
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceThe design of magneto-plasmonic nanostructures formed by magnetic Prussian Blue-type nanocrystals decorated with Au nanoparticles†
Roger
Sanchis-Gual
 a,
Isidora
Susic
a,
Ramón
Torres-Cavanillas
a,
Daniel
Arenas-Esteban
b,
Sara
Bals
b,
Talal
Mallah
c,
Marc
Coronado-Puchau
*a and
Eugenio
Coronado
*a
a,
Isidora
Susic
a,
Ramón
Torres-Cavanillas
a,
Daniel
Arenas-Esteban
b,
Sara
Bals
b,
Talal
Mallah
c,
Marc
Coronado-Puchau
*a and
Eugenio
Coronado
*a
aInstituto de Ciencia Molecular, Universitat de València, Catedrático José Beltran 2, 46980, Paterna, Spain. E-mail: Marc.Coronado@uv.es; Eugenio.Coronado@uv.es
bElectron Microscopy for Materials Science (EMAT) and NANOlab Center of Excellence, University of Antwerp, Groenenborgerlaan 171, 2020 Antwerp, Belgium
cInstitut de Chimie Moléculaire et des Matériaux d’Orsay, Université Paris-Saclay, CNRS, 91405 Orsay Cedex, France
First published on 18th January 2021
Abstract
We have developed a general protocol for the preparation of hybrid nanostructures formed by nanoparticles (NPs) of molecule-based magnets based on Prussian Blue Analogues (PBAs) decorated with plasmonic Au NPs of different shapes. By adjusting the pH, Au NPs can be attached preferentially along the edges of the PBA or randomly on the surface. The protocol allows tuning the plasmonic properties of the hybrids in the whole visible spectrum.
Magneto-plasmonic nanoparticles (NPs) are one of the emerging multifunctional materials in the fields of physics1 and nanomedicine.2 Still, most of the work in this topic has been focused on the design of core@shell NPs formed by an inorganic magnetic core (typically a metal oxide) and a gold plasmonic shell. An interesting alternative is replacing the magnetic inorganic core by a molecule-based magnet instead of a pure inorganic one since the former can provide some advantages in terms of chemical versatility, tunability of the properties and optical transparency.3 In spite of these features, this possibility has remained so far almost unexplored.
Thus, the only example of a hybrid molecular-plasmonic nanostructure is that made of a gold core and a shell of Prussian Blue (Au@PB), where PB refers to a bimetallic cyanide-bridged cubic network formulated as FeIII4[FeII(CN)6]3 (in short FeFe).4 Note that PB is the first member of an extensive family of coordination polymers of the general formula AaMb[M′(CN)6]c·nH2O (A = alkali cation, M and M′ = transition metal ions) known as Prussian Blue Analogues (PBAs), which have aroused strong interest in molecular magnetism,5,6 materials science7–9 and medicine.10 In magnetism this family is very appealing since it provides unique examples of high-Tc magnets11 and photomagnets.12 However, the chemical approach required to prepare the core@shell (Au@PB) nanostructures is quite limited since it is restricted to use as shell PB (FeFe), or the reduced form of the NiFe PBA derivative, only.13–16 These two compounds undergo long-range magnetic ordering at low temperatures only (Tc = 5.6 and 2 K, respectively). Hence, they are not very useful to investigate the enhanced magneto-optical (MO) properties that may arise in these systems as a result of the coupling of the magnetic and the plasmonic components.17,18 To overcome this limitation, other PBAs exhibiting higher Tc values need to be grown directly on the Au core. However, this still requires the insertion of a NiFe layer in between the Au and the PBA magnetic shell, which strongly limits the interaction between the magnetic layer and the plasmonic core and prevents the appearance of noticeable magneto-plasmonic effects.19 A second limitation of this procedure is that it is restricted to spherical Au NPs because anisotropic shapes (such as nanorods (NRs) or nanostars (NSs)) are too reactive towards cyanide. Still, these limitations have not been too problematic for using this kind of nanostructure in biomedicine (dual function NPs for imaging, photothermal therapy, or biosensing)20,21 and electrocatalysis.22,23
We propose here a simple and versatile approach to synthesize hybrid Au-PBA nanostructures formed by a PBA cubic NP surrounded by Au NPs of different shapes and connected through a bridging molecule. Thanks to the different plasmonic NPs, the protocol allows tuning the plasmon band position in the whole range of the visible spectrum.
The synthetic strategy is illustrated in Fig. 1 using NPs of the NiCr magnetic PBA derivative. These PBA NPs are colorless, negatively charged, stable in water in the absence of any coating agent and may have a Tc as large as 90 K in their bulk form.24 They are obtained by mixing the corresponding precursors (equimolar 10−3 M aqueous solutions of K3[Cr(CN)6] and NiCl2·(H2O)6) under vigorous stirring at room temperature. Cubic shape NPs of K0.07Ni[Cr(CN)6]0.69 were obtained with a mean size of 140 ± 40 nm (Fig. S2, ESI†) and a negative ζ-potential value of −37 ± 11 mV. This size provides NPs that are big enough to clearly observe the Au decoration while they exhibit high colloidal stability which facilitates the reaction. The first step consists of exchanging, in aqueous solution the citrate molecules that coat the Au NPs, by the heteroditopic thiol-polyethyleneglycol-amine (HS-PEG-NH2) ligand. In a second step, the as-obtained Au NPs are anchored to the PBA NPs. Such a decoration takes place due to: (i) the strong affinity of the thiol group to the metal NP25 and (ii) the strong interaction between the amine group and the negatively charged PBA NPs. It is worth noting that using the citrate-stabilized Au NPs, the formation of the final hybrid is not possible due to the electrostatic repulsion between the two negatively charged components (see Fig. S7, ESI†). That is why the first step requires a ligand exchange of the starting citrate-stabilized Au NPs of 12.4 ± 1.0 nm (see Fig. S3, ESI†)26 with HS-PEG-NH2. This ligand exchange was monitored by ζ-potential (see Table S1, ESI†) and by attenuated total reflectance Fourier-transform infrared (ATR-FTIR, Fig. S17, ESI†). The ζ-potential varies from a negative value, corresponding to the citrate capping, to a positive value, related to the HS-PEG-NH2. In addition, the appearance in the ATR-FTIR of bands corresponding to C–H and C–O–C vibration modes of the PEG molecule and the decrease of the intensity of the citrate bands indicates a major substitution of citrate by the heteroditopic ligand.
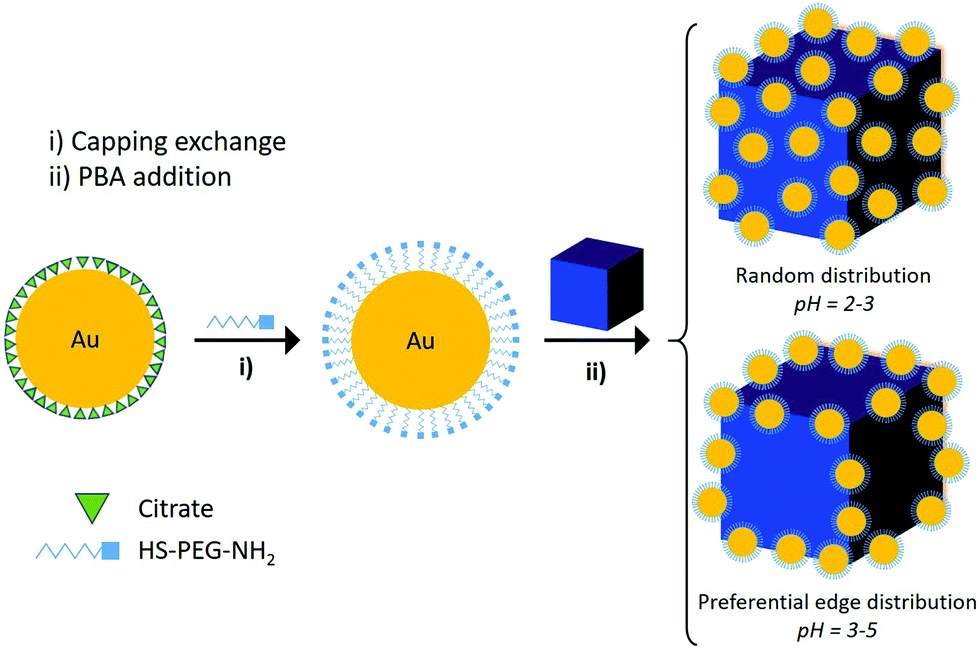 | ||
| Fig. 1 Schematic illustration of the preparation of heterostructures formed by Au nanospheres decorating PBA NPs. The first step involves a capping substitution and the second step the attachment of the Au onto the PBA surface. Two different Au decorations can be achieved by adjusting the pH: a random and a preferential distribution on the edges. | ||
In a further step, the solution of Au NPs stabilized with HS-PEG-NH2 was adjusted at pH between 2 and 4 in order to protonate the dangling amine group (ζ-potential values vary from 9 ± 6 mV to >20 mV). Mixing this solution with PBA NPs resulted in the decoration of the PBA NPs by the Au NPs. pH plays an important role in the whole process because at low pH (below 2) the Au NPs tend to aggregate (strong ionic force), while at pH above 5 a poor decoration of the PBA NPs is observed (weak electrostatic interaction because of the weak positively charged Au NPs) (Fig. S1, ESI†). Hence, to optimize the decoration, the pH value was kept in the range of 2–5.
When the pH is in the 2–3 range, a random decoration of the PBA-NPs by Au is obtained, as demonstrated by TEM imaging. (Fig. 2a and Fig. S8, ESI†). An increase of the pH value (3–5) leads to a preferential binding of the Au NPs along the edges of the PBA cube (Fig. 2b and Fig. S8, ESI†). TEM images of the preferential edge decoration taken at different incidence angles are shown in Fig. S9 (ESI†). In addition, 3D imaging by tomography of both heterostructures was performed to visualize these complex morphologies. These images confirm the preferential pH-dependent decoration of the edges of the PBA cube by Au NPs (see Fig. S10 and videos of the 3D reconstructions in the ESI†).
 | ||
| Fig. 2 TEM images obtained for a single NP of the heterostructure with (a) random and (b) preferential edge decoration onto NiCr PBA. (c) UV-Vis spectra of Au NPs and Au-PBA heterostructures. Inset: Magnified image of the plasmon bands. (d) Hysteresis loop at 2 K of PBA and Au-PBA. | ||
Energy-dispersive X-ray spectroscopy mapping (Fig. S11, ESI†) confirms the presence of Cr and Ni at the center of the NP and Au at the cube edges. The selective decoration of the edges at a pH ranging from 3–5 is due to the larger negative charge density on (and near) the cube edges than on its faces. Indeed, as it can be observed from its crystalline structure (Fig. S18, ESI†), on the particles' surface, each Cr(CN)63− species is surrounded by 5 Ni2+ (four belonging to the surface and one inside the NP), while on the edges each hexacyanometalate is surrounded by only 4 Ni2+ (two belonging to the surface and two to the particles' edge). At a pH of 3–5, the positive charge density of the Au NPs is weaker, therefore, promoting electrostatic interaction with the PBA NPs edges where the negative charge density is larger. This effect is enhanced due to the possible presence of a larger amount of defects in the vicinity of the cube edges than on its faces. It is worth mentioning that the Au decoration (and thus the electrostatic interaction) is maintained after redispersing the hybrid in water at pH 7 (Fig. S12, ESI†).
An optimum decoration of the PBA-NPs can be achieved by tuning the ratio (Au:PBA) of the two components for the two pH ranges. For the low pH range (2–3, random distribution), we carried out the experiments with an Au:PBA molar ratio in the range 0.31 to 0.50. In this range, the higher the ratio is, the higher the decoration is (Fig. S13, ESI†). For high pH range (3–5, edge distribution), we investigated three different ratios, namely 0.28, 0.41 and 1.12. TEM imaging seems to indicate that the best results are obtained with the molar ratio of 0.41 (Fig. S14, ESI†). In order to assess the efficiency of the interaction between the two types of NPs, we determined the molar ratio of two thoroughly washed powder samples obtained with Au![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :PBA = 0.51 (low pH range) and 0.41 (high pH range) using inductively coupled plasma (ICP) measurements. The values of the Au:PBA ratio of 0.43 and 0.26 were obtained for the former and latter cases respectively, allowing the determination of the optimum ratio for the two pH ranges.
:PBA = 0.51 (low pH range) and 0.41 (high pH range) using inductively coupled plasma (ICP) measurements. The values of the Au:PBA ratio of 0.43 and 0.26 were obtained for the former and latter cases respectively, allowing the determination of the optimum ratio for the two pH ranges.
We have presented a new synthetic approach that allows an optimum selective decoration (random vs. preferential) of PBA NPs with Au NPs. Let us now discuss the plasmonic and magnetic properties of these hybrid nanostructures.
The optical properties of Au NPs arise from the Localized Surface Plasmon Resonance (LSPR). Hence, the plasmon band is expected to be sensitive to the interaction between Au and PBA NPs. In Fig. 2c, we observe that the UV-vis absorption spectrum is dominated by the plasmon band. For the functionalized Au-PEG NPs, the maximum of this band is located at 520 nm and shifts to 526 nm when Au-PEG is attached to the PBA-NPs, independently on the type of decoration. The plasmon band shift is related to the change of the dielectric properties and, in particular, to the local refractive index at the surface of the plasmonic NPs.27 It is worth noting that no shift is observed in the control experiment where the HS-PEG-NH2 molecule was not used (see Fig. S8, ESI†). Therefore, this shift is a clear indication of the PBA NPs decoration by the functionalized Au NPs. However, we also observe a progressive decay of the absorbance of the plasmonic band after 2 hours that becomes large after 48 h (Fig. S19, ESI†). A decrease of intensity of the plasmonic band is usually associated with the aggregation of the colloidal suspension but may be also due to Au degradation in the present case. Indeed, cyanide dissolution of spherical Au colloids usually takes place quickly;28 thus, in order to check the chemical stability of the hybrid materials, X-ray powder diffraction study was performed on the pure PBA NPs and the hybrid material after leaving the suspensions to stand for 1 week in water (see Fig. S20, ESI†). The detection of the characteristic Au diffraction peaks (at 38.2 and 44.3 degrees), together with unaltered PBA peaks indicates that the decrease of the intensity of the plasmonic band is due to the aggregation of the colloidal suspension with time and not to chemical degradation of the hybrid materials.
As far as the magnetic properties of the hybrid nanostructures are concerned, we observe that they are almost identical to those measured for pristine NiCr PBA NPs (Fig. S21, S22, ESI,† and Fig. 2d).29 Thus, a ferromagnetic behaviour below 68 K and a weak hysteresis in the magnetization of ca. 60 Oe at 2 K is observed, in agreement with the soft character of this PBA derivative (Fig. 2d). The only noticeable difference between hybrid and pristine NPs is the lower value for the saturation magnetization. This is due to the contribution of Au in the total weight of the nanostructure. In fact, if the presence of Au is corrected using the estimated amount of Au extracted from ICP measurements, such a difference disappears.
The generality of this approach is further illustrated by the fact that any plasmonic NP able to form thiol bonds with HS-PEG-NH2 in acid conditions can be potentially used. This possibility opens the door to tune at will the plasmonic properties of the hybrid nanostructure simply by choosing the appropriate plasmonic NP. In this manner, citrate-stabilized spherical Ag NPs were used instead of Au NPs following the same experimental procedure in the pH range of 3–5 (see Fig. S4 and Table S1, ESI†). The obtained results are shown in Fig. 3a and b. The plasmon band of the Ag NPs is now located at 410 nm (to be compared with 520 for the Au NPs) and exhibits a red-shift of ca. 7 nm. This shift is similar to that observed in Au NPs, as it mainly depends on the shape of the NP, being independent of the metal.30 Other Au NPs with different shapes can also be used to decorate the NiCr PBA. Thus, we have chosen anisotropic Au NPs (NRs and NSs; see Fig. S5 and S6, ESI†) which exhibit an intense plasmon band located at ca. 800 nm and ca. 700 nm respectively. In these cases, the plasmon band can be tuned in a broad range, by changing the ratio length/width in the AuNRs,31 and the size of the branches and the core for the AuNSs.32 In these two cases the optimal ratio of Au:PBA changes with respect to that of the Au spheres, being 0.55:1 and 1.25:1 for AuNRs–PBA and AuNSs–PBA, respectively. This variation can be attributed to the different contact areas obtained for the different structures.
 | ||
| Fig. 3 TEM images and UV-Vis spectra obtained for the heterostructures: Ag-PBA, AuNRs-PBA and AuNSs-PBA. The insets show the plasmon band shift. These heterostructures were obtained at pH 3–5. | ||
Fig. 3 shows the resulting nanostructures and plasmonic properties. As in the previous case, the synthesis was followed by UV-Vis spectra, searching for the plasmon shift as a proof of the interaction between Au and PBA NPs. In both cases, a shift in the plasmon band was noticed. Nevertheless, these shifts were more pronounced than the one observed for the Au spheres, due to the higher sensibility of these plasmons.27 Concretely, AuNR and AuNS hybrid nanostructures show plasmon red-shifts of ca. 9 nm and 22 nm, respectively.
The possibility to cover the whole visible spectrum by choosing the appropriate plasmonic NP may be very interesting for improving the magneto-plasmonic properties of these hybrids since the MO response of the magnet is enhanced when its maximum matches in energy that of the plasmon band.17 Since this synthetic strategy enables the preparation of many combinations of plasmonic NP/PBA heterostructures which remain stable at neutral pH, it may also be suitable for biomedical applications, where a NIR plasmon needs to be combined with an efficient PBA contrast agent.
Finally, it can be noted that this procedure can also be extended to other PBA NPs. For example, NiCr can be changed by PB (FeFe). Fig. S15a and b (ESI†) show the nanostructures achieved, as well as the corresponding shift in the UV-Vis spectrum of ca. 6 nm. Therefore, following this approach it is possible to attach Au NPs in any negatively charged PB or PBA NP without a capping agent, such as NiFe, CuCr or CoFe. These hybrids can be observed in Fig. S15 (ESI†). Notice however that the colloidal stability of these systems is limited by the size of the PBA since, if they are too small (below 25 nm), the NPs tend to aggregate (see Fig. S16, ESI†) and if they are too big (above 300 nm) they tend to precipitate.
In summary, we have developed a simple and general synthetic procedure to prepare, in aqueous solution, hybrid magneto-plasmonic nanostructures formed by metallic NPs decorating negatively charged magnetic NPs based on PBA. By adjusting the pH, it is possible to control the location of the Au NPs on the PBA cubic NP. Indeed, Au can be attached randomly over the whole cubic surface or preferentially on the edges. This general methodology was first tested with spherical Au NPs and then extended to different plasmonic NPs of various shapes. It permitted tuning the plasmon band energy in a broad range of the visible spectrum, thus providing a versatile platform to investigate the enhancement of the MO properties of the hybrids thanks to their coupling with the plasmons. This aspect, fully unexplored in molecular-based magnets, will be addressed in the future combining photomagnetism with plasmonics.
The authors acknowledge funding from the EU (COST Action MOLSPIN CA15128, ERC Advanced Grant Mol-2D 788222, ERC Consolidator Grant REALNANO 815128 and Grant Agreement No. 731019 (EUSMI)), the Spanish MCIU (Unit of Excellence “Maria de Maeztu” CEX2019-000919-M and Project MAT2017-89993-R co-financed by FEDER), the Generalitat Valenciana (Prometeo Programme PROMETEO/2017/066 and iDiFEDER/2018/061) and the Ministry of Education and Science of Russian Federation (Agreement No. 14.W03.31.0001). R. S.-G. thanks the Spanish MCIU for a F. P. U. fellowship.
Conflicts of interest
There are no conflicts to declare.Notes and references
- I. S. Maksymov, Rev. Phys., 2016, 1, 36–51 CrossRef.
- T. T. Nguyen, F. Mammeri and S. Ammar, Nanomaterials, 2018, 8, 149 CrossRef.
- E. Coronado, Nat. Rev. Mater., 2020, 5, 87–104 CrossRef.
- L. Catala and T. Mallah, Coord. Chem. Rev., 2017, 346, 32–61 CrossRef CAS.
- E. Coronado, M. Makarewicz, J. P. Prieto-Ruiz, H. Prima-García and F. M. Romero, Adv. Mater., 2011, 23, 4323–4326 CrossRef CAS.
- S. H. Lapidus, A. G. Graham, C. M. Kareis, C. G. Hawkins, P. W. Stephens and J. S. Miller, J. Am. Chem. Soc., 2019, 141, 911–921 CrossRef CAS.
- S. ichi Ohkoshi, A. Namai and H. Tokoro, Coord. Chem. Rev., 2019, 380, 572–583 CrossRef.
- K. Hurlbutt, S. Wheeler, I. Capone and M. Pasta, Joule, 2018, 2, 1950–1960 CrossRef CAS.
- M. Morant-Giner, R. Sanchis-Gual, J. Romero, A. Alberola, L. García-Cruz, S. Agouram, M. Galbiati, N. M. Padial, J. C. Waerenborgh, C. Martí-Gastaldo, S. Tatay, A. Forment-Aliaga and E. Coronado, Adv. Funct. Mater., 2017, 1706125, 1–11 Search PubMed.
- Z. Qin, Y. Li and N. Gu, Adv. Healthcare Mater., 2018, 7, 1–13 Search PubMed.
- S. Ferlay, T. Mallah, R. Ouahès, P. Veillet and M. Verdaguer, Nature, 1995, 378, 701–703 CrossRef CAS.
- L. Trinh, S. Zerdane, S. Mazérat, N. Dia, D. Dragoe, C. Herrero, E. Rivière, L. Catala, M. Cammarata, E. Collet and T. Mallah, Inorg. Chem., 2020, 59, 13153–13161 CrossRef CAS.
- G. Maurin-Pasturel, J. Long, Y. Guari, F. Godiard, M. G. Willinger, C. Guerin and J. Larionova, Angew. Chem., Int. Ed., 2014, 53, 3872–3876 CrossRef CAS.
- L. Cheng, H. Gong, W. Zhu, J. Liu, X. Wang, G. Liu and Z. Liu, Biomaterials, 2014, 35, 9844–9852 CrossRef CAS.
- Y. Yin, Q. Li, S. Ma, H. Liu, B. Dong, J. Yang and D. Liu, Anal. Chem., 2017, 89, 1551–1557 CrossRef CAS.
- J. D. Qiu, H. Z. Peng, R. P. Liang, J. Li and X. H. Xia, Langmuir, 2007, 23, 2133–2137 CrossRef CAS.
- M. Caminale, L. Anghinolfi, E. Magnano, F. Bondino, M. Canepa, L. Mattera and F. Bisio, ACS Appl. Mater. Interfaces, 2013, 5, 1955–1960 CrossRef CAS.
- G. Armelles, A. Cebollada, A. García-Martín and M. U. González, Adv. Opt. Mater., 2013, 1, 2 CrossRef.
- G. Shemer and G. Markovich, J. Phys. Chem. B, 2002, 106, 9195–9197 CrossRef CAS.
- L. Jing, X. Liang, Z. Deng, S. Feng, X. Li and M. Huang, Biomaterials, 2014, 35, 5814–5821 CrossRef CAS.
- G. Maurin-Pasturel, E. Rascol, M. Busson, S. Sevestre, J. Lai-Kee-Him, P. Bron, J. Long, J. Chopineau, J.-M. Devoisselle, Y. Guari and J. Larionova, Inorg. Chem. Front., 2017, 4, 1737–1741 RSC.
- F. N. Crespilho, V. Zucolotto, C. M. A. Brett, O. N. Oliveira and F. C. Nart, J. Phys. Chem. B, 2006, 110, 17478–17483 CrossRef CAS.
- M. H. Xue, Q. Xu, M. Zhou and J. J. Zhu, Electrochem. Commun., 2006, 8, 1468–1474 CrossRef CAS.
- V. Gadet, T. Mallah, I. Castro, M. Verdaguer and P. Veillet, J. Am. Chem. Soc., 1992, 114, 9213–9214 CrossRef CAS.
- H. Häkkinen, Nat. Chem., 2012, 4, 443–455 CrossRef.
- J. Turkevich, Gold Bull., 1985, 18, 125–131 CrossRef CAS.
- K. M. Mayer and J. H. Hafner, Chem. Rev., 2011, 111, 3828–3857 CrossRef CAS.
- N. R. Jana, L. Gearheart, S. O. Obare and C. J. Murphy, Langmuir, 2002, 18, 922–927 CrossRef CAS.
- M. F. Dumont, E. S. Knowles, A. Guiet, D. M. Pajerowski, A. Gomez, S. W. Kycia, M. W. Meisel and D. R. Talham, Inorg. Chem., 2011, 50, 4295–4300 CrossRef CAS.
- K. S. Lee and M. A. El-Sayed, J. Phys. Chem. B, 2006, 110, 19220–19225 CrossRef CAS.
- Y.-Y. Yu, S.-S. Chang, C.-L. Lee and C. R. C. Wang, J. Phys. Chem. B, 1997, 101, 6661–6664 CrossRef CAS.
- S. Barbosa, A. Agrawal, L. Rodríguez-Lorenzo, I. Pastoriza-Santos, R. A. Alvarez-Puebla, A. Kornowski, H. Weller and L. M. Liz-Marzán, Langmuir, 2010, 26, 14943–14950 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0cc08034a |
| This journal is © The Royal Society of Chemistry 2021 |