
Upconversion in molecular hetero-nonanuclear lanthanide complexes in solution†
Richard C.
Knighton‡
 a,
Lohona K.
Soro‡
a,
Alexandre
Lecointre
a,
Guillaume
Pilet
b,
Alexandra
Fateeva
b,
Laurie
Pontille
b,
Laura
Francés-Soriano
c,
Niko
Hildebrandt
cd and
Loïc J.
Charbonnière
*a
a,
Lohona K.
Soro‡
a,
Alexandre
Lecointre
a,
Guillaume
Pilet
b,
Alexandra
Fateeva
b,
Laurie
Pontille
b,
Laura
Francés-Soriano
c,
Niko
Hildebrandt
cd and
Loïc J.
Charbonnière
*a
aEquipe de synthèse pour l’analyse (SynPA), Institut Pluridisciplinaire Hubert Curien (IPHC), UMR 7178, CNRS/Université de Strasbourg, ECPM, 25 rue Becquerel, Strasbourg cedex 67087, France. E-mail: l.charbonn@unistra.fr
bLaboratoire des Multimatériaux et Interfaces (LMI) UMR 5615, Université Claude Bernard Lyon 1, Avenue du 11 novembre 1918, Villeurbanne cedex 69622, France
cNanoFRET.com, Laboratoire COBRA (Chimie Organique, Bioorganique, Réactivite et Analyse), UMR 6014, CNRS, Université de Rouen Normandie, INSA, Mont-Saint-Aignan cedex 76821, France
dUniversité Paris-Saclay, Saint-Aubin 91190, France
First published on 8th December 2020
Abstract
Here we show that nonanuclear lanthanide complexes respresent a new class of solution state upconversion (UC) molecules. For a composition of one Tb per eight Yb the nonanuclear complexes display a very efficient UC phenomenon with Tb luminescence in the visible region upon 980 nm NIR excitation of Yb. An unprecedented value of 1.0 × 10−7 was obtained for the UC efficiency at only 2.86 W cm−2, demonstrating these new molecular complexes to be up to 26 times more efficient than the best current molecular systems, the UC being observed down to a concentration of 10 nM.
While UC has been observed and studied for more than sixty years,1 it is only in the last decade that researchers have been playing with the coordination chemistry toolkit in an attempt to observe it at the molecular scale in solution.2 The main difficulty of the molecular approach relies on the presence of organic matter constituting the ligands and the solvent molecules. The multistep mechanisms of UC require that the NIR intermediate excited state persists sufficiently long enough to enable the further climbing of the energy ladder. As the main source of non-radiative deactivation of excited states is attributed to energy transfer to the overtones of vibrational bands in the NIR,3 decreasing this quenching is of paramount importance in molecular UC systems. However, by combining a judicious choice of energy donors and acceptors, a few discrete coordination complexes have been revealed as UC molecular systems in the solid state,4 but also in diluted solutions.2,5–7
In 1994, Auzel proposed the formation of “ion clusters” as being responsible for solid state cooperative UC processes observed in some doped glasses.8 Such clusters result from ion pairing at very short distances (less than 5 Å) and were later confirmed to be the origin of green cooperative UC luminescence of Tb observed in some doped Yb/Tb solids.9 We thus hypothesized that chemical engineering of equivalent complexes might lead to the observation of the cooperative photosensitization process in discrete molecular entities in solution.
Among known lanthanide (Ln) complexes,10 a family based on β-diketonate ligands attracted our attention as a result of their straightforward synthesis,11 versatility in nuclearity12 and composition,13 and short inter-Ln distances.14 The simple case of acetylacetonate ligands forming nonanuclear complexes12 particularly piqued our interest with the close proximity of up to nine Ln atoms in a very small volume and intermetallic distances shorter than 5 Å. It is noteworthy that a controlled doping of such complexes might allow the formation of entities containing up to eight sensitizing ions for one emitting UC center, a situation considered to be optimum for maximizing energy transfer UC.15 Taking advantage of these prior studies, we show that the structures obtained in the solid state retain their integrity in solution and that the chemical composition can be tuned to obtain mixed Yb/Tb nonanuclear complexes exhibiting outstanding UC properties in solution.
The nonanuclear complexes of general composition [Ln9L16(OH)10](OH) (L = Acac = acetylacetonate, yttrium (Y) will be integrated in the abbreviation of Ln for brevity) were obtained by adaptation of the literature procedures (Scheme 1a).11 Full synthetic details and characterization of the complexes can be found in the ESI.† The solid state structure of the Ln9 complexes12 can be viewed as two pentanuclear square pyramids sharing the apical Ln atom, with a torsion angle of approximately 45° between the two pyramids, resulting in square anti-prismatic geometry at the central Ln (Scheme 1a). The eight triangular faces of the pyramids are capped by μ3-OH groups linked to the three Ln at the edges of the triangles, while the four Ln atoms of the two square faces are connected by μ4-OH bonds. All Ln atoms except for the central one are linked to two acetylacetonate ligands.
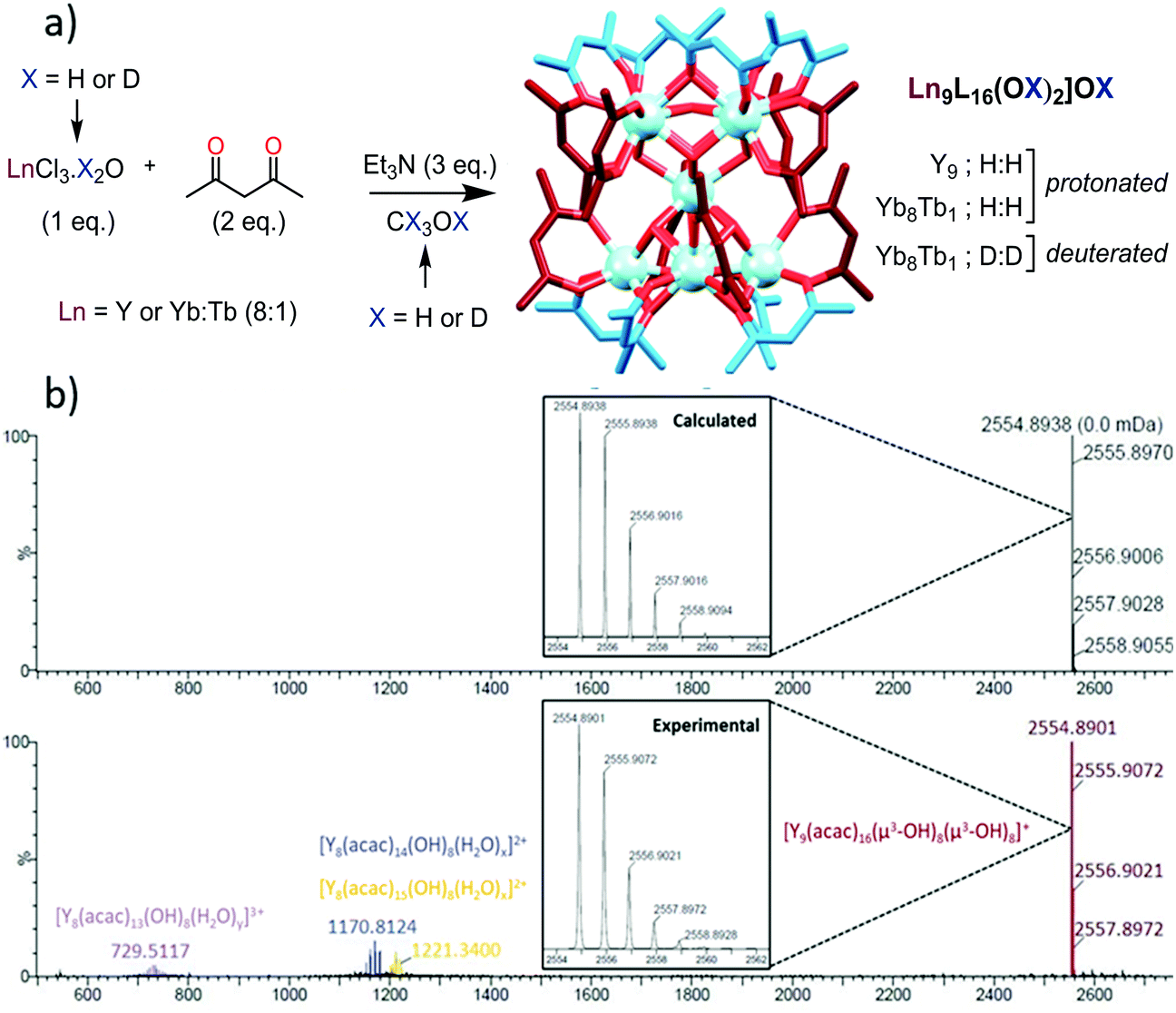 | ||
| Scheme 1 (a) Synthetic protocol for the preparation of the nonanuclear complexes. (b) ES/MS spectra of the Y nonanuclear cluster showing a major peak at 2554.8901 and the expansion of this peak with the calculated isotopic distribution. | ||
For the study in solution, the Y9 complex was taken as a reference due to (a) the chemical similarity of Y(III) with Ln(III) cations of the end of the Ln series (ionic radii of 1.027, 1.019 and 1.015 Å respectively for Dy(III), Y(III) and Ho(III) for a coordination number of 8)16 and (b) its diamagnetic nature allowing for an easy interpretation of NMR experiments. The complex was redissolved in CH3OH and first studied by electrospray mass spectrometry (Scheme 1b). The spectrum displays one major peak at 2554.89 m/z units corresponding to the monocharged [Y9L16(OH)10]+ complex and minor peaks were observed attributed to doubly charged polyhydrated fragments having lost one YL1 unit or one YL2 unit and triply charged polyhydrated complexes missing one YL3− unit (Fig. S1, ESI†).
The 1H-NMR spectrum of the Y cluster in CDCl3 (Fig. S2, ESI†) presents time-averaged C4 symmetry, with two sets of methyl groups (48H each) and two sets of CH (8H each) for the Acac ligands, and two broad singlets integrating for 8H and 2H corresponding to the two sets of OH bonds observed in the solid state structure (vide supra). Different NMR experiments (13C, HSQC, HMQC, ROESY and DOSY Fig. S2–S7, ESI†) pointed to the two sets of Acac ligands originating from two families of independent ligands, which were ascribed to eight Acac ligands capping the top and the bottom of the cluster (exo ligands, blue in Scheme 1a) and eight Acac ligands positioned at the equatorial plane of the cluster (endo ligands, red in Scheme 1a). Each of these ligands can easily rotate to exchange the positions of the two methyl groups, but the exchange between the exo and endo positions is slow on the NMR time scale at 298 K. The 2D-DOSY NMR spectrum was particularly informative (Fig. S7, ESI†), as the average value of the diffusion coefficients at 4.37 × 10−10 m2 s−1 represents a volume of 3270 ± 150 Å3, in good agreement with the crystal structure of the complex in the solid state (2847(1) Å3).
Considering that the maximization of the number of sensitizing ions per emitting centers (activators) is expected to optimize the UC process,15 we then prepared mixed complexes containing 8 Yb donors and one Tb acceptor by simply starting from a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :8 Tb:Yb mixture. The complexes were obtained from CH3OH and LnCl3·6H2O, or from CD3OD and LnCl3·D2O starting salts, respectively referred to H:H or D:D. The solid-state infrared (IR) spectra of the novel complexes display a net shift of the μx-OH(D) vibrational band from 3316 cm−1 to 2439 cm−1 upon deuteration (Fig. S8, ESI,† for the IR spectra). Powder XRD of the H:H complex (Fig. S9, ESI†) clearly demonstrated that, in the solid state, the synthesized [Yb8Tb1] cluster is isostructural to the reported [Tb9] ones.12 TEM-EDX analysis was performed on 10 different points of the monocrystals of H:H, giving a composition of 89.6 ± 2.4% of Yb and 10.4 ± 2.4% of Tb, in good agreement with the 89.1% of Yb and 10.9% of Tb, introduced in the synthesis. Alternatively, the Yb and Tb contents of the heteronuclear complexes were determined by ICP-AES measurements confirming the Yb8Tb1 composition with an estimated error of ± 0.1 atomic unit.
:8 Tb:Yb mixture. The complexes were obtained from CH3OH and LnCl3·6H2O, or from CD3OD and LnCl3·D2O starting salts, respectively referred to H:H or D:D. The solid-state infrared (IR) spectra of the novel complexes display a net shift of the μx-OH(D) vibrational band from 3316 cm−1 to 2439 cm−1 upon deuteration (Fig. S8, ESI,† for the IR spectra). Powder XRD of the H:H complex (Fig. S9, ESI†) clearly demonstrated that, in the solid state, the synthesized [Yb8Tb1] cluster is isostructural to the reported [Tb9] ones.12 TEM-EDX analysis was performed on 10 different points of the monocrystals of H:H, giving a composition of 89.6 ± 2.4% of Yb and 10.4 ± 2.4% of Tb, in good agreement with the 89.1% of Yb and 10.9% of Tb, introduced in the synthesis. Alternatively, the Yb and Tb contents of the heteronuclear complexes were determined by ICP-AES measurements confirming the Yb8Tb1 composition with an estimated error of ± 0.1 atomic unit.
The UV-Vis-NIR absorption spectrum of the H:H complex in CH3OH is presented in Fig. 1 (Fig. S10, ESI,† for the D:D complex). It is composed of a broad absorption band in the UV region at 291 nm (ε = 14.8 × 104 M−1 cm−1), attributed to transitions centered on the Acac ligands.17 In the NIR, the spectrum displays a broad absorption band at 975 nm (ε = 46.9 M−1 cm−1 in CH3OH; ε = 51.7 M−1 cm−1 in CD3OD) attributed to the 2F7/2 → 2F5/2 transition of Yb. At high concentrations, a very weak absorption band can be observed around 485 nm (ε = 0.055 M−1 cm−1, Fig. S10, ESI†) attributed to the 7F6 → 5D4 transition of Tb.
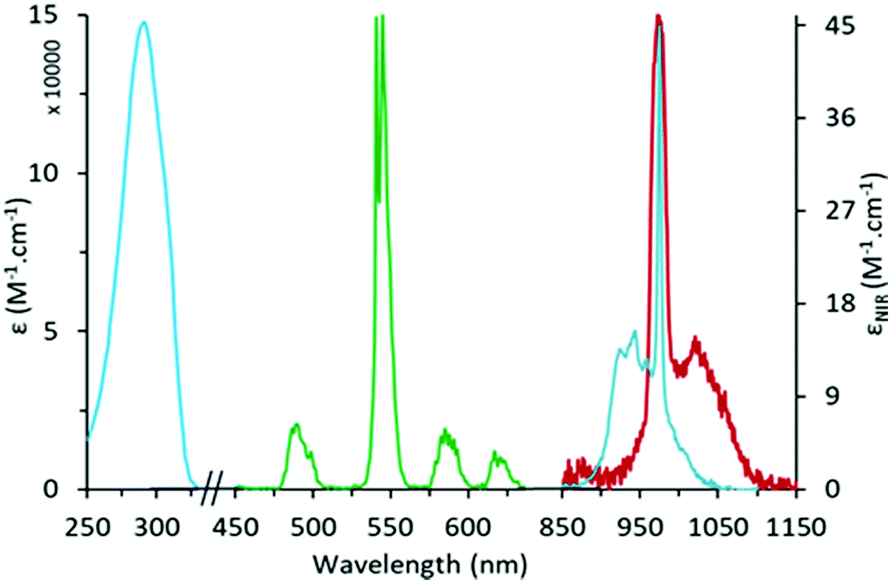 | ||
| Fig. 1 UV-Vis-NIR absorption spectrum (blue) and normalized emission spectra (λexc = 292 nm) in the visible (green) and NIR (red) domains for the H:H [Yb8TbL16(OH)10](OH) cluster in CH3OH. | ||
Upon excitation into the Acac absorption band (292 nm), the complexes display two sets of emission bands (Fig. 1), those in the visible region corresponding to the 5D4 → 7FJ (J = 6 to 3) transitions of Tb,18 (ΦTb = 1.5% in CH3OH), and a broad emission band peaking at 975 nm corresponding to the 2F5/2 → 2F7/2 transition of Yb (ΦYb = 0.08%). The corresponding luminescence lifetimes were 0.95 ms for Tb and <1 μs for Yb in CH3OH. For the D:D complex in CD3OD, the lifetimes increase to 16.6 μs for Yb and 1.12 ms (95%) and 350 μs (5%) for Tb, indicating two different chemical environments. From previous site specific doping experiments,11 the larger Tb ion is expected to be predominantly positioned at the central site of the cluster and one might relate this to the longer lifetime value regarding the relative intensities of the two components. However, the excitation through the ligand absorption bands is expected to favor the emission from the cations positioned at the periphery of the cluster to which Acac ligands are directly bonded and the attribution of the lifetimes to the two different sites is still subject to doubt.
From the NIR absorption band of Yb and weighting the absorption coefficients by the number of Yb atoms (8), it was possible to calculate the radiative lifetime of Yb, τrad, according to the methodology described by Werts and coworkers.19 Full experimental description of the method is given in the ESI,† (Section S3). Values of τrad between 578 and 683 μs were obtained (Table S1, ESI†) for the H:H and D:D complexes in CH3OH or CD3OD, affording a mean value of 639 ± 28 μs. The Yb lifetimes being too short to be measured with our setup in CH3OH, only values in CD3OD for the D:D complex were used (Table S1, ESI†) to calculate a Yb centered luminescence quantum yield of 2.6% with a modest sensitization efficiency of 22% by excitation through the Acac ligands.
The most striking feature of the mixed cluster was observed when the D:D complex in CD3OD was excited in the NIR absorption band of Yb at 980 nm (Fig. 2a), resulting in a strong emission in the visible region with the typical spectral signature of the Tb emission.
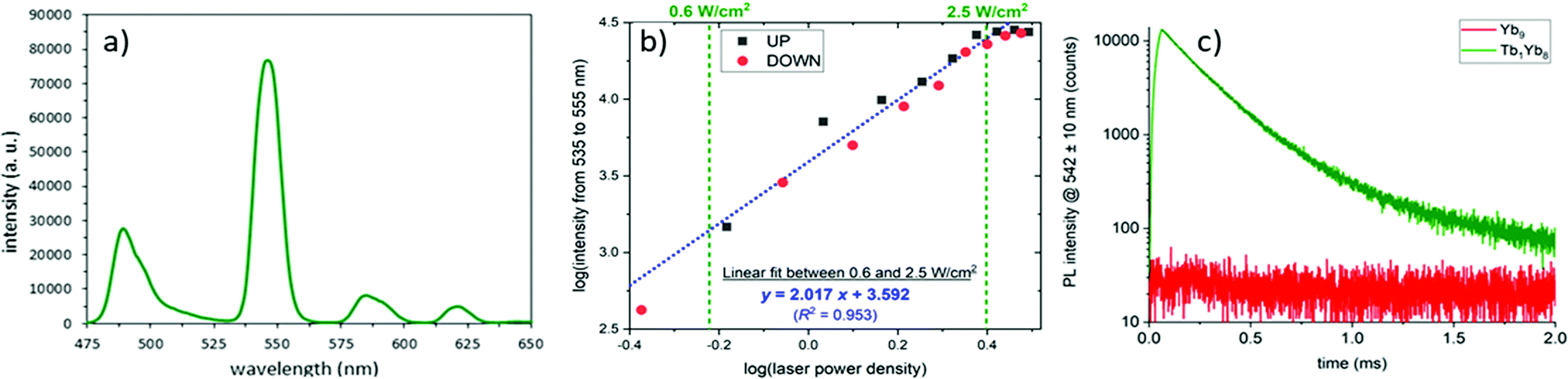 | ||
| Fig. 2 (a) UC emission spectrum of the (D:D) complex in CD3OD (c = 2.0 mM, P = 2.86 W cm−2, λexc = 980 nm). (b) UC intensity as a function of the incident pump power density in a log/log scale (squares correspond to increasing pump intensities and red dots for decreasing ones). Blue dotted line represents the linear regression of the data. (c) Temporal evolution of the UC emission at 542 nm upon 60 μs pulsed excitation at 980 nm for complexes of Yb8Tb1 (green) and Yb9 (red) composition. | ||
The luminescence intensity was recorded as a function of the incident pump power density and the logarithmic representation (Fig. 2b) presents a quasi linear profile with a slope of two, accrediting the two photon UC process.20,21
Analysis of the temporal evolution of the UC intensity at 542 nm upon pulsed excitation (Fig. 2c) evidences a first slow increase during the 60 μs pumping period, indicative of a cooperative photosensitization mechanism,7,21 followed by a bi-exponential decay (τ1 = 170 μs (93%); τ2 = 390 μs (7%)). The same experiment performed on the Yb9 cluster resulted in the absence of the green emission, confirming that the emission arises from the Tb atoms.
The proposed mechanism (Fig. 3) entails a first absorption of a photon by a Yb atom, leading to a [Yb7Yb*Tb] species, the absorption of a second photon forming the [Yb6Yb*2Tb1] intermediate, and cooperative energy transfer to Tb to form the [Yb8Tb1*] excited states which decay to the ground state by emission of visible light.
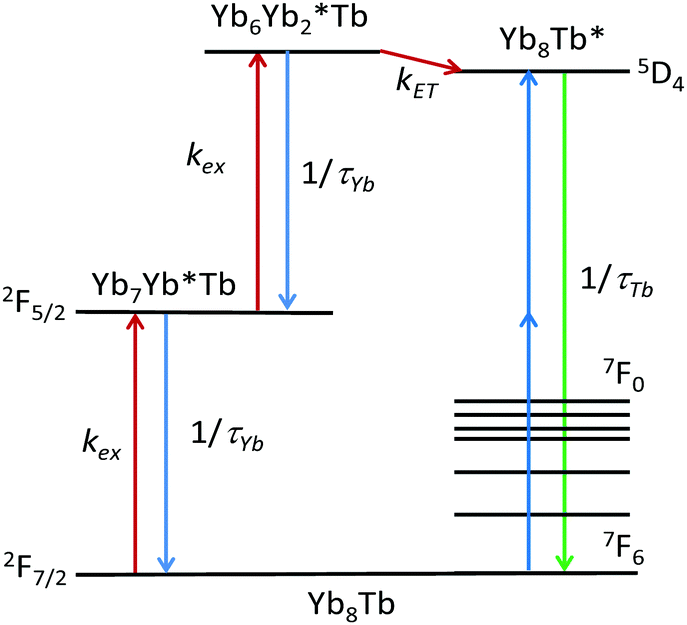 | ||
| Fig. 3 Proposed cooperative photosensitization process UC mechanism. | ||
The UC quantum yield (ΦUC), calculated following published procedures,22 was determined to be 1 × 10−7 (@2.86 W cm−2). As ΦUC is directly proportional to the power of the excitation source for a two-photon process,22a and the calculations of the ΦUC do not take into account the factor 2 sometimes used to account for the two-photon process,5 this value is 92 to 377 times larger than that of the best UC Er complexes (1.95 × 10−9 < ΦUC < 8 × 10−9@21 W cm−2),5 and 26 times larger than previous heteropolynuclear Yb/Tb UC complexes (1.4 × 10−8 @10.3 W cm−2).6 The importance of reducing excited-state deactivation by OH oscillators is exemplified by the 18-fold larger ΦUC for the D:D cluster compared to its H:H congener (Table S1, ESI†).
The sensitivity of the UC emission was investigated by decreasing the concentration of the solution down to the lowest observable emission. At micromolar concentrations, the Tb UC emission signal was still fully resolvable and the lower limit of detection with our setup at full power (P = 10.3 W cm−2) was determined to be 10 nM (Fig. S11, ESI†).
In conclusion, the doping of Yb nonanuclear complexes by Tb ions afforded heteronuclear [Yb8Tb(Acac)16(OH)10](OH) complexes which display a cooperative UC photosensitization process demonstrated to be more than one order of magnitude more efficient than previous molecular or supramolecular UC probes,2,5–7 being observable at concentration as low as 10 nM. The tremendous potential of readily accessible β-diketonate ligands, coupled with the wide variety of Ln donor–acceptor dyads and the structural diversity of such complexes, opens avenues to even more efficient UC systems, with applications in luminescence bio-analytical applications.23
Financial support is gratefully acknowledged from the French Ministère de l’Education Nationale et de la Recherche and the French Canada Research Fund and the LabEx CSC (ANR-10-LABX-0026-CSC). Dr Jean-Marc Strub and Dr Bruno Vincent are acknowledged for technical support.
Conflicts of interest
There are no conflicts to declare.Notes and references
- (a) P. A. Franken, A. E. Hill, C. W. Peters and G. Weinreich, Phys. Rev. Lett., 1961, 7, 118 CrossRef; (b) F. Auzel, Chem. Rev., 2004, 104, 139 CrossRef CAS PubMed.
- (a) L. Aboshyan-Sorgho, C. Besnard, P. Pattison, K. R. Kittilstved, A. Aebischer, J.-C. G. Bünzli, A. Hauser and C. Piguet, Angew. Chem., Int. Ed., 2011, 50, 4108 ( Angew. Chem. , 2011 , 123 , 4194–4198 ) CrossRef CAS PubMed; (b) A. Nonat, C. F. Chan, T. Liu, C. Platas-Iglesias, K.-L. Wong and L. J. Charbonnière, Nat. Commun., 2016, 7, 11978 CrossRef CAS PubMed.
- (a) C. Doffek, N. Alzakhem, C. Bischof, J. Wahsner, T. Güden-Silber, J. Lügger, C. Platas-Iglesias and M. Seitz, J. Am. Chem. Soc., 2012, 134, 16413 CrossRef CAS PubMed; (b) A. Beeby, I. M. Clarkson, R. S. Dickins, S. Faulkner, D. Parker, L. Royle, A. S. de Sousa, J. A. G. Williams and M. Woods, J. Chem. Soc., Perkin Trans. 2, 1999, 493 RSC.
- (a) J. Kalmbach, C. Wang, Y. You, C. Förster, H. Schubert, K. Heinze, U. Resch-Genger and M. Seitz, Angew. Chem., Int. Ed., 2020, 59, 18804 CrossRef CAS PubMed; (b) J. T. Mo, Z. Wang, P. Y. Fu, L. Y. Zhang, Y. N. Fan, M. Pan and C. Y. Su, CCS Chem., 2020, 2, 729 Search PubMed.
- B. Golesorkhi, A. Fürstenberg, H. Nozary and C. Piguet, Chem. Sci., 2019, 10, 6876 RSC.
- N. Souri, P. Tian, C. Platas-Iglesias, K.-L. Wong, A. Nonat and L. J. Charbonnière, J. Am. Chem. Soc., 2017, 139, 1456 CrossRef CAS PubMed.
- A. Nonat, S. Bahamyirou, A. Lecointre, F. Przybilla, Y. Mély, C. Platas-Iglesias, F. Camerel, O. Jeannin and L. J. Charbonnière, J. Am. Chem. Soc., 2019, 141, 1568 CrossRef CAS PubMed.
- F. Auzel, D. Meichenin, F. Pellé and P. Goldner, Opt. Mater., 1994, 4, 35 CrossRef CAS.
- (a) G. M. Salley, R. Valiente and H. U. Güdel, Phys. Rev. B: Condens. Matter Mater. Phys., 2003, 67, 134111 CrossRef; (b) G. M. Salley, R. Valiente and H. U. Güdel, J. Lumin., 2001, 94–95, 305 CrossRef.
- J. Kobylarczyk, E. Kuzniak, M. Liberka, S. Chorazy, B. Sieklucka and R. Podgajny, Coord. Chem. Rev., 2020, 419, 213394 CrossRef CAS.
- F. Baril-Robert, S. Petit, G. Pilet, G. Chastanet, C. Reber and D. Luneau, Inorg. Chem., 2010, 49, 10970 CrossRef CAS PubMed.
- S. Petit, F. Baril-Robert, G. Pilet, C. Reber and D. Luneau, Dalton Trans., 2009, 6809 RSC.
- D. Guettas, C. M. Balogh, C. Sonneville, Y. Malicet, F. Lepoivre, E. Onal, A. Fateeva, C. Reber, D. Luneau, O. Maury and G. Pilet, Eur. J. Inorg. Chem., 2016, 3932 CrossRef CAS.
- G. Xu, Z.-M. Wang, Z. He, Z. Lu, C.-S. Liao and C.-H. Yan, Inorg. Chem., 2002, 41, 6802 CrossRef CAS PubMed.
- D. Zare, Y. Suffren, L. Guénée, S. V. Eliseeva, H. Nozary, L. Aboshyan-Sorgho, S. Petoud, A. Hauser and C. Piguet, Dalton Trans., 2015, 44, 2529 RSC.
- R. D. Shannon, Acta Crystallogr., Sect. A: Cryst. Phys., Diffr., Theor. Gen. Crystallogr., 1976, 32, 751 CrossRef.
- G. Napier, J. D. Neilson and T. M. Sheperd, Chem. Phys. Lett., 1975, 31, 328–333 CrossRef CAS.
- J.-C. G. Bünzli, Chem. Rev., 2010, 110, 2729 CrossRef PubMed.
- M. H. V. Werts, R. T. F. Jukes and J. W. Verhoeven, Phys. Chem. Chem. Phys., 2002, 4, 1542 RSC.
- M. Pollnau, D. R. Gamelin, S. R. Lüthi, H. U. Güdel and M. P. Hehlen, Phys. Rev. B: Condens. Matter Mater. Phys., 2000, 61, 3337 CrossRef CAS.
- G. M. Salley, R. Valiente and H. U. Güdel, J. Phys.: Condens. Matter, 2002, 14, 5461 CrossRef CAS.
- (a) G. Chen, H. Qiu, P. N. Prasad and X. Chen, Chem. Rev., 2014, 114, 5161 CrossRef CAS PubMed; (b) N. Weibel, L. J. Charbonnière, M. Guardigli, A. Roda and R. Ziessel, J. Am. Chem. Soc., 2004, 126, 4888 CrossRef CAS PubMed.
- L. Sun, R. Wei, J. Feng and H. Zhang, Coord. Chem. Rev., 2018, 364, 10 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Full experimental details, NMR, ESI and photophysical spectra (PDF). See DOI: 10.1039/d0cc07337g |
| ‡ These authors have contributed equally. |
| This journal is © The Royal Society of Chemistry 2021 |