
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceTuning the mechanistic pathways of peptide self-assembly by aromatic interactions†
Goutam
Ghosh
 *,
Kalathil K.
Kartha
and
Gustavo
Fernández
*
*,
Kalathil K.
Kartha
and
Gustavo
Fernández
*
Organisch-Chemisches Institut, Westfälische Wilhelms-Universität Münster, Correnstraße 36, 48149 Münster, Germany. E-mail: gghosh.chem@gmail.com; fernandg@uni-muenster.de
First published on 19th January 2021
Abstract
Herein, we have unravelled the key influence of aromatic interactions on the mechanistic pathways of peptide self-assembly by introducing suitable chromophores (pyrene vs. naphthalene). Although both self-assembled peptides are indistinguishable in their morphologies, this minor structural difference strongly affects the packing modes (parallel vs. antiparallel) and the corresponding self-assembly mechanism (cooperative vs. isodemsic).
Self-assembly is a ubiquitous phenomenon that regulates key processes and functions in living organisms.1 Inspired by these sophisticated architectures and with the aim of addressing crucial issues in biology and materials science, a wide variety of artificial building blocks have been explored for constructing programmable self-assembled nanostructures.2 In this regard, peptides are archetypal building blocks because of their biocompatibility, biodegradability and tunable self-assembly process and corresponding nanostructures.3 Additionally, experimental variables such as pH, temperature, light, ionic strength, solvent interactions and salt concentration are known to strongly affect the aggregation pathways, mechanism and morphology of peptide assemblies.4 More importantly, the self-assembly of peptides is largely dominated by the subtle interplay between different attractive non-covalent interactions (such as aromatic, van der Waals, hydrogen bonding (H-bonding) and electrostatic interactions) and repulsive interactions (such as electrostatic interactions among similar charges and steric effects).5 Among all non-covalent interactions, aromatic interactions not only play a significant role in self-assembly and gelation processes of peptides, but they can also balance the hydrophobicity of the systems.6 Recently, peptide amphiphiles bearing chromophores such as pyrene,7a naphthalene,7b,c anthracene,7d perylene diimide7e and naphthalene diimide7f have received considerable attention for the bottom-up construction of tunable luminescent nanomaterials. However, understanding the relationship between aromatic interactions and mechanistic pathways of peptide self-assembly remains elusive.8
To address this issue, we herein unravel the role of aromatic interactions on tuning the self-assembly mechanism of peptide assemblies in aqueous media. To demonstrate this approach, we have rationally designed a well-known dipeptide sequence “Phe-Phe” (FF)9 and conjugated it with different π-chromophores such as pyrene (Py-FF) and naphthalene (Nap-FF) (Scheme 1). Furthermore, a control molecule without chromophore (Ac-FF) has also been prepared (Scheme S3 in ESI†). Detailed experimental studies of both peptides unveiled a dramatic change in their mechanistic pathways of supramolecular polymerization (SP) depending on the extent of aromatic interactions (cooperative SP for Py-FFvs. isodesmic SP for Nap-FF). Intriguingly, despite the different assembly mechanism, both molecules self-assemble into the same aggregate morphology (1D nanofibers), which is a rare phenomenon in self-assembly.10
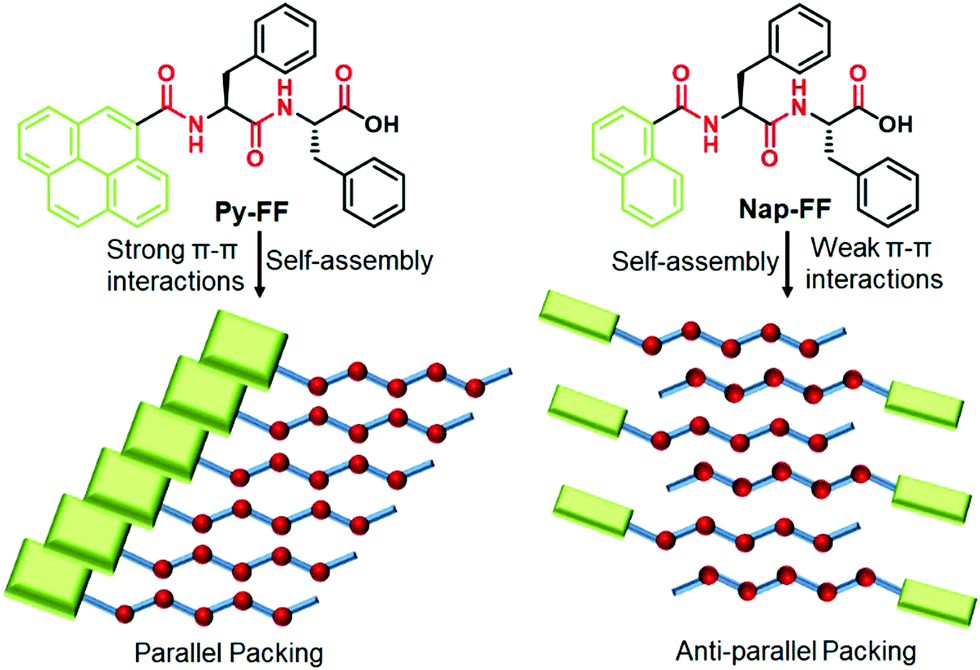 | ||
| Scheme 1 Chemical structures of Py-FF and Nap-FF (top) and schematic representation of their packing modes upon aqueous self-assembly (bottom). | ||
The comparative self-assembly studies of Py-FF and Nap-FF were investigated by different spectroscopic and microscopic techniques such as UV-vis, fluorescence, circular dichroism (CD), Fourier-transform infrared spectroscopy (FTIR), nuclear magnetic resonance (NMR) and atomic force microscopy (AFM). The UV-vis spectra of Py-FF at 1 × 10−4 M and room temperature (RT) in a ‘good’ solvent such as tetrahydrofuran (THF) shows three sharp bands at 341, 276 and 243 nm (Fig. 1a), indicating a monomeric state.11 On the other hand, a broad UV-vis spectrum with significant reduction of absorption is observed for Py-FF in aqueous media (H2O/THF = 9/1), suggestive of a plausible self-assembly through strong π–π interactions (Fig. 1a). By contrast, the UV-vis spectra of Nap-FF in THF and H2O (or in H2O/THF = 9/1) at 1 × 10−4 M and RT exhibit no significant changes, suggesting weak aromatic interactions (Fig. 1e). Fluorescence studies of monomeric Py-FF in THF reveal two strong emission bands at 385 and 404 nm that are considerably reduced upon aggregation in H2O/THF (9/1) (Fig. 1b), suggesting aggregation-caused quenching (ACQ) due to strong π-stacking.12 This behaviour strongly differs for Nap-FF in H2O, where an enhanced emission intensity was observed compared to THF (Fig. 1f). This behaviour may possibly result from aggregation-induced emission (AIE)12 owing to weak aromatic interactions between the chromophores, which further corroborates the findings noticed by UV-vis spectroscopy. The significant differences in the strength of aromatic interactions for both peptides are further supported by the observation of up field shifts for the aromatic protons of Py-FF in solvent-dependent 1H NMR studies (Fig. S7, ESI†), whereas no such shifts were noticed for Nap-FF (Fig. S8, ESI†). FT-IR spectra of both Py-FF and Nap-FF in THF reveal two sharp peaks at 3569 and 3511 cm−1, corresponding to two different non-hydrogen bonded NH protons (Fig. 1c and g). Notably, these two bands merge into a single band at lower frequency (3410 cm−1) in D2O, demonstrating the involvement of strong H-bonding. The self-assembly of Py-FF and Nap-FF was also investigated by CD spectroscopy (Fig. 1d and h). While the solution of monomeric Py-FF in THF is nearly CD silent, strong negative CD signals at 340, 285, 242 and 216 nm were observed upon aggregation in H2O/THF (9/1). The bands at 340, 285 and 242 nm correspond to absorption bands noticed in UV-vis spectroscopy, indicating strong exciton coupling among the pyrene chromophores. The additional intense negative band at 216 nm is suggestive of the formation of β-sheet rich secondary structures. This is further proven by the appearance of two intense peaks at 1634 and 1669 cm−1 in the amide I region in FT-IR studies (Fig. S9, ESI†).13,14 Additionally, a thioflavin T (ThT) assay was also performed to probe β-sheet formation (Fig. S10, ESI†). On the other hand, the CD spectrum of Nap-FF in H2O exhibits an intense single positive band at 222 nm, whereas only residual bands are observed in the region where the naphthalene dye absorbs (250–300 nm; Fig. 1h). These results suggest negligible exciton coupling of the naphthalene chromophores during aqueous self-assembly. The CD signal of Nap-FF originates from the Ac-FF motif, as a similar positive band at 221 nm arises for the latter in water (Fig. S11, ESI†), suggesting a similar type of orientation. As expected, the nearly CD silent spectrum observed for Nap-FF in THF indicates a lack of aggregation in this media (Fig. 1h). Moreover, the CD spectra and ThT assay (Fig. S12, ESI†) of Nap-FF rule out the possibility of β-sheet formation, which is further supported by the position of the amide I bands obtained in FT-IR spectra (Fig. S13, ESI†).14,15 The FT-IR bands of Ac-FF (Fig. S14, ESI†) exhibit similar trends as those found for Nap-FF, further indicating a similar type of packing. Comparison of the CD spectra and ThT assay of Py-FF and Nap-FF discloses clear differences in the supramolecular organization for both systems,16 which agrees well with the observations described earlier.
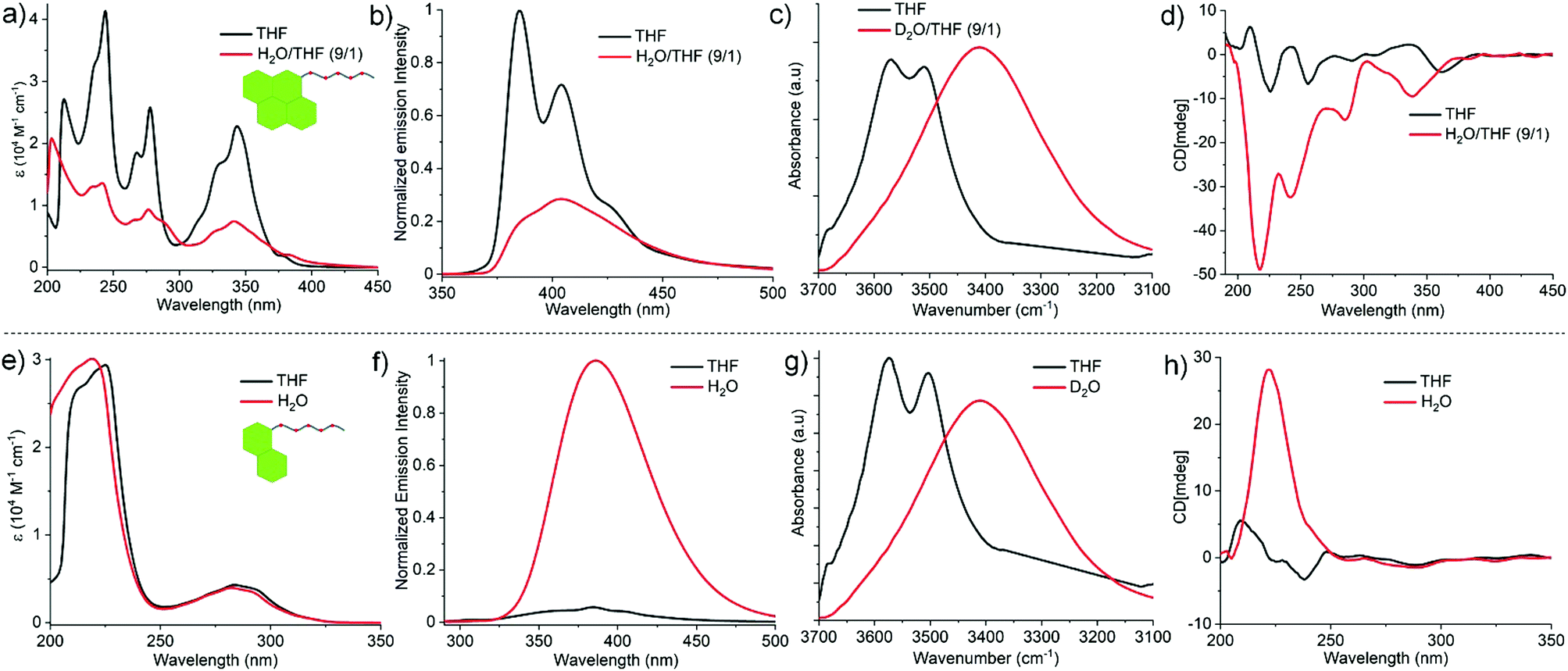 | ||
| Fig. 1 Solvent-dependent UV-vis spectra of (a) Py-FF and (e) Nap-FF (the peak below 250 nm corresponds to n–π* band of amides of FF motif); fluorescence spectra of (b) Py-FF (excitation wavelength = 340 nm) and (f) Nap-FF (excitation wavelength = 290 nm); FTIR spectra of (c) Py-FF and (g) Nap-FF; CD spectra of (d) Py-FF and (h) Nap-FF [C = 1 × 10−4 M; T = 298 K]. | ||
From these observations, we conclude that the self-assembly of Py-FF occurs by the synergistic effect of strong π–π interactions between the pyrene motifs and H-bonding involving the peptide groups via a parallel molecular organization (Scheme 1). On the other hand, the self-assembly of Nap-FF is largely stabilized by H-bonding between the peptide moieties, which have to arrange in a way that no effective aromatic interactions occur between the naphthalene moieties. The arrangement of the Nap-FF monomers in an antiparallel fashion, as shown in Scheme 1, might be one plausible possibility that accounts for the experimental observations.
In order to investigate the mechanistic pathways of self-assembly for both Py-FF and Nap-FF in aqueous media, variable temperature (VT) experiments were carried out by monitoring the UV-vis, fluorescence and CD spectral changes. For Py-FF, cooling the solution from 343 to 288 K at a rate of 1 K min−1 leads to similar spectral changes to those observed when comparing the UV-vis spectra in THF and aqueous solution (see Fig. 1a), indicating a transition from monomeric to aggregated state (Fig. 2a). The obtained non-sigmoidal cooling curves at different concentrations (Fig. 2b) clearly suggest a cooperative self-assembly pathway driven by synergistic aromatic and intermolecular H-bonding interactions. Global fitting of the cooling curves to the nucleation–elongation (cooperative) model17 (Fig. 2b) yields the following parameters: ΔH° = −64.45 kJ mol−1, ΔS° = −127.31 J mol−1 K−1, ΔG° = −26.49 kJ mol−1 and degree of cooperativity (σ) = 4.5 × 10−5 (Table 1). The cooperative mechanism was also supported by the non-sigmoidal cooling curves extracted from VT-CD (Fig. S16, ESI†) and VT-fluorescence studies (Fig. S17, ESI†).
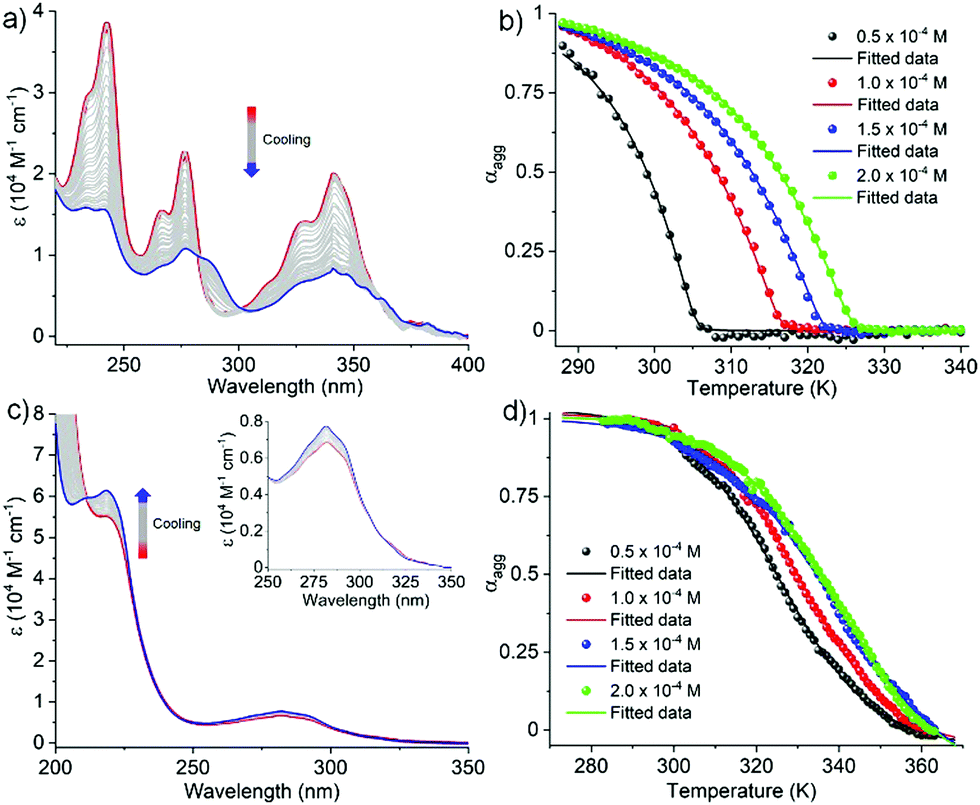 | ||
| Fig. 2 (a) VT UV-vis spectra of Py-FF in H2O/THF (9/1); (b) cooling curves of Py-FF monitored at 243 nm and fitted to the cooperative model; (c) VT UV-vis spectra of Nap-FF in H2O (inset image corresponds to zoomed area of the absorption of naphthalene moiety); (d) cooling curves of Nap-FF monitored at 219 nm and fitted to the isodesmic model [cooling rate = 1 K min−1; C = 1 × 10−4 M]. | ||
| Compound | ΔH° [kJ mol−1] | ΔS° [J mol−1 K−1] |
|
T e/K | ΔG° [kJ mol−1] | K el/M−1 [298 K] | K nucl/M#x2212;1 [298 K] | σ |
|---|---|---|---|---|---|---|---|---|
| Py-FF | −64.45 | −127.31 | −24.81 | 316.1 | −26.49 | 4.4 × 104 | 1.98 | 4.5 × 10−5 |
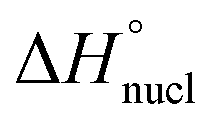
| ΔH [kJ mol−1] | ΔS [J mol−1 K−1] | ΔG° [kJ mol−1] | K a/M−1 [298 K] | |
|---|---|---|---|---|
| Nap-FF | −74.19 | −151.31 | −29.10 | 1.7 × 105 |
In contrast to Py-FF, VT-UV-vis cooling experiments for Nap-FF (from 363 to 283 K; 1 K min−1) disclose clear sigmoidal curves at different concentrations (Fig. 2d), indicating an isodesmic supramolecular polymerization.18 This switch in mechanism from cooperative (for Py-FF) to isodesmic (for Nap-FF) can be rationalized by the lack of synergistic non-covalent interactions for the latter, i.e. largely driven by intermolecular H-bonding.19 Application of the isodesmic model to the experimental data allows the derivation of the corresponding thermodynamic parameters (Fig. S19, ESI†): ΔH = −74.19 kJ mol−1, ΔS = −151.31 J mol−1 K−1, ΔG° = −29.1 kJ mol−1 (Table 1). In analogy to Py-FF, the isodesmic mechanism for Nap-FF was further supported by VT-CD (Fig. S20, ESI†) and VT-fluorescence studies (Fig. S21, ESI†).
Interestingly, in stark contrast to most examples of supramolecular assemblies reported to date, the different assembly mechanism for Py-FF and Nap-FF is not accompanied by a significant change in aggregate morphology. Microscopic studies by AFM on mica disclose similar type of fiber-like 1D morphologies for both compounds. Whereas Py-FF self-assembles into 1D nanofibers (height 2.5 ± 0.3 nm and width 60-70 nm) with lengths of several micrometers (Fig. 3a and Fig. S22, ESI†), bundled 1D nanofibers (Fig. 3b; height 3.0 ± 0.5 nm and width 35 nm of single fiber, Fig. S23, ESI†) are formed by Nap-FF. The higher tendency of Nap-FF to bundle might be explained by the difficulty in shielding the hydrophobic naphthalene core from the polar solvent molecules in the proposed antiparallel arrangement (see Scheme 1), leading to a more pronounced lateral growth.
 | ||
| Fig. 3 AFM images of (a) Py-FF and (b) Nap-FF [C = 1 × 10−4 M]. | ||
In summary, we have rationally designed two peptide amphiphiles with different chromophores (pyrene: Py-FF and naphthalene: Nap-FF) and studied their comparative aqueous self-assembly by various techniques. Interestingly, the different extent of aromatic interactions of the pyrene and naphthalene groups governs the packing modes (parallel for Py-FFvs. antiparallel for Nap-FF) and the resulting self-assembly mechanism (cooperative for Py-FFvs. isodesmic for Nap-FF). Despite these marked differences, both molecules form the same type of aggregate morphology (1D nanofibers), a phenomenon that is rare in self-assembly. Our experimental findings highlight the importance of aromatic interactions in controlling the mechanism of peptide self-assembly and, consequently, the properties of the secondary structures, which may have important implications in the biomedical field.
G. G., K. K. K. and G. F. thank the European Commission (European Research Council) for funding (ERC-StG-2016 SUPRACOP-715923).
Conflicts of interest
There are no conflicts to declare.Notes and references
- G. M. Whitesides, J. P. Mathias and C. T. Seto, Science, 1991, 254, 1312 CrossRef CAS.
- (a) T. Aida, E. W. Meijer and S. Stupp, Science, 2012, 335, 813 CrossRef CAS; (b) C. Rest, R. Kandanelli and G. Fernández, Chem. Soc. Rev., 2015, 44, 2543 RSC; (c) Z. Chen, A. Lohr, C. R. Saha-Möller and F. Würthner, Chem. Soc. Rev., 2009, 38, 564 RSC; (d) S. S. Babu, V. K. Praveen and A. Ajayaghosh, Chem. Rev., 2014, 114, 1973 CrossRef CAS; (e) G. Ghosh, P. Dey and S. Ghosh, Chem. Commun., 2020, 56, 6757 RSC.
- (a) F. Qiu, Y. Chen, C. Tang and X. Zhao, Int. J. Nanomed., 2018, 13, 5003 CrossRef CAS; (b) W. K. Restu, Y. Nishida, S. Yamamoto, J. Ishii and T. Maruyama, Langmuir, 2018, 34, 8065 CrossRef CAS; (c) E. Radvar and H. S. Azevedo, Macromol. Biosci., 2019, 19, 1800221 CrossRef; (d) D. E. Clarke, C. D. J. Parmenter and O. A. Scherman, Angew. Chem., Int. Ed., 2018, 57, 7709 CrossRef CAS; (e) Q. Meng, Y. Kou, X. Ma, Y. Liang, L. Guo, C. Ni and K. Liu, Langmuir, 2012, 28, 5017 CrossRef CAS.
- (a) G. Ghosh, R. Barman, J. Sarkar and S. Ghosh, J. Phys. Chem. B, 2019, 123, 5909 CrossRef CAS; (b) G. Ghosh and G. Fernández, Beilstein J. Org. Chem., 2020, 16, 2017 CrossRef; (c) H. Frisch, Y. Nie, S. Raunser and P. Besenius, Chem. – Eur. J., 2015, 21, 3304 CrossRef CAS; (d) K. Bauri, M. Nandi and P. De, Polym. Chem., 2018, 9, 1257 RSC; (e) N. Falcone and H. B. Kraatz, Chem. – Eur. J., 2018, 24, 14316 CrossRef CAS; (f) P. A. Korevaar, C. J. Newcomb, E. W. Meijer and S. I. Stupp, J. Am. Chem. Soc., 2014, 136, 8540 CrossRef CAS; (g) D. Spitzer, L. L. Rodrigues, D. Straßburger, M. Mezger and P. Besenius, Angew. Chem., Int. Ed., 2017, 56, 15461 CrossRef CAS.
- (a) J. Wang, K. Liu, R. Xing and X. Yan, Chem. Soc. Rev., 2016, 45, 5589 RSC; (b) R. Otter and P. Besenius, Org. Biomol. Chem., 2019, 17, 6719 RSC.
- (a) A. Dasgupta and D. Das, Langmuir, 2019, 35, 10704 CrossRef CAS; (b) S. Fleming and R. V. Ulijn, Chem. Soc. Rev., 2014, 43, 8150 RSC; (c) A. Ustinov, H. Weissman, E. Shirman, I. Pinkas, X. Zuo and B. Rybtchinski, J. Am. Chem. Soc., 2011, 133, 16201 CrossRef CAS.
- (a) B. Pramanik, N. Singha and D. Das, ACS Appl. Polym. Mater., 2019, 1, 833 CrossRef CAS; (b) L. Chen, K. Morris, A. Laybourn, D. Elias, M. R. Hicks, A. Rodger, L. Serpell and D. J. Adams, Langmuir, 2010, 26, 5232 CrossRef CAS; (c) A. Das and S. Ghosh, Chem. – Eur. J., 2010, 16, 13622 CrossRef CAS; (d) P. K. Gavel, D. Dev, H. S. Parmar, S. Bhasin and A. K. Das, ACS Appl. Mater. Interfaces, 2018, 10, 10729 CrossRef CAS; (e) S. Ahmed, K. N. Amba Sankar, B. Pramanik, K. Mohanta and D. Das, Langmuir, 2018, 34, 8355 CrossRef CAS; (f) N. Singha, P. Gupta, B. Pramanik, S. Ahmed, A. Dasgupta, A. Ukil and D. Das, Biomacromolecules, 2017, 18, 3630 CrossRef CAS.
- S. Ogi, K. Matsumoto and S. Yamaguchi, Angew. Chem., Int. Ed., 2018, 57, 2339 CrossRef CAS.
- (a) M. Reches and E. Gazit, Science, 2003, 300, 625 CrossRef CAS; (b) S. Marchesan, A. V. Vargiu and K. E. Styan, Molecules, 2015, 20, 19775 CrossRef CAS.
- N. M. Casellas, S. Pujals, D. Bochicchio, G. M. Pavan, T. Torres, L. Albertazzi and M. García-Iglesias, Chem. Commun., 2018, 54, 4112 RSC.
- J. Duhamel, Polymers, 2012, 4, 211 CrossRef CAS.
- (a) F. Würthner, Angew. Chem., Int. Ed., 2020, 59, 14192 CrossRef; (b) Z. Zhao, H. Zhang, J. W. Y. Lam and B. Z. Tang, Angew. Chem., Int. Ed., 2020, 59, 9888 CrossRef CAS.
- (a) P. I. Haris and D. Chapman, Biopolymers, 1995, 37, 251 CrossRef CAS; (b) M. Jackson and H. H. Mantsch, Crit. Rev. Biochem. Mol. Biol., 1995, 30, 95 CrossRef CAS; (c) J. P. Schneider, D. J. Pochan, B. Ozbas, K. Rajagopal, L. Pakstis and J. Kretsinger, J. Am. Chem. Soc., 2002, 124, 15030 CrossRef CAS; (d) H. Yan, A. Saiani, J. E. Gough and A. F. Miller, Biomacromolecules, 2006, 7, 2776 CrossRef CAS.
- (a) N. Yamada, K. Ariga, M. Naito, K. Matsubara and E. Koyama, J. Am. Chem. Soc., 1998, 120, 12192 CrossRef CAS; (b) Y. Zou, Y. Li, W. Hao, X. Hu and G. Ma, J. Phys. Chem. B, 2013, 117, 4003 CrossRef CAS.
- The absence of amide I bands below 1640 cm−1 and above 1680 cm−1 for Nap-FF supports its inability to form antiparallel β-sheets (for details, see ref. 14).
- (a) Y. Zhang, H. Gu, Z. Yang and B. Xu, J. Am. Chem. Soc., 2003, 125, 13680 CrossRef CAS; (b) P. G. Argudo, R. Contreras-Montoya, L. Á. de Cienfuegos, J. M. Cuerva, M. Cano, D. Alba-Molina, M. T. Martín-Romero, L. Camacho and J. J. Giner-Casares, Soft Matter, 2018, 14, 9343 RSC.
- (a) H. M. M. ten Eikelder, A. J. Markvoort, T. F. A. de Greef and P. A. J. Hilbers, J. Phys. Chem. B, 2012, 116, 5291 CrossRef CAS; (b) A. J. Markvoort, H. M. M. ten Eikelder, P. A. J. Hilbers, T. F. A. de Greef and E. W. Meijer, Nat. Commun., 2011, 2, 509 CrossRef.
- M. M. J. Smulders, M. M. L. Nieuwenhuizen, T. F. A. de Greef, P. van der Schoot, A. P. H. J. Schenning and E. W. Meijer, Chem. – Eur. J., 2010, 16, 362 CrossRef CAS.
- (a) M. M. L. Nieuwenhuizen, T. F. A. de Greef, R. L. J. van der Bruggen, J. M. J. Paulusse, W. P. J. Appel, M. M. J. Smulders, R. P. Sijbesma and E. W. Meijer, Chem. – Eur. J., 2010, 16, 1601 CrossRef CAS; (b) W. P. J. Appel, M. M. L. Nieuwenhuizen and E. W. Meijer, in Supramolecular Polymer Chemistry, ed. A. Harada, Wiley-VCH, Germany, 2012, ch. 1, vol. 1, pp. 1–28 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0cc07199d |
| This journal is © The Royal Society of Chemistry 2021 |