
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePEGylated sequence-controlled macromolecules using supramolecular binding to target the Taspase1/Importin α interaction†
Peter
Pasch‡
a,
Alexander
Höing‡
 b,
Serap
Ueclue
a,
Matthias
Killa
c,
Jens
Voskuhl
c,
Shirley K.
Knauer
*b and
Laura
Hartmann
*a
b,
Serap
Ueclue
a,
Matthias
Killa
c,
Jens
Voskuhl
c,
Shirley K.
Knauer
*b and
Laura
Hartmann
*a
aDepartment for Organic Chemistry and Macromolecular Chemistry, Heinrich Heine University Düsseldorf, Universitätsstraße 1, Düsseldorf 40225, Germany. E-mail: laura.hartmann@hhu.de
bDepartment for Molecular Biology II, Center of Medical Biotechnology (ZMB) University Duisburg-Essen, Universitätsstrasse 5, Essen, 45117, Germany. E-mail: shirley.knauer@uni-due.de
cFaculty of chemistry (Organic Chemistry), University of Duisburg-Essen, Universitätsstraße 7, 45141 Essen, Germany
First published on 12th February 2021
Abstract
A novel strategy to inhibit the oncologically relevant protease Taspase1 is explored by developing PEGylated macromolecular ligands presenting the supramolecular binding motif guanidiniocarbonylpyrrole (GCP). Taspase1 requires interaction of its nuclear localization signal (NLS) with import receptor Importin α. We show the synthesis and effective interference of PEGylated multivalent macromolecular ligands with Taspase1–Importin α-complex formation.
Proteins are an important class of biomacromolecules and their interactions play key roles in almost every process of a living organism. Understanding and manipulating protein interactions offers the opportunity to treat or fight diseases.1 Many protein–protein interactions rely on so-called multivalent binding events where multiple sites of the proteins have to interact simultaneously in order to create a strong binding.2,3 Accordingly, synthetic molecules to interfere with protein binding often are multivalent constructs as well, consisting of a synthetic scaffold presenting multiple binding units. The design of multivalent molecules is as diverse as the protein targets they address – one important class of scaffolds being polymers due to their synthetic ease and variability.3,4
Today, polymer chemistry offers a new tool: the synthesis of sequence-controlled macromolecules. Different synthetic strategies have been introduced for the synthesis of sequence-controlled polymers and give access to multifunctional macromolecules with high levels of structural and thereby potentially also functional control.5 We have developed the so-called solid phase polymer synthesis, where we employ standard peptide chemistry and tailor-made non-natural building blocks to generate sequence-defined macromolecules presenting different binding units such as carbohydrates, peptides or catechols. We have successfully demonstrated that through control over the monomer sequence and thereby parameters such as the number and position of binding units, architecture and conformation of the macromolecule, new and improved modulators of protein interactions are accessible.6
In this work, we extend on our previous concept with a non-natural supramolecular binding motif, the guanidiniocarbonyl-pyrrole (GCP) motif,7 an arginine mimetic that binds oxoanions via a hydrogen-bond-assisted ion pairing8 and shows significantly stronger affinity than natural amino acids.9 Our goal is to create macromolecular inhibitors of Taspase1 protease (Fig. 1). From proliferation to differentiation right up to apoptosis, almost every cellular process is regulated by or involves proteases.10,11 One of the 28 threonine proteases encoded in the human genome is the tumor-relevant Threonine aspartase 1 (Taspase1).10,12 It is usually expressed during embryonic development, but it is re-expressed in many tumor cell lines, and a knockout decreases proliferation and promotes apoptosis in correlation to their potential drug target in tumor therapy. Surprisingly, earlier studies revealed that Taspase1 is not affected by general former Taspase1 expression levels13,14 making Tasapse1 a protease inhibitor.12,14,15 Previous studies focused on the enzymatic activity of Taspase1.13,15–17 In this study, we aim at a different inhibition mechanism for Taspase1 by targeting functionally relevant interactions with the import receptor Importin α.18,19 While Taspase1 effectively cleaves other pro-enzymes as a heterodimer consisting of the subunits α (25 kDa) and β (20 kDa), Taspase1 itself is also expressed as an inactive α/β-monomer (45 kDa) and undergoes autoproteolytic activation.12,20,21 Autoproteolysis is supposed to take place inside the nucleus where Taspase1 is transported by interaction of its bipartite nuclear localization signal (NLS) located in the Taspase1 α-subunit (Fig. 1B) with Importin α.18,19,21 In the nucleus, the Taspase1 monomer undergoes autoproteolysis, and the two subunits reassemble to form the active Taspase1 heterodimer.12,21 Thus, interaction of the Taspase1 NLS with Importin α is pivotal for activation. Here, we aim at developing ligands that effectively block the NLS and thereby inhibit Importin α-complex formation as the first step of Taspase1 activation. Since basic amino acid clusters constituting the bipartite NLS of Taspase1 are flanked by multiple anionic amino acids such as aspartic and glutamic acids (see Fig. 1C), we envision that macromolecules presenting multiple oxo-anion binding motifs, GCP, should allow for binding to this site of the protein. It was previously shown that multivalent GCP ligands allow for the design of high affinity ligands by addressing multiple binding sites within a protein structure and can be used for stabilization of protein–protein complexes.22 Here we now want to realize both high affinity binding to the NLS and at the same time effective inhibition of binding to Importin α. We rationalize that in order to achieve both, we require two features of the macromolecular ligand – one segment presenting multiple GCP motifs able to address anionic amino acids in the NLS domain but that do not mediate binding with Importin α, and a second segment, ideally non-binding and sterically demanding to shield the NLS domain from any further interaction. This design is thus based on the general concept of sterical shielding for multivalent ligands to achieve inhibition.3,23
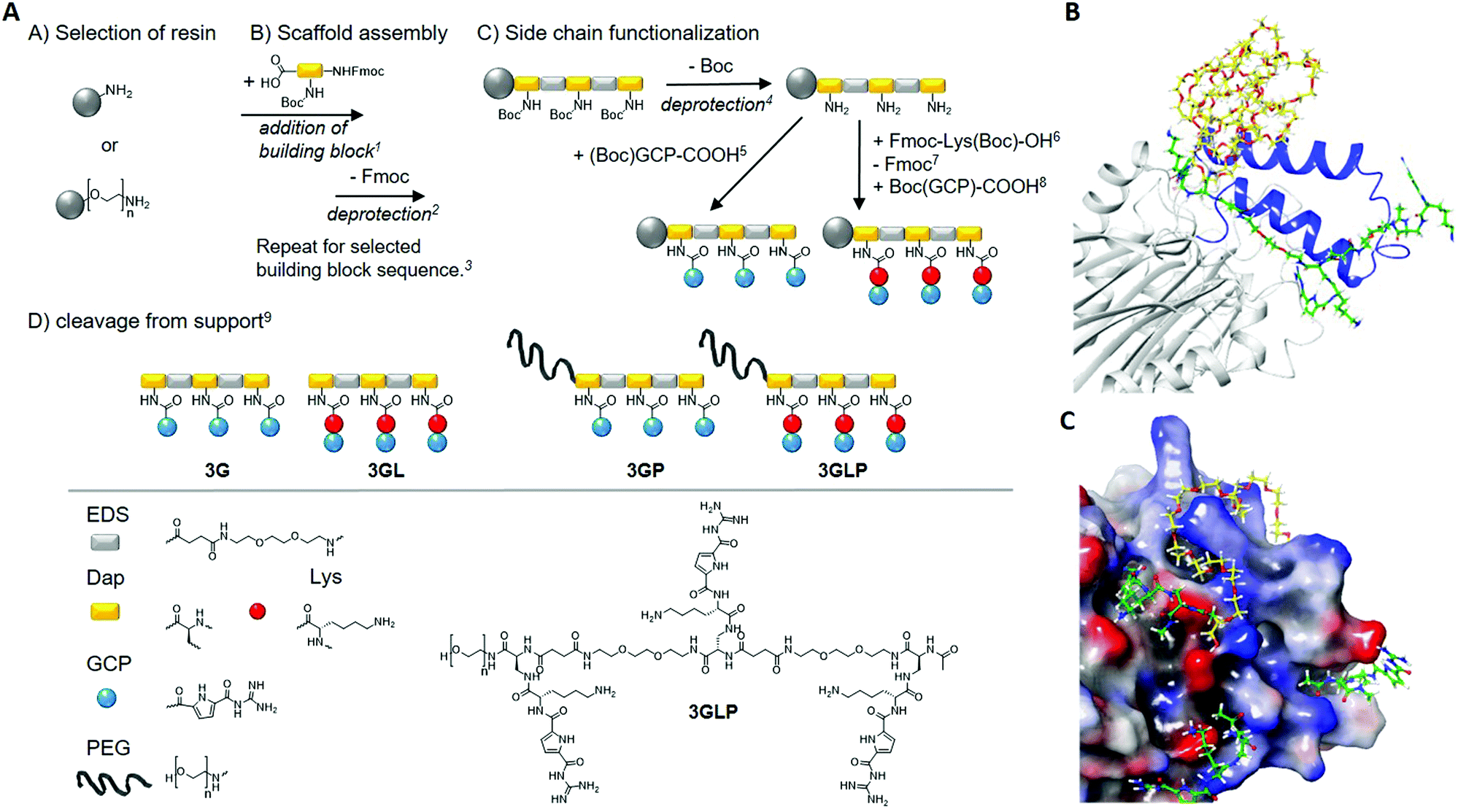 | ||
Fig. 1 Synthesis of GCP macromolecules using solid phase polymer synthesis (left) and a model of a supramolecular ligand blocking the NLS of Taspase1 (right). (A) Reaction conditions: 15 eq. building block, 5 eq. PyBOP, 10 eq. DIPEA in DMF, 90 min, 225v% piperidine in DMF, 20 min, 3Next Fmoc-deprotection and acetylation of N-terminus (Ac2O, 20 min), 44 M HCl in dioxane, 20 min (on-resin cleavage of Boc), 55 eq. (Boc)GCP-COOH, 5 eq. PyBOP, 10 eq. DIPEA in DMF, 90 min (double coupling), 65 eq. Fmoc-Lys(Boc)-OH, 5 eq. PyBOP, 10 eq. DIPEA in DMF, 90 min (double coupling), 725v% piperidine in DMF, 20 min, 85 eq. (Boc)GCP-COOH, 5 eq. PyBOP, 10 eq. DIPEA in DMF, 90 min (double coupling), 9TentaGel® S RAM: 5% triisopropylsilane, 95% TFA, 90 min, TentaGel® PAP: TFA/thioanisole (95![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :5), 24 hours. PEG chain n = 70 (MW = 3106.7 g mol−1). (B) Model of a supramolecular ligand blocking the NLS of Taspase1. (C) Schematic illustration of 3GLP (green) as it addresses carboxylates near the loop (blue) while the PEG (yellow) masks the cationic loop. (bottom): the carboxylates (red) are addressed with GCP (green) and the cationic loop (blue) is masked by the PEG chain (yellow). The model is based on the literature.28 :5), 24 hours. PEG chain n = 70 (MW = 3106.7 g mol−1). (B) Model of a supramolecular ligand blocking the NLS of Taspase1. (C) Schematic illustration of 3GLP (green) as it addresses carboxylates near the loop (blue) while the PEG (yellow) masks the cationic loop. (bottom): the carboxylates (red) are addressed with GCP (green) and the cationic loop (blue) is masked by the PEG chain (yellow). The model is based on the literature.28 | ||
For the first segment, we employ the previously established synthesis of sequence-controlled macromolecules (Fig. 1A).24,25Via stepwise addition on a solid support, a monodisperse, sequence-controlled scaffold is assembled and used for site selective attachment of GCP motifs.26 Here we used a previously developed EDS building block (4-((2-(2-(2-aminoethoxy)ethoxy)ethyl)-amino)-4-oxobutanoic) introducing hydrophilic ethylene glycol units within the backbone.25 Fmoc-Dap(Boc)-OH (Nα-Fmoc-Nβ-Boc-L-2,3-diaminopropionic acid) was applied for attachment of GCP on the side chains: Dap side chains were deprotected on the solid support cleaving the Boc protecting groups and releasing primary amines for further functionalization with carboxylated GCP-derivative (see the ESI†). In order to further increase the affinity of GCP towards anionic amino acids, lysine as a cationic amino acid was added next to the GCP side chain by including an additional Fmoc-Lys(Boc)-OH during side chain assembly. Two different macromolecules were synthesized introducing three GCP side chains (3G) as well as Lys-GCP side chains (3GL). Model calculations suggest that multiple amino acids could be addressed via trivalent GCP macromolecules with one EDS as a spacer in between the binding motifs (Fig. 1). As our second segment in order to create GCP macromolecule inhibitors, we chose poly(ethylene glycol) (PEG) of 3 kDa that can be easily installed by starting the solid phase assembly from a PEG-preloaded resin giving PEGylated GCP macromolecules 3GP and 3GLP. PEG is well known as a so-called stealth polymer to minimize non-specific interaction with proteins and to act as a steric shield blocking protein–protein interactions.27 All macromolecules were cleaved off the resin, purified by preparative HPLC, isolated by freeze drying with relative purities >95% (as determined by RP-HPLC) and further characterized by 1H-NMR, UHRMS and MALDI-ToF analysis (see the ESI†).
First, we looked at the direct binding of our ligands to Taspase1 by successfully setting up a surface plasmon resonance (SPR) assay (see the ESI†). Applying isothermal calorimetry was not successful at this time (see the ESI†). Monovalent macromolecules presenting only one GCP unit showed no binding and thus were omitted from any further testing (see the ESI†). For the trivalent macromolecules, binding is in the μM range as was expected based on previous GCP ligands (Fig. 2).29
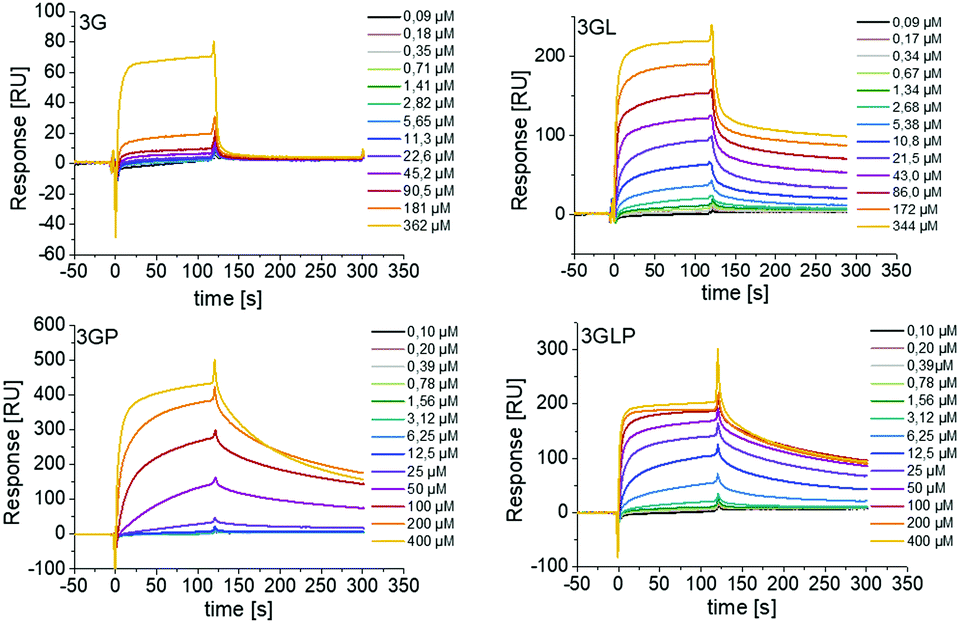 | ||
| Fig. 2 SPR sensograms showing direct binding of trivalent GCP-ligands with and without PEG to immobilized Taspase1. Monovalent GCP-ligands showed no binding (see the ESI†). | ||
We observe a clear increase in binding upon introduction of the lysine residues next to the GCP unit, indicating an increase in affinity through the additional cationic moieties. Surprisingly, for 3G we see an increase in binding upon introduction of the PEG block (3GP) which might be attributed to the higher molecular weight of this ligand and slower diffusion, as PEG itself showed no binding (see the ESI†). However, we did not see such increase for 3GLP.
We next performed an in vitro pull-down assay to investigate the proposed inhibitory effect on the interaction between Taspase1 and Importin α. For this, we used recombinant GST-Importin α protein bound to a GSH matrix and added Taspase1-His pre-incubated with the respective ligands (see the ESI†). Unbound protein was removed and the Tasapse1 bound to the matrix via its interaction with Importin α eluted. The samples were then analyzed by SDS-PAGE and Western Blotting. To validate our working hypothesis that only the PEGylated compounds will disrupt the protein interaction, we first compared the ligands (3GP, 3GLP) and the controls (PEG alone, the non-PEGylated ligands 3G and 3GL) directly (Fig. 3). Indeed, the interaction between Taspase1 and Importin α was effectively disrupted by the pre-incubation of Taspase1 with the PEGylated GCP-ligands. Interestingly, ligands missing the PEG stealth block failed to interfere with Taspase1–Importin α complex formation and so did PEG itself. This suggests that the GCP-motif guides the ligand to Taspase1, but is not able to shield the NLS directly, in line with our model (Fig. 1). Furthermore, the PEG block itself does not affect the interaction and therefore does not ensnare the NLS while unguided. Thus, the introduction of the stealth block PEG to the guiding GCP-block is a necessary step for the ligands to act as inhibitors. Densiometric quantification of Western Blot analysis revealed a slightly increased effect of the lysine-containing ligand 3GLP (40% Taspase bound) compared to 3GP (55% Taspase1 bound) (see the ESI†). To further compare the PEGylated ligands that differ in their binding motifs (GCP and GCP plus lysine) 3GP and 3GLP were tested at different ligand concentrations ranging from 0 μM to 200 μM in the pull-down assay and the results were again quantified (Fig. 3). 3GP effectively hampered the interaction already at 50 μM (87% Taspase1 bound). Increasing the concentration to more than 100 μM (59% Taspase1 bound) was not able to additionally fortify its effect. As seen in the direct binding study via SPR, addition of lysine next to the GCP motifs increased the apparent affinity. We hypothesize that this, when combined with the PEG segment, should also give more efficient inhibitors. Indeed, 3GLP was effective already at 10 μM (82% Taspase1 bound), and its potency reached a limit at a concentration of 50 μM (41% Taspase1 bound). Importantly, we did not observe a comparable effect when using the PEG control or the non-PEGylated ligands (see the ESI†), nor was the binding of Importin α to the column affected by the PEGylated ligand (see the ESI†).
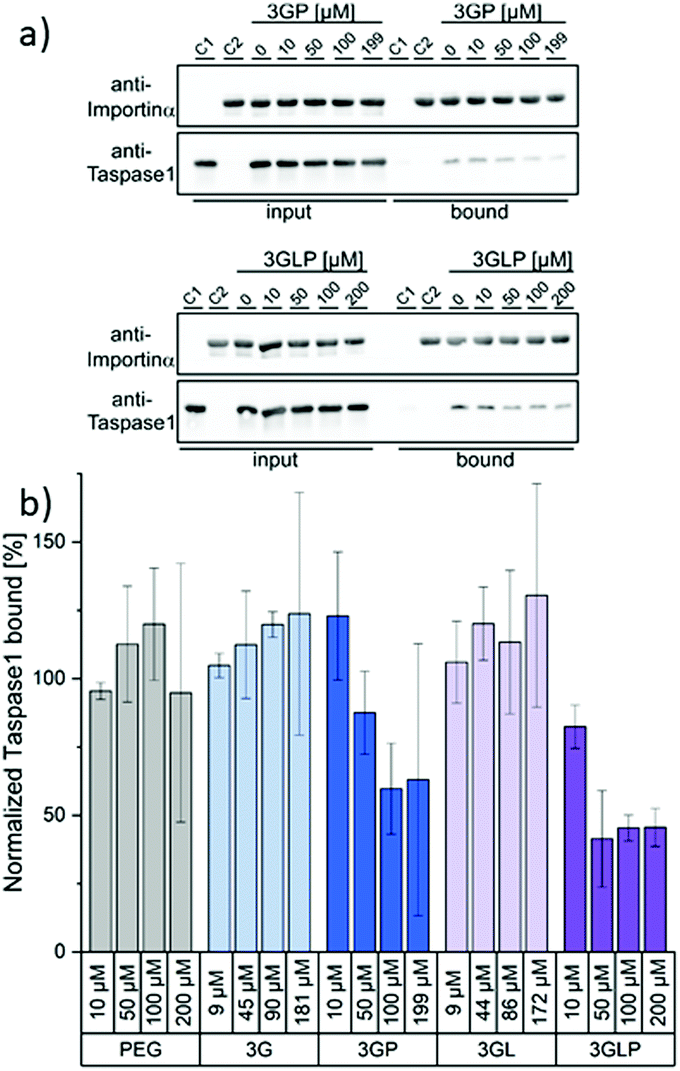 | ||
| Fig. 3 The interaction between Taspase1 and Importin α is effectively disrupted by PEGylated GCP-ligands: (a) Exemplary blots from pull-down assays with increasing concentrations of the PEGylated GCP-ligands 3GP and 3GLP (see the ESI† for other samples). Column-bound protein fraction after the pull-down in comparison to the input initially present on the column. Controls: only Taspase 1 (C1) or GST-Importin α (C2) were added to the column. (b) Densiometric quantification of pull-down assays, comprising the mean of three replicates ± standard deviation. | ||
In conclusion, we explored the concept of multivalent ligands for sterical shielding and developed structure-guided PEGylated sequence-controlled macromolecules for Taspase1 using GCP as a binding motif. We further showed that these ligands could effectively be used to disrupt the functionally relevant interaction with Importin α in a concentration-dependent manner, thereby exploiting a novel inhibition mechanism for this protease. Future studies will include investigations concerning the potential selectivity of the ligands as well as their potential for cellular studies. As a first prerequisite, the ligands were tested in a cell viability assay and showed no toxicity (see the ESI†).
The authors acknowledge the CRC 1093 funded by the “Deutsche Forschungsgemeinschaft”
Conflicts of interest
There are no conflicts to declare.Notes and references
- H. Lu, Q. Zhou, J. He, Z. Jiang, C. Peng, R. Tong and J. Shi, Signal Transduction Targeted Ther., 2020, 5, 213 CrossRef.
- (a) E. Mahon and M. Barboiu, Org. Biomol. Chem., 2015, 13, 10590–10599 RSC; (b) C. T. Varner, T. Rosen, J. T. Martin and R. S. Kane, Biomacromolecules, 2015, 16(1), 43–55 CrossRef CAS.
- M. Mammen, S.-K. Choi and G. M. Whitesides, Angew. Chem., Int. Ed., 1998, 37, 2754–2794 CrossRef.
- (a) C. Fasting, C. A. Schalley, M. Weber, O. Seitz, S. Hecht, B. Koksch, J. Dernedde, C. Graf, E.-W. Knapp and R. Haag, Angew. Chem., Int. Ed., 2012, 51, 10472–10498 CrossRef CAS; (b) C. R. Becer, Macromol. Rapid Commun., 2012, 33, 742–752 CrossRef CAS.
- (a) S. C. Solleder, R. V. Schneider, K. S. Wetzel, A. C. Boukis and M. A. R. Meier, Macromol. Rapid Commun., 2017, 38, 1600711 CrossRef; (b) S. A. Hill, C. Gerke and L. Hartmann, Chem. – Asian J., 2018, 13, 3611 CrossRef CAS; (c) J. F. Lutz, M. Ouchi, D. R. Liu and M. Sawamoto, Science, 2013, 341(6146), 1238149 CrossRef.
- (a) L. Soria-Martinez, S. Bauer, M. Giesler, S. Schelhaas, J. Materlik, K. Janus, P. Pierzyna, M. Becker, N. L. Snyder, L. Hartmann and M. Schelhaas, J. Am. Chem. Soc., 2020, 142(11), 5252–5265 CrossRef CAS; (b) S. Boden, F. Reise, J. Kania, T. K. Lindhorst and L. Hartmann, Macromol. Biosci., 2019, 19, 1800425 CrossRef; (c) L. Fischer, R. C. Steffens, T. J. Paul and L. Hartmann, Polym. Chem., 2020, 11, 6091–6096 RSC; (d) T. Freichel, V. Heine, D. Laaf, E. E. Mackintosh, S. Sarafova, L. Elling, N. L. Snyder and L. Hartmann, Macromol. Biosci., 2020, 20, 2000163 CrossRef CAS.
- (a) C. Schmuck and M. J. Schwegmann, J. Am. Chem. Soc., 2005, 127, 3373–3379 CrossRef CAS; (b) D. Maity, A. Gigante, P. A. Sánchez-Murcia, E. Sijbesma, M. Li, D. Bier, S. Mosel, S. Knauer, C. Ottmann and C. Schmuck, Org. Biomol. Chem., 2019, 17, 4359–4363 RSC.
- (a) C. Schmuck, Coord. Chem. Rev., 2006, 250, 3053–3067 CrossRef CAS; (b) J. Matić, F. Šupljika, T. Tandarić, M. Dukši, P. Piotrowski, R. Vianello, A. Brozovic, I. Piantanida, C. Schmuck and M. R. Stojković, Int. J. Biol. Macromol., 2019, 134, 422–434 CrossRef.
- (a) C. Schmuck and M. Heil, ChemBioChem, 2003, 4(11), 1232–1238 CrossRef CAS; (b) X. Liu, K. Wang, M. Externbrink, J. Niemeyer, M. Giese and X. Y. Hu, Chin. Chem. Lett., 2020, 31(5), 1239–1242 CrossRef CAS.
- C. López-Otín and L. M. Matrisian, Nat. Rev. Cancer, 2007, 7, 800–808 CrossRef.
- C. López-Otín and J. S. J. Bond, Biol. Chem., 2008, 283, 30433–30437 CrossRef.
- J. J.-D. Hsieh, E. H.-Y. Cheng and S. J. Korsmeyer, Cell, 2003, 115, 293–303 CrossRef CAS.
- D. Y. Chen, Y. Lee, B. A. van Tine, A. C. Searleman, T. D. Westergard, H. Liu, H.-C. Tu, S. Takeda, Y. Dong and D. R. Piwnica-Worms, Cancer Res., 2012, 72, 736–746 CrossRef CAS.
- D. Y. Chen, H. Liu, S. Takeda, H.-C. Tu, S. Sasagawa, B. A. van Tine, D. Lu, E. H.-Y. Cheng and J. J.-D. Hsieh, Cancer Res., 2010, 70, 5358–5367 CrossRef CAS.
- S. K. Knauer, V. Fetz, J. Rabenstein, S. Friedl, B. Hofmann, S. Sabiani, E. Schröder, L. Kunst, E. Proschak and E. Thines, PLoS One, 2011, 6, 18253 CrossRef.
- J. T. Lee, D. Y. Chen, Z. Yang, A. D. Ramos, J. J.-D. Hsieh and M. Bogyo, Bioorg. Med. Chem. Lett., 2009, 19, 5086–5090 CrossRef CAS.
- J. van den Boom, M. Mamić, D. Baccelliere, S. Zweerink, F. Kaschani, S. Knauer, P. Bayer and M. Kaiser, ChemBioChem, 2014, 15, 2233–2237 CrossRef CAS.
- Y. Miyamoto, K. Yamada and Y. Yoneda, J. Biochem., 2016, 160, 2728–2735 CrossRef.
- C. Bier, S. K. Knauer, D. Docter, G. Schneider, O. H. Kramer and R. H. Stauber, Traffic, 2011, 12, 703–714 CrossRef CAS.
- D. Wünsch, A. Hahlbrock, S. Jung, T. Schirmeister, J. van den Boom, O. Schilling, S. K. Knauer and R. H. Stauber, Oncogene, 2016, 35, 3351–3364 CrossRef.
- A. Corbett, K. Mills, B. A. Lange, M. Stewart, E. Devine and J. C. J. Lange, Biol. Chem., 2007, 282, 5101–5105 CrossRef.
- L. Bartsch, M. Bartel, A. Gigante, J. Iglesias-Fernández, Y. B. Ruiz-Blanco, C. Beuck, J. Briels, N. Toetsch, P. Bayer, E. Sanchez-Garcia, C. Ottmann and C. Schmuck, ChemBioChem, 2019, 20(23), 2921–2926 CrossRef CAS.
- F. Jacobi, D. Wilms, T. Seiler, T. Queckbörner, M. Tabatabai, L. Hartmann and S. Schmidt, Biomacromolecules, 2020, 21(12), 4850–4856 CrossRef CAS.
- (a) F. Wojcik, S. Mosca and L. Hartmann, J. Org. Chem., 2012, 77(9), 4226–4234 CrossRef CAS; (b) D. Ponader, F. Wojcik, F. Beceren-Braun, J. Dernedde and L. Hartmann, Biomacromolecules, 2012, 13(6), 1845–1852 CrossRef CAS.
- C. Gerke, M. F. Ebbesen, D. Jansen, S. Boden, T. Freichel and L. Hartmann, Biomacromolecules, 2017, 18, 787–796 CrossRef CAS.
- C. Schmuck, V. Bickert, M. Merschky, L. Geiger, D. Rupprecht, J. Dudaczek, P. Wich, T. Rehm and U. Machon, Eur. J. Org. Chem., 2008, 324–329 CrossRef CAS.
- (a) S. Schöttler, G. Becker, S. Winzen, T. Steinbach, K. Mohr, K. Landfester, V. Mailänder and F. R. Wurm, Nat. Nanotechnol., 2016, 11, 372–377 CrossRef; (b) S. De Santis, R. Chiaraluce, V. Consalvi, F. Novelli, M. Petrosino, P. Punzi, F. Sciubba, C. Giordano, G. Masci and A. Scipioni, ChemPlusChem, 2017, 82, 241–250 CrossRef CAS; (c) S. Akocak, M. R. Alam, A. M. Shabana, R. Kishore, K. Sanku, D. Vullo, H. Thompson, E. R. Swenson, C. T. Supuran and M. A. J. Ilies, Med. Chem., 2016, 59, 5077–5088 CrossRef CAS; (d) K. Chitphet, S. M. Geary, C. H. F. Chan, A. L. Simons, G. J. Weiner and A. K. Salem, Biomater. Sci. Eng., 2020, 6(5), 2659–2667 CrossRef CAS.
- J. van den Boom, F. Trusch, L. Hoppstock, C. Beuck and P. Bayer, PLoS One, 2016, 11(3), 1–13 CrossRef.
- C. Schmuck and V. Bickert, J. Org. Chem., 2007, 72(18), 6832–6839 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0cc07139k |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2021 |