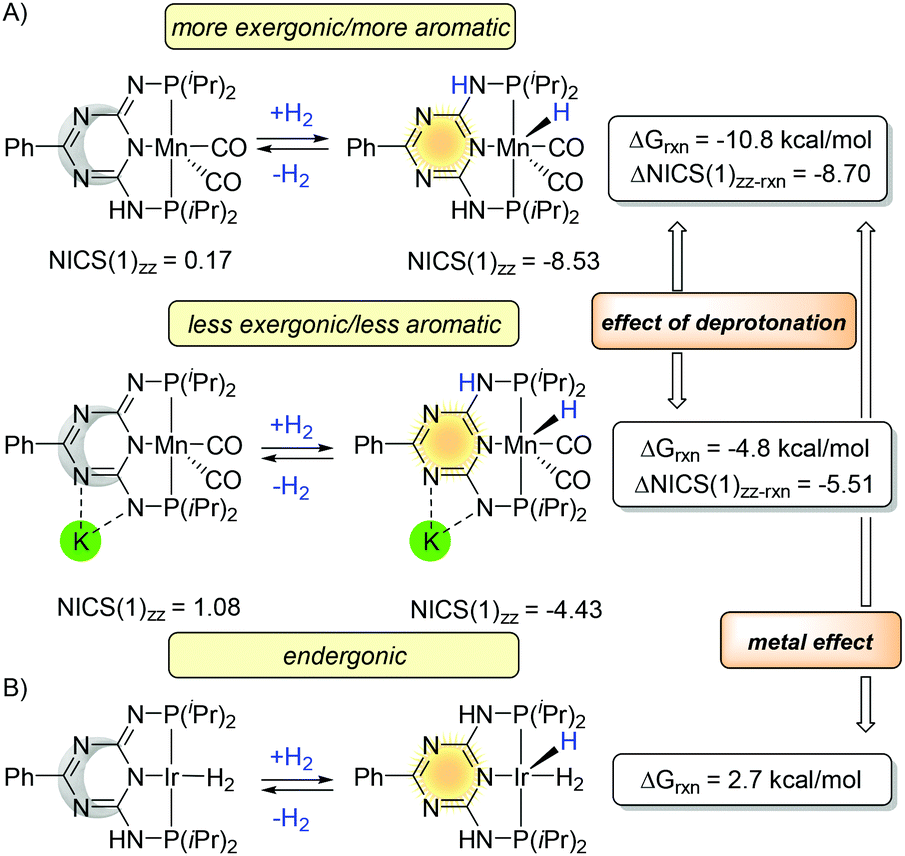 | ||
| Fig. 21 Trends in aromaticity and thermodynamics for the triazine-based PN3P-pincer catalysts. (A) Mn triazine-based PN3P-pincer complexes and (B) Ir triazine-based PN3P-pincer complexes. | ||
Similar trends are observed for the side reaction in catalytic processes. For example, addition of alcohol and formation of alkoxo complexes have remarkably different outcomes for Ru and Ir (Fig. 22).74 The Ru–O bond is stronger (126 ± 10 kcal mol−1) than Ir–O (99 ± 10 kcal mol−1), and the difference is larger than C–H/N–H bond energy, taking lead in the energy bond balance.128 Although these two platforms are different, on the basis of our previous discussion, one can conclude that Milstein system gains more energy from aromatization and more energy from bond formation (dominated by Ir/Ru–O) than those of trazine-based systems.
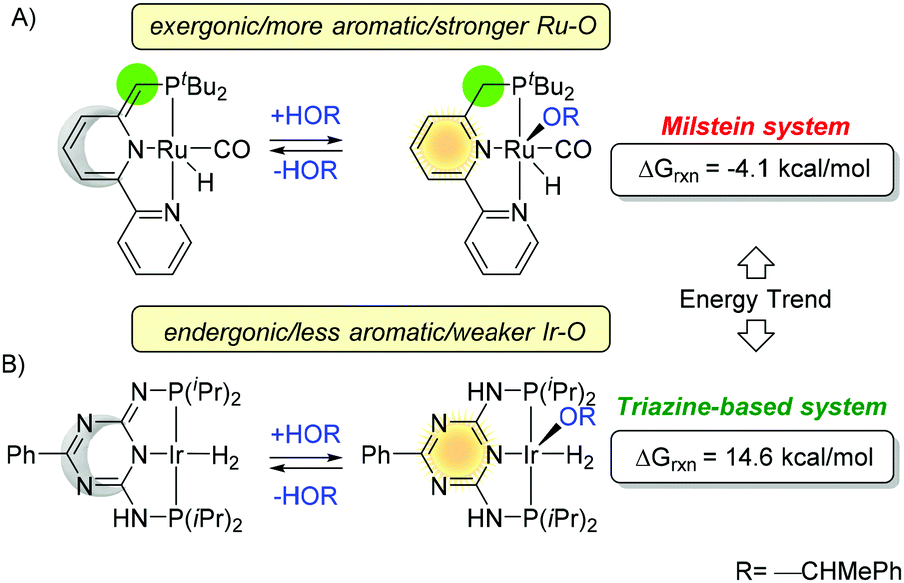 | ||
| Fig. 22 Energetic comparison of the formation of alkoxo complexes in catalytic side reactions. | ||
It is also expected that the arm with the N–H spacer will interact more strongly with solvents than a less polar C–H spacer. Nevertheless, there is only −0.77 kcal mol−1 energy difference between interaction of ammonia with benzene (H2N–H–π = −2.22 kcal mol−1), and methane and benzene (H3C–H/π = −1.45 kcal mol−1).129 Thus, in toluene, a difference in solvation of the NH and CH2 arm is expected to be less than 1 kcal mol−1. The H2N–H–THF interaction energy is much higher, 3.3 kcal mol−1, as expected from more polar solvents and hydrogen bonding interaction by Sahu et al.130 The solvent–solute interaction will also influence the aromaticity of the ring. The HOMA index of the aniline decreases slightly from 0.966 to 0.959 if water solvent as discovered by Zborowski et al.131 As a result, solvation effects will favor hydrogenation of the PN3P platform but at the same time decrease aromaticity of the ring will disfavor the reaction. Thus, balance between both these should be carefully addressed to make a fair comparison.
4. Perspectives
Metal–ligand cooperative catalysis and reactivities offer several unique features for activation and transformations of molecules. MLC processes involving aromatization–dearomatization steps represent a new paradigm for the design of transition metal catalysts with a number of unprecedented reactivities demonstrated, which may be challenging when the metal center or the ligand acts alone. Using the Milstein and the Huang pincer systems as examples, we highlighted the employment of aromaticity descriptors and demonstrated the correlation between the reaction aromatic stabilization energy (ΔEAroma) and ΔNICS(1)zz and the experimental observations. Although a deep fundamental understanding of aromaticity is a challenging task, it can be applied practically in, but not limited to, Shvo, Milstein, and Huang platforms. The aromatic stabilization energy contributes to the thermodynamics of the reaction and it should be taken into account in the mechanistic discussions. The bond formed/broken and aromatization/dearomatization phenomena reflect the reaction energy and are essential for understanding the catalytic cycles. Importantly, in addition to the bifunctional activities, the exceptional (pseudo)dearomatized character presents ligand centered reactivities that should open up a new direction for future catalyst development. As the field continues to grow and advance, practical applications are conceivable.Conflicts of interest
K.-W. H. declares the following competing financial interests: Huang is one of the inventors of US Patent # 8598351 “Phospho-amino pincer-type ligands and catalytic metal complexes thereof”, US Patent # 10300469 and CN Patent # 106413891 “Metal-ligand cooperative catalysis through NH arm deprotection/pyridine dearomatization for efficient hydrogen generation from formic acid”, and US Patent # 10399072 “Novel Imines With Tunable Nucleophilicity and Steric Properties Through Metal Coordination: Applications as Ligands and Metalloorganocatalysts”, all assigned to KAUST.Acknowledgements
Financial support and the service of Ibex, Shaheen 2 High Performance Computing Facilities were provided by King Abdullah University of Science and Technology (KAUST).Notes and references
- R. H. Crabtree, The Organometallic Chemistry of the Transition Metals, Wiley, 6th edn, 2014 Search PubMed.
- J. Hartwig, Organotransition Metal Chemistry: From Bonding to Catalysis, University Science Books, 1st edn, 2009 Search PubMed.
- W. N. Lipscomb and N. Sträter, Chem. Rev., 1996, 96, 2375–2434 CrossRef CAS PubMed.
- G. Parkin, Chem. Rev., 2004, 104, 699–768 CrossRef CAS PubMed.
- M. D. Wodrich and X. Hu, Nat. Rev. Chem., 2018, 2, 0099 CrossRef CAS.
- Y. Blum and Y. Shvo, Isr. J. Chem., 1984, 24, 144–148 CrossRef CAS.
- Y. Blum, D. Czarkie, Y. Rahamim and Y. Shvo, Organometallics, 1985, 4, 1459–1461 CrossRef CAS.
- Y. Shvo and D. Czarkie, J. Organomet. Chem., 1986, 315, C25–C28 CrossRef CAS.
- Y. Shvo, D. Czarkie, Y. Rahamim and D. F. Chodosh, J. Am. Chem. Soc., 1986, 108, 7400–7402 CrossRef CAS.
- S. Hashiguchi, A. Fujii, J. Takehara, T. Ikariya and R. Noyori, J. Am. Chem. Soc., 1995, 117, 7562–7563 CrossRef CAS.
- A. Fujii, S. Hashiguchi, N. Uematsu, T. Ikariya and R. Noyori, J. Am. Chem. Soc., 1996, 118, 2521–2522 CrossRef CAS.
- T. Ohkuma, H. Ooka, M. Yamakawa, T. Ikariya and R. Noyori, J. Org. Chem., 1996, 61, 4872–4873 CrossRef CAS.
- N. Uematsu, A. Fujii, S. Hashiguchi, T. Ikariya and R. Noyori, J. Am. Chem. Soc., 1996, 118, 4916–4917 CrossRef CAS.
- R. Noyori, M. Kitamura and T. Ohkuma, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 5356 CrossRef CAS PubMed.
- T. Ikariya and M. Shibasaki, Bifunctional Molecular Catalysis, Springer, Berlin, Heidelberg, 2011 Search PubMed.
- R. Noyori, M. Koizumi, D. Ishii and T. Ohkuma, Pure Appl. Chem., 2001, 73, 227–232 CAS.
- H. Grützmacher, Angew. Chem., Int. Ed., 2008, 47, 1814–1818 CrossRef PubMed.
- C. Gunanathan and D. Milstein, Top. Organomet. Chem., 2011, 37, 55–84 CrossRef CAS.
- B. Zhao, Z. Han and K. Ding, Angew. Chem., Int. Ed., 2013, 52, 4744–4788 CrossRef CAS PubMed.
- R. Noyori and T. Ohkuma, Angew. Chem., Int. Ed., 2001, 40, 40–73 CrossRef CAS PubMed.
- M. Kitamura and R. Noyori, Ruthenium in Organic Synthesis, Wiley-VCH Verlag GmbH & Co. KGaA, 2005, pp. 3–52 Search PubMed.
- T. Ohkuma, N. Utsumi, K. Tsutsumi, K. Murata, C. Sandoval and R. Noyori, J. Am. Chem. Soc., 2006, 128, 8724–8725 CrossRef CAS PubMed.
- T. Ohkuma, K. Tsutsumi, N. Utsumi, N. Arai, R. Noyori and K. Murata, Org. Lett., 2007, 9, 255–257 CrossRef CAS PubMed.
- H. G. Nedden, A. Zanotti-Gerosa and M. Wills, Chem. Rec., 2016, 16, 2623–2643 CrossRef PubMed.
- J. R. Khusnutdinova and D. Milstein, Angew. Chem., Int. Ed., 2015, 54, 12236–12273 CrossRef CAS PubMed.
- J. Zhang, G. Leitus, Y. Ben-David and D. Milstein, J. Am. Chem. Soc., 2005, 127, 10840–10841 CrossRef CAS PubMed.
- D. Milstein, Top. Catal., 2010, 53, 915–923 CrossRef CAS.
- C. Gunanathan and D. Milstein, Acc. Chem. Res., 2011, 44, 588–602 CrossRef CAS PubMed.
- T. Shimbayashi and K.-i. Fujita, Catalysts, 2020, 10, 635 CrossRef CAS.
- L.-P. He, T. Chen, D. Gong, Z. Lai and K.-W. Huang, Organometallics, 2012, 31, 5208–5211 CrossRef CAS.
- L.-P. He, T. Chen, D.-X. Xue, M. Eddaoudi and K.-W. Huang, J. Organomet. Chem., 2012, 700, 202–206 CrossRef CAS.
- K.-W. Huang, T. Chen, L. He, D. Gong, W. Jia and L. Yao, Phospho-amino pincer-type ligands and catalytic metal complexes thereof, US Pat., US 8598351 B2, 2013 Search PubMed.
- T. P. Gonçalves and K.-W. Huang, J. Am. Chem. Soc., 2017, 139, 13442–13449 CrossRef PubMed.
- R. Karvembu, R. Prabhakaran and K. Natarajan, Coord. Chem. Rev., 2005, 249, 911–918 CrossRef CAS.
- J. S. M. Samec, J.-E. Bäckvall, P. G. Andersson and P. Brandt, Chem. Soc. Rev., 2006, 35, 237–248 RSC.
- B. L. Conley, M. K. Pennington-Boggio, E. Boz and T. J. Williams, Chem. Rev., 2010, 110, 2294–2312 CrossRef CAS PubMed.
- C. P. Casey, S. W. Singer, D. R. Powell, R. K. Hayashi and M. Kavana, J. Am. Chem. Soc., 2001, 123, 1090–1100 CrossRef CAS PubMed.
- C. P. Casey and J. B. Johnson, Can. J. Chem., 2005, 83, 1339–1346 CrossRef CAS.
- J. B. Johnson and J.-E. Bäckvall, J. Org. Chem., 2003, 68, 7681–7684 CrossRef CAS PubMed.
- C. P. Casey and H. Guan, J. Am. Chem. Soc., 2007, 129, 5816–5817 CrossRef CAS PubMed.
- H. J. Knölker, E. Baum, H. Goesmann and R. Klauss, Angew. Chem., Int. Ed., 1999, 38, 2064–2066 CrossRef.
- T. Liu, X. Wang, C. Hoffmann, D. L. DuBois and R. M. Bullock, Angew. Chem., Int. Ed., 2014, 53, 5300–5304 CrossRef CAS PubMed.
- X. Lu, Y. Zhang, P. Yun, M. Zhang and T. Li, Org. Biomol. Chem., 2013, 11, 5264–5277 RSC.
- S. Kusumoto, M. Akiyama and K. Nozaki, J. Am. Chem. Soc., 2013, 135, 18726–18729 CrossRef CAS PubMed.
- S. Kusumoto and K. Nozaki, Nat. Commun., 2015, 6, 6296 CrossRef PubMed.
- C. Boga, A. C. Bonamartini, L. Forlani, V. Modarelli, L. Righi, P. Sgarabotto and P. E. Todesco, Eur. J. Org. Chem., 2001, 1175–1182 CrossRef CAS.
- P. J. Giordano, C. R. Bock and M. S. Wrighton, J. Am. Chem. Soc., 1978, 100, 6960–6965 CrossRef CAS.
- Y. Himeda, N. Onozawa-Komatsuzaki, H. Sugihara, H. Arakawa and K. Kasuga, Organometallics, 2004, 23, 1480–1483 CrossRef CAS.
- Y. Himeda, N. Onozawa-Komatsuzaki, H. Sugihara and K. Kasuga, J. Am. Chem. Soc., 2005, 127, 13118–13119 CrossRef CAS PubMed.
- Y. M. Badiei, W.-H. Wang, J. F. Hull, D. J. Szalda, J. T. Muckerman, Y. Himeda and E. Fujita, Inorg. Chem., 2013, 52, 12576–12586 CrossRef CAS PubMed.
- W.-H. Wang, J. T. Muckerman, E. Fujita and Y. Himeda, ACS Catal., 2013, 3, 856–860 CrossRef CAS.
- K.-i. Fujita, N. Tanino and R. Yamaguchi, Org. Lett., 2007, 9, 109–111 CrossRef CAS PubMed.
- R. Kawahara, K.-i. Fujita and R. Yamaguchi, J. Am. Chem. Soc., 2012, 134, 3643–3646 CrossRef CAS PubMed.
- E. Stepowska, H. Jiang and D. Song, Chem. Commun., 2010, 46, 556–558 RSC.
- R. Langer, I. Fuchs, M. Vogt, E. Balaraman, Y. Diskin-Posner, L. J. W. Shimon, Y. Ben-David and D. Milstein, Chem. – Eur. J., 2013, 19, 3407–3414 CrossRef CAS PubMed.
- W. S. J. Kelly, G. H. Ford and S. M. Nelson, J. Chem. Soc. A, 1971, 388–396 RSC.
- C. J. Moulton and B. L. Shaw, J. Chem. Soc., Dalton Trans., 1976, 1020–1024 RSC.
- A. Sacco, G. Vasapollo, C. F. Nobile, A. Piergiovanni, M. A. Pellinghelli and M. Lanfranchi, J. Organomet. Chem., 1988, 356, 397–409 CrossRef CAS.
- J. Zhang, G. Leitus, Y. Ben-David and D. Milstein, Angew. Chem., Int. Ed., 2006, 45, 1113–1115 CrossRef CAS PubMed.
- C. Gunanathan, Y. Ben-David and D. Milstein, Science, 2007, 317, 790–792 CrossRef CAS PubMed.
- S. W. Kohl, L. Weiner, L. Schwartsburd, L. Konstantinovski, L. J. W. Shimon, Y. Ben-David, M. A. Iron and D. Milstein, Science, 2009, 324, 74–77 CrossRef CAS.
- E. Balaraman, B. Gnanaprakasam, L. J. W. Shimon and D. Milstein, J. Am. Chem. Soc., 2010, 132, 16756–16758 CrossRef CAS PubMed.
- E. Balaraman, C. Gunanathan, J. Zhang, L. J. W. Shimon and D. Milstein, Nat. Chem., 2011, 3, 609 CrossRef CAS.
- H. Li, X. Wang, F. Huang, G. Lu, J. Jiang and Z.-X. Wang, Organometallics, 2011, 30, 5233–5247 CrossRef CAS.
- H. Li, M. Wen and Z.-X. Wang, Inorg. Chem., 2012, 51, 5716–5727 CrossRef CAS PubMed.
- C. Gunanathan and D. Milstein, Chem. Rev., 2014, 114, 12024–12087 CrossRef CAS.
- W. Schirmer, U. Flörke and H.-J. Haupt, Z. Anorg. Allg. Chem., 1987, 545, 83–97 CrossRef CAS.
- D. Benito-Garagorri, E. Becker, J. Wiedermann, W. Lackner, M. Pollak, K. Mereiter, J. Kisala and K. Kirchner, Organometallics, 2006, 25, 1900–1913 CrossRef CAS.
- H. Li, B. Zheng and K.-W. Huang, Coord. Chem. Rev., 2015, 293–294, 116–138 CrossRef CAS and references therein.
- H. Li, T. o. P. Gonçalves, D. Lupp and K.-W. Huang, ACS Catal., 2019, 9, 1619–1629 CrossRef CAS.
- T. Chen, L.-P. He, D. Gong, L. Yang, X. Miao, J. Eppinger and K.-W. Huang, Tetrahedron Lett., 2012, 53, 4409–4412 CrossRef CAS.
- G. Zeng, T. Chen, L. He, I. Pinnau, Z. Lai and K.-W. Huang, Chem. – Eur. J., 2012, 18, 15940–15943 CrossRef CAS PubMed.
- T. Chen, H. Li, S. Qu, B. Zheng, L. He, Z. Lai, Z.-X. Wang and K.-W. Huang, Organometallics, 2014, 33, 4152–4155 CrossRef CAS.
- S. Qu, Y. Dang, C. Song, M. Wen, K.-W. Huang and Z.-X. Wang, J. Am. Chem. Soc., 2014, 136, 4974–4991 CrossRef CAS.
- E. C. H. Constable, Molecules, 2020, 25, 2623 CrossRef CAS PubMed.
- M. Faraday, Philos. Trans. R. Soc. London, 1825, 115, 440–466 CrossRef.
- F. A. Kekulé, Bull. Soc. Chim., 1865, 3, 98–110 Search PubMed.
- E. Erlenmeyer, Ann. Chem. Pharm., 1866, 137, 327–359 CrossRef.
- E. Hückel, Z. Phys., 1931, 70, 204–286 CrossRef.
- E. Hückel, Z. Phys., 1931, 72, 310–337 CrossRef.
- L. Pauling and J. Sherman, J. Chem. Phys., 1933, 1, 606–617 CrossRef CAS.
- G. B. Kistiakowsky, J. R. Ruhoff, H. A. Smith and W. E. Vaughan, J. Am. Chem. Soc., 1936, 58, 146–153 CrossRef CAS.
- J. A. N. F. Gomes and R. B. Mallion, Chem. Rev., 2001, 101, 1349–1384 CrossRef CAS.
- T. M. Krygowski and M. K. Cyrański, Chem. Rev., 2001, 101, 1385–1420 CrossRef CAS PubMed.
- Z. Chen, C. S. Wannere, C. Corminboeuf, R. Puchta and P. v. R. Schleyer, Chem. Rev., 2005, 105, 3842–3888 CrossRef CAS PubMed.
- T. M. Krygowski, H. Szatylowicz, O. A. Stasyuk, J. Dominikowska and M. Palusiak, Chem. Rev., 2014, 114, 6383–6422 CrossRef CAS.
- F. Feixas, E. Matito, J. Poater and M. Solà, Chem. Soc. Rev., 2015, 44, 6434–6451 RSC.
- R. Gershoni-Poranne and A. Stanger, Chem. Soc. Rev., 2015, 44, 6597–6615 RSC.
- T. M. Krygowski and H. Szatylowicz, Chemtexts, 2015, 1, 12 CrossRef CAS PubMed.
- M. Solà, Front. Chem., 2017, 5, 22 Search PubMed.
- D. Yu, C. Rong, T. Lu, F. De Proft and S. Liu, ACS Omega, 2018, 3, 18370–18379 CrossRef CAS.
- J. Kruszewski and T. M. Krygowski, Tetrahedron Lett., 1972, 13, 3839–3842 CrossRef.
- P. v. R. Schleyer, C. Maerker, A. Dransfeld, H. Jiao and N. J. R. v. E. Hommes, J. Am. Chem. Soc., 1996, 118, 6317–6318 CrossRef CAS.
- H. Fallah-Bagher-Shaidaei, C. S. Wannere, C. Corminboeuf, R. Puchta and P. V. R. Schleyer, Org. Lett., 2006, 8, 863–866 CrossRef CAS PubMed.
- A. Stanger, J. Org. Chem., 2006, 71, 883–893 CrossRef CAS PubMed.
- P. v. R. Schleyer, M. Manoharan, Z.-X. Wang, B. Kiran, H. Jiao, R. Puchta and N. J. R. van Eikema Hommes, Org. Lett., 2001, 3, 2465–2468 CrossRef CAS PubMed.
- C. Corminboeuf, T. Heine, G. Seifert, P. v. R. Schleyer and J. Weber, Phys. Chem. Chem. Phys., 2004, 6, 273–276 RSC.
- M. K. Cyrañski, T. M. Krygowski, A. R. Katritzky and P. V. R. Schleyer, J. Org. Chem., 2002, 67, 1333–1338 CrossRef.
- I. Fernandez and G. Frenking, Faraday Discuss., 2007, 135, 403–421 RSC.
- I. Fernández and G. Frenking, Open Org. Chem. J., 2011, 5, 79–86 CrossRef.
- Z. Rashid, J. H. van Lenthe and R. W. A. Havenith, J. Phys. Chem. A, 2012, 116, 4778–4788 CrossRef CAS PubMed.
- Z. Mucsi, G. A. Chass and I. G. Csizmadia, J. Phys. Chem. B, 2009, 113, 10308–10314 CrossRef CAS PubMed.
- C.-X. Zhuo, C. Zheng and S.-L. You, Acc. Chem. Res., 2014, 47, 2558–2573 CrossRef CAS PubMed.
- W. C. Wertjes, E. H. Southgate and D. Sarlah, Chem. Soc. Rev., 2018, 47, 7996–8017 RSC.
- C. Zheng and S.-L. You, Chem, 2016, 1, 830–857 CAS.
- K. Li, T. P. Gonçalves, K.-W. Huang and Y. Lu, Angew. Chem., Int. Ed., 2019, 58, 5427–5431 CrossRef CAS.
- T. Kakeshpour, J. I. Wu and J. E. Jackson, J. Am. Chem. Soc., 2016, 138, 3427–3432 CrossRef CAS.
- C.-H. Wu, Y. Zhang, K. van Rickley and J. I. Wu, Chem. Commun., 2018, 54, 3512–3515 RSC.
- H. R. Paudel, R. Das, C.-H. Wu and J. I. Wu, Org. Biomol. Chem., 2020, 18, 1078–1081 RSC.
- Y. Zhang, C.-H. Wu and J. I. C. Wu, Org. Biomol. Chem., 2019, 17, 1881–1885 RSC.
- J. I. Wu, J. E. Jackson and P. V. R. Schleyer, J. Am. Chem. Soc., 2014, 136, 13526–13529 CrossRef CAS.
- H. Li, A. Al-Dakhil, D. Lupp, S. S. Gholap, Z. Lai, L.-C. Liang and K.-W. Huang, Org. Lett., 2018, 20, 6430–6435 CrossRef CAS PubMed.
- M. J. Cook, A. R. Katritzky, P. Linda and R. D. Tack, J. Chem. Soc. D, 1971, 510–511 RSC.
- M. J. Cook, A. R. Katritzky, P. Linda and R. D. Tack, J. Chem. Soc., Perkin Trans. 2, 1972, 1295–1301 RSC.
- G. A. DiLabio, D. A. Pratt, A. D. LoFaro and J. S. Wright, J. Phys. Chem. A, 1999, 103, 1653–1661 CrossRef CAS.
- S. W. Benson, J. Chem. Educ., 1965, 42, 502 CrossRef CAS.
- Z. Sun, K.-W. Huang and J. Wu, J. Am. Chem. Soc., 2011, 133, 11896–11899 CrossRef CAS.
- Y. Li, W.-K. Heng, B. S. Lee, N. Aratani, J. L. Zafra, N. Bao, R. Lee, Y. M. Sung, Z. Sun, K.-W. Huang, R. D. Webster, J. T. López Navarrete, D. Kim, A. Osuka, J. Casado, J. Ding and J. Wu, J. Am. Chem. Soc., 2012, 134, 14913–14922 CrossRef CAS.
- P. Hu, S. Lee, T. S. Herng, N. Aratani, T. P. Gonçalves, Q. Qi, X. Shi, H. Yamada, K.-W. Huang, J. Ding, D. Kim and J. Wu, J. Am. Chem. Soc., 2016, 138, 1065–1077 CrossRef CAS PubMed.
- W. Zeng, Z. Sun, T. S. Herng, T. P. Gonçalves, T. Y. Gopalakrishna, K. W. Huang, J. Ding and J. Wu, Angew. Chem., Int. Ed., 2016, 55, 8615–8619 CrossRef CAS.
- H. Li, T. P. Gonçalves, J. Hu, Q. Zhao, D. Gong, Z. Lai, Z. Wang, J. Zheng and K.-W. Huang, J. Org. Chem., 2018, 83, 14969–14977 CrossRef CAS PubMed.
- H. Li, T. P. Gonçalves, Q. Zhao, D. Gong, Z. Lai, Z. Wang, J. Zheng and K.-W. Huang, Chem. Commun., 2018, 54, 11395–11398 RSC.
- D. Oren, Y. Diskin-Posner, L. Avram, M. Feller and D. Milstein, Organometallics, 2018, 37, 2217–2221 CrossRef CAS PubMed.
- C. A. Tolman, Chem. Rev., 1977, 77, 313–348 CrossRef CAS.
- M.-H. Huang, T. P. Gonçalves and K.-W. Huang, J. Chin. Chem. Soc., 2019, 66, 455–458 CrossRef CAS.
- R. Fertig, T. Irrgang, F. Freitag, J. Zander and R. Kempe, ACS Catal., 2018, 8, 8525–8530 CrossRef CAS.
- D. Lupp and K.-W. Huang, Organometallics, 2020, 39, 18–24 CrossRef CAS.
- Y.-R. Luo and Y.-R. Luo, Comprehensive handbook of chemical bond energies, CRC Press, Boca Raton, 2007 Search PubMed.
- S. Tsuzuki, K. Honda, T. Uchimaru, M. Mikami and K. Tanabe, J. Am. Chem. Soc., 2000, 122, 11450–11458 CrossRef CAS.
- P. K. Sahu and S.-L. Lee, J. Chem. Phys., 2005, 123, 044308 CrossRef PubMed.
- K. K. Zborowski, H. Szatyłowicz and T. M. Krygowski, Struct. Chem., 2020, 31, 1717–1728 CrossRef CAS.
- F.-T. Sheng, J.-Y. Wang, W. Tan, Y.-C. Zhang and S. Feng, Progresses in organocatalytic asymmetric dearomatization reactions of indole derivatives, Org. Chem. Front., 2020, 7, 3967–3998 RSC.
Footnotes |
| † This work is dedicated to Professor Tien-Yau Luh on the occasion of his 75th birthday. |
| ‡ These two authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2021 |