
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceLeader peptide exchange to produce hybrid, new-to-nature ribosomal natural products†
Laura
Franz
 a and
Jesko
Koehnke
*ab
a and
Jesko
Koehnke
*ab
aWorkgroup Structural Biology of Biosynthetic Enzymes, Helmholtz Institute for Pharmaceutical Research Saarland (HIPS), Helmholtz Centre for Infection Research (HZI), Saarland University, Campus Geb. E8.1, Saarbrücken 66123, Germany
bSchool of Chemistry, University of Glasgow, Glasgow, UK. E-mail: Jesko.koehnke@glasgow.ac.uk
First published on 24th May 2021
Abstract
Ribosomal natural products contain exquisite post-translational peptide modifications that are installed by a range of pathway-specific enzymes. We present proof of principle for a Sortase A-based approach that enables peptide modification by enzymes from unrelated pathways. This allowed the one-pot synthesis of a new-to-nature, hybrid ribosomal natural product.
Natural products and their derivatives have been an invaluable, rich source for drugs.1,2 In recent years, the fast-expanding superfamily of ribosomally synthesized and post-translationally modified peptide (RiPP) natural products has been recognised as a major source for novel compounds with potent biological (e.g. antibiotics) activities.3,4 RiPP biosynthesis begins with the production of a short, ribosomally synthesized precursor peptide (PP), usually comprised of a N-terminal leader peptide (LP) and a C-terminal core peptide (CP) (Scheme 1).3,4 While the LP is essential for binding (and activation) of the biosynthetic enzymes and typically discarded during biosynthesis, the CP is transformed into the mature RiPP. Primary RiPP enzymes require specific parts of the LP, or recognition sequences (RSs), for activity and introduce characteristic, RiPP class-defining post-translational modifications (PTMs) in the CP. The spatial separation of substrate recognition (LP) and the site of catalysis (CP) permits primary RiPP enzymes to be very promiscuous with respect to the CP sequence.3–11 Secondary RiPP enzymes do not depend on the LP and are responsible for specific tailoring steps. The PTMs introduced by primary enzymes are incredibly diverse and expand the chemical space accessible to RiPPs far beyond the 20 canonical amino acids that comprise the PP (ribosomal expression).3,4 They include, but are not limited to, azol(in)e heterocycles, lanthionine-, sactionine-, C–C and ester-crosslinks, thioamidation, and N-methylation of the CP backbone.3,4 These PTMs endow RiPPs with several desirable properties, including potent bioactivities. The ability to combine the primary enzymes from unrelated pathways could facilitate the generation of RiPP-inspired natural product libraries for drug discovery. It would require the attachment of different LPs to a given CP.
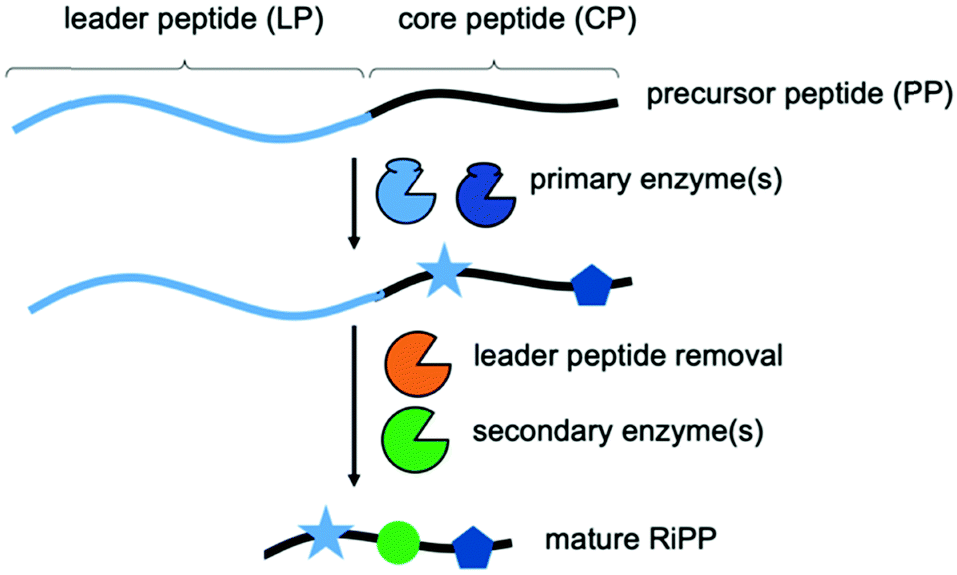 | ||
| Scheme 1 Overview of RiPP biosynthesis. | ||
To accomplish this feat, the initial focus had been on establishing conditions for LP-independent peptide processing. This was attempted by primary RiPP enzyme engineering, or by supplying the LP in trans.12–17 Recently, the improved understanding of RiPP enzymology was used in an inspired approach to engineer new to nature, hybrid RiPPs.18,19 By packaging two RSs from different RiPP families into a single, chimeric LP it was possible to generate unprecedented molecules (Scheme S1, ESI†). Thus far chimeric LPs have been limited to two RSs and the approach requires a thorough understanding of the substrate recognition requirements of each enzyme. We sought a less involved method, that would allow us to simply swap LPs between biosynthetic steps. This approach would allow iterative processing and not require a detailed understanding of the substrate recognition sequences required by the enzymes used, potentially making it plug-and-play. Our leader peptide exchange (LPX) technique is based on sortase A (SrtA)-mediated transpeptidation (Scheme 2).
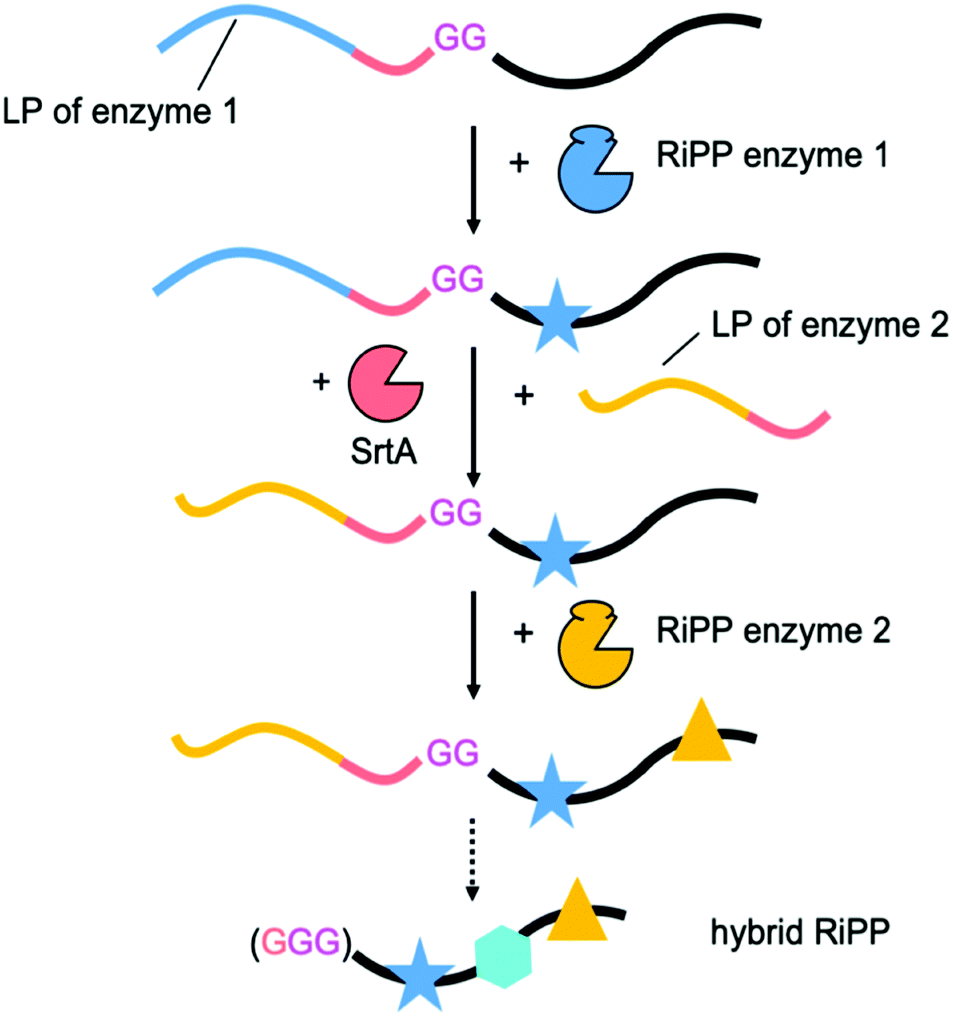 | ||
| Scheme 2 The leader peptide exchange (LPX) strategy enables Sortase A (SrtA, red)-mediated LPX and thus iterative core peptide processing by biosynthetic enzymes of different RiPP classes. A final step using SrtA or another protease liberates the hybrid RiPP. SrtA recognition sequence is shown in red, di-Gly linker in magenta. Symbols (e.g. stars) represent PTMs. | ||
To demonstrate the utility and feasibility of our idea we decided to generate a new-to-nature RiPP by combining enzymes from the unrelated cyanobactin and microviridin pathways (Fig. S1 and S2, ESI†). The enzyme LynD is found in a cyanobactin pathway discovered in Lyngbya sp. and converts cysteine residues into thiazoline heterocycles during aesturamide biosynthesis (Fig. S1, ESI†).13,20 We chose to combine this heterocyclase with MdnC of the microviridin J (derived from PP MdnA) pathway found in Microcystis aeruginosa.21,22 This enzyme catalyses the formation of two ω–ester bonds from Ser/Thr and Asp/Glu side chains, which leads to the formation of macrocycles (Fig. S2, ESI†). An engineered variant of SrtA, SrtA7m,23 was chosen to facilitate LPX. We designed two synthetic peptides: peptide 1 was comprised of a truncated cyanobactin LP suitable for LynD, followed by the SrtA recognition motif (LPXTG), a di-glycine and the MdnA core peptide harbouring two point mutations (Ser2 and Pro7 to Cys) (Fig. S3, ESI†). Peptide 2 consisted of a truncated MdnA LP containing the MdnC RS and a C-terminal SrtA recognition motif (Fig. S3, ESI†). To probe SrtA-mediated LPX, we incubated peptide 1 (All calculated and observed masses including mass errors can be found in Table S1, ESI†) and peptide 2 with SrtA7m (2 h, 37 °C) and analysed the reaction products by liquid chromatography-electrospray ionization-mass spectrometry (LC-ESI-MS). We readily detected the desired reaction product, peptide 3 (Fig. S4, ESI†). Since the SrtA7m reaction is an equilibrium, we also detected the other expected masses (Fig. S4, ESI†). Several techniques have been developed to pull the SrtA7m equilibrium into the desired direction and may be employed to optimize this system in the future.24,25
With LPX successfully established, we next tested the tolerance of LynD and MdnC for the presence of the SrtA recognition sequence that we had introduced. Incubation of peptide 1 with LynD in the presence of ATP/Mg2+ led to an observed loss of 36 Da, which is in agreement with the desired two cyclodehydration reactions required to convert the two CP Cys residues into thiazolines (Fig. S5, ESI†). Treatment of this reaction product with iodoacetamide, which alkylates free Cys residues, did not result in a mass shift. This strongly implies the conversion of the two Cys residues to thiazolines in the LynD-treated peptide (4) (Fig. S5, ESI†). Incubation of peptide 3 with MdnC in the presence of ATP/Mg2+ also resulted in the expected loss of 36 Da. This is in agreement with the formation of two ω–ester bonds in peptide 3 (Fig. S6, ESI†). These data confirm the compatibility of both enzymes with the SrtA recognition sequence and highlights the malleability of RiPP systems.
With all components of our model system tested, we combined them in one pot. After incubation of peptide 1 with LynD and ATP/Mg2+, SrtA7m was added and we rapidly observed the liberation of the core peptide with two thiazolines (6) (Fig. 1-III and Fig. S7, ESI†). After addition of peptide 2, the SrtA-mediated ligation product with 6 was observed (7, Fig. 1-IV and Fig. S7, ESI†). Subsequent addition of MdnC led to another loss of 36 Da, indicating that MdnC tolerates the two non-natural thiazolines in the core peptide and introduces two ω–esters into the peptide (8, Fig. 1-V and Fig. S7, ESI†). Since SrtA7m was not removed, the MdnA LP was cleaved by SrtA7m and we obtained a new to nature, hybrid RiPP 9 (Fig. 1-V and Fig. S7, ESI†). Verification of the ω–ester connectivity was attempted via methanolysis of 9, but the peptide was completely insoluble in methanol. Very extensive peptide MS/MS did not resolve this issue either, presumable because the combination of heterocycles with ω–ester bonds made the peptide very resistant to fragmentation. We therefore view the structure of 9 as tentative. Reactions without the addition of SrtA stalled after the introduction of the two thiazolines and did not result in ω–ester formation, or the production of 9. To the best of our knowledge, 9 represents the first heterocycle-containing graspetide, the newly coined name for microviridins and related compounds.4
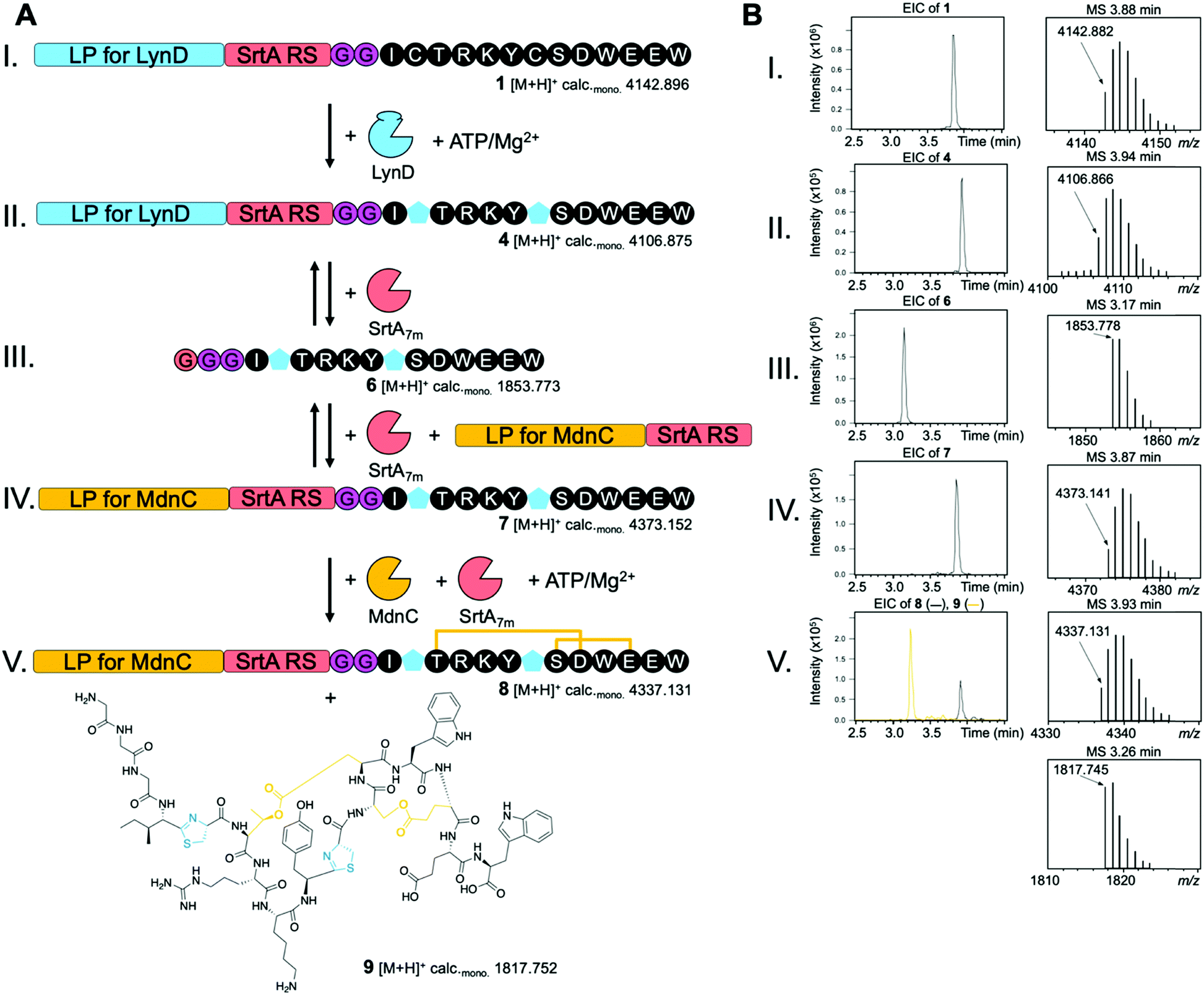 | ||
| Fig. 1 Combination of the cyanobactin heterocyclase LynD and the microviridn J ATP-grasp ligase MdnC using the LPX strategy to produce a heterocycle-containing graspetide (9). (A) Schematic of the LPX model system. After introduction of the thiazolines (blue pentagons) in 1 by LynD, the LynD LP is exchanged for the MdnA LP (2) using SrtA-mediated transpeptidation yielding 7. LPX allows the modification of the core peptide (black) by MdnC that introduces two ω–ester cross-links (8). SrtA finally cleaves the MdnA LP yielding the new-to-nature hybrid RiPP 9. (B) Extracted ion chromatograms (EICs, ±5 ppm) and mass spectra for the desired products of each step shown in (A). Shown are the masses of the singly-charged monoisotopic ions [M + H]+. An extensive presentation of all products and secondary products (SrtA reactions are reversible) in all steps (I.–V.) is given in the ESI,† Fig. S7. | ||
We have demonstrated that the SrtA-based LPX technique is compatible with RiPP enzymes. Since our approach dispenses with the need for a detailed understanding of enzyme-substrate recognition for a particular enzyme/pathway, it has the potential to greatly expedite the creation of hybrid RiPPs. It is going to be important to consider the combination of enzymes used when designing this approach. In the case presented here we used LynD before MdnC, because it was reasonable to assume that the topology introduced by MdnC would preclude processing of the cysteine residues by LynD. In addition, we selected LynD because it is specific to cysteine residues and leaves Ser/Thr available for MdnC. The use of other heterocyclases that are able to convert Ser/Thr residues to oxazolines would preclude MdnC from forming ω–esters. Depending on the timing of addition, this could also lead to competition for reaction sites that would reduce yields and complicate analysis. It could of course also be viewed as an opportunity to generate a set of products from a single reaction, which may yield unexpected, interesting candidates for a given activity screen. The protection of side-chains may offer a solution to this issue, but would then require the purification of intermediates prior to addition of the next RiPP enzyme. In this context it is going to be interesting to trial this approach with RiPPs that depend on a particular sequence N-terminal of the core peptide for proper processing, such as cyclization, for example in azacycle formation during thiopeptide biosynthesis. Depending on the desired outcome, it may be necessary to include the native protease site between the SrtA recognition sequence and the core peptide. It may be of particular interest to use the LPX approach in combination with self-sacrificing RiPP enzymes, such as the backbone N-methylating enzyme OphA,26 which cannot be combined with other leader-dependent RiPP enzymes through hybrid leader peptides.
We focussed our efforts on proof-of-concept and thus did not attempt to maximise reaction yields. The two main aspects that would improve yields are 1. The use of optimized SrtA substrates, which have been specifically developed to drive the ligation reaction in the desired reaction and 2. The inactivation or removal of SrtA or the SrtA-substrate complex prior to addition of the second enzyme (compare Fig. S7, ESI†). A large variety of tools to accomplish the former task have been developed and established for SrtA and will be used in future studies. The latter point arose because the active SrtA7m in the reaction led to a competition with MdnC.
Truncated leader peptides were used here to reduce synthesis costs, but full-length leader peptides are of course compatible. Intriguingly, the peptides used as starting material in this study can of course be produced ribosomally, for example through expression in Escherichia coli. As a consequence the entire system could be moved into an in vivo setting. While the yields of in vivo ligation reactions using SrtA are not ideal, it would allow the leverage of genetics to create large libraries based on core peptide randomization. In targeted bioactivity screens the in vivo efficiency of SrtA may well prove sufficient to discover novel, exciting molecules.
We thank Dr Sebastian Adam for the expression and purification of MdnC. We thank the German Research Foundation for an Emmy Noether Fellowship (KO 4116/3-1) and the Boehringer Ingelheim Foundation (BIS) for an Exploration Grant.
Conflicts of interest
There are no conflicts to declare.Notes and references
- D. J. Newman and G. M. Cragg, J. Nat. Prod., 2020, 83, 770–803 CrossRef CAS PubMed.
- L. Katz and R. H. Baltz, J. Ind. Microbiol. Biotechnol., 2016, 43, 155–176 CrossRef CAS PubMed.
- P. G. Arnison, M. J. Bibb, G. Bierbaum, A. A. Bowers, T. S. Bugni, G. Bulaj, J. A. Camarero, D. J. Campopiano, G. L. Challis, J. Clardy, P. D. Cotter, D. J. Craik, M. Dawson, E. Dittmann, S. Donadio, P. C. Dorrestein, K.-D. Entian, M. A. Fischbach, J. S. Garavelli, U. Göransson, C. W. Gruber, D. H. Haft, T. K. Hemscheidt, C. Hertweck, C. Hill, A. R. Horswill, M. Jaspars, W. L. Kelly, J. P. Klinman, O. P. Kuipers, A. J. Link, W. Liu, M. A. Marahiel, D. A. Mitchell, G. N. Moll, B. S. Moore, R. Müller, S. K. Nair, I. F. Nes, G. E. Norris, B. M. Olivera, H. Onaka, M. L. Patchett, J. Piel, M. J. T. Reaney, S. Rebuffat, R. P. Ross, H.-G. Sahl, E. W. Schmidt, M. E. Selsted, K. Severinov, B. Shen, K. Sivonen, L. Smith, T. Stein, R. D. Süssmuth, J. R. Tagg, G.-L. Tang, A. W. Truman, J. C. Vederas, C. T. Walsh, J. D. Walton, S. C. Wenzel, J. M. Willey and W. A. van der Donk, Nat. Prod. Rep., 2013, 30, 108–160 RSC.
- M. Montalbán-López, T. A. Scott, S. Ramesh, I. R. Rahman, A. J. van Heel, J. H. Viel, V. Bandarian, E. Dittmann, O. Genilloud, Y. Goto, M. J. Grande Burgos, C. Hill, S. Kim, J. Koehnke, J. A. Latham, A. J. Link, B. Martínez, S. K. Nair, Y. Nicolet, S. Rebuffat, H.-G. Sahl, D. Sareen, E. W. Schmidt, L. Schmitt, K. Severinov, R. D. Süssmuth, A. W. Truman, H. Wang, J.-K. Weng, G. P. van Wezel, Q. Zhang, J. Zhong, J. Piel, D. A. Mitchell, O. P. Kuipers and W. A. van der Donk, Nat. Prod. Rep., 2021, 38(1), 130–239 RSC.
- T. J. Oman and W. A. van der Donk, Nat. Chem. Biol., 2010, 6, 9–18 CrossRef CAS PubMed.
- S. Schmitt, M. Montalbán-López, D. Peterhoff, J. Deng, R. Wagner, M. Held, O. P. Kuipers and S. Panke, Nat. Chem. Biol., 2019, 15, 437–443 CrossRef CAS PubMed.
- K. J. Hetrick, M. C. Walker and W. A. van der Donk, ACS Cent. Sci., 2018, 4, 458–467 CrossRef CAS PubMed.
- J. H. Urban, M. A. Moosmeier, T. Aumüller, M. Thein, T. Bosma, R. Rink, K. Groth, M. Zulley, K. Siegers, K. Tissot, G. N. Moll and J. Prassler, Nat. Commun., 2017, 8, 1500 CrossRef PubMed.
- E. Oueis, M. Jaspars, N. J. Westwood and J. H. Naismith, Angew. Chem., Int. Ed., 2016, 55, 5842–5845 CrossRef CAS PubMed.
- A. A. Vinogradov, M. Shimomura, Y. Goto, T. Ozaki, S. Asamizu, Y. Sugai, H. Suga and H. Onaka, Nat. Commun., 2020, 11, 2272 CrossRef CAS PubMed.
- S. J. Pan and A. J. Link, J. Am. Chem. Soc., 2011, 133, 5016–5023 CrossRef CAS PubMed.
- E. Reyna-González, B. Schmid, D. Petras, R. D. Süssmuth and E. Dittmann, Angew. Chem., Int. Ed., 2016, 55, 9398–9401 CrossRef PubMed.
- J. Koehnke, G. Mann, A. F. Bent, H. Ludewig, S. Shirran, C. Botting, T. Lebl, W. Houssen, M. Jaspars and J. H. Naismith, Nat. Chem. Biol., 2015, 11, 558–563 CrossRef CAS PubMed.
- W. Gu, D. Sardar, E. Pierce and E. W. Schmidt, J. Am. Chem. Soc., 2018, 140, 16213–16221 CrossRef CAS PubMed.
- T. J. Oman, P. J. Knerr, N. A. Bindman, J. E. Velásquez and W. A. van der Donk, J. Am. Chem. Soc., 2012, 134, 6952–6955 CrossRef CAS PubMed.
- Y. Goto, Y. Ito, Y. Kato, S. Tsunoda and H. Suga, Chem. Biol., 2014, 21, 766–774 CrossRef CAS PubMed.
- R. Khusainov and O. P. Kuipers, ChemBioChem, 2012, 13, 2433–2438 CrossRef CAS PubMed.
- B. J. Burkhart, N. Kakkar, G. A. Hudson, W. A. van der Donk and D. A. Mitchell, ACS Cent. Sci., 2017, 3, 629–638 CrossRef CAS PubMed.
- S. R. Fleming, T. E. Bartges, A. A. Vinogradov, C. L. Kirkpatrick, Y. Goto, H. Suga, L. M. Hicks and A. A. Bowers, J. Am. Chem. Soc., 2019, 141, 758–762 CrossRef CAS PubMed.
- J. Martins and V. Vasconcelos, Mar. Drugs, 2015, 13(11), 6910–6946 CrossRef CAS PubMed.
- N. Ziemert, K. Ishida, A. Weiz, C. Hertweck and E. Dittmann, Appl. Environ. Microbiol., 2010, 76, 3568–3574 CrossRef CAS PubMed.
- K. Li, H. L. Condurso, G. Li, Y. Ding and S. D. Brunauer, Nat. Chem. Biol., 2016, 12, 973–979 CrossRef CAS PubMed.
- H. Hirakawa, S. Ishikawa and T. Nagamune, Biotechnol. J., 2015, 10, 1487–1492 CrossRef CAS PubMed.
- F. Liu, E. Y. Luo, D. B. Flora and A. R. Mezo, J. Org. Chem., 2014, 79, 487–492 CrossRef CAS PubMed.
- J. M. Antos, G.-L. Chew, C. P. Guimaraes, N. C. Yoder, G. M. Grotenbreg, M. W.-L. Popp and H. L. Ploegh, J. Am. Chem. Soc., 2009, 131, 10800–10801 CrossRef CAS PubMed.
- N. S. van der Velden, N. Kälin, M. J. Helf, J. Piel, M. F. Freeman and M. Künzler, Nat. Chem. Biol., 2017, 13, 833–835 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0cc06889f |
| This journal is © The Royal Society of Chemistry 2021 |