
Reactivity of 3-nitroindoles with electron-rich species
Batoul
Rkein
,
Antoine
Bigot
 ,
Léo
Birbaum
,
Maxime
Manneveau
,
Michaël
De Paolis
,
Julien
Legros
and
Isabelle
Chataigner†
*
,
Léo
Birbaum
,
Maxime
Manneveau
,
Michaël
De Paolis
,
Julien
Legros
and
Isabelle
Chataigner†
*
Normandie Université, INSA Rouen, UNIROUEN, CNRS, COBRA Laboratory, F-76000 Rouen, France. E-mail: isabelle.chataigner@univ-rouen.fr
First published on 24th November 2020
Abstract
The indol(in)e building block is one of the “privileged-structures” for the pharmaceutical industry since this fragment plays a central role in drug discovery. While the electron-rich character of the indole motif has been investigated for decades, exploiting the electrophilic reactivity of 3-nitroindole derivatives has recently been put at the heart of a wide range of new, albeit challenging, chemical reactions. In particular, dearomatization processes have considerably enriched the scope of C2![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C3 functionalizations of these scaffolds. This feature article showcases this remarkable electrophilic reactivity of 3-nitroindoles with electron-rich species and highlights their value in generating diversely substituted indol(in)es. This compilation underlines how these heteroaromatic templates have gradually become model substrates for electron-poor aromatic compounds in dearomatization strategies.
C3 functionalizations of these scaffolds. This feature article showcases this remarkable electrophilic reactivity of 3-nitroindoles with electron-rich species and highlights their value in generating diversely substituted indol(in)es. This compilation underlines how these heteroaromatic templates have gradually become model substrates for electron-poor aromatic compounds in dearomatization strategies.
 Batoul Rkein | Batoul Rkein studied chemistry at the University of Rennes 1 (France), acquiring her master degree in 2016. She then received her doctoral degree in organic chemistry at the University of Rouen Normandy (France) in 2019. Her PhD work focused on dearomatization of electron-poor arenes under the guidance of Pr. I. Chataigner and Dr M. Durandetti. In 2020, she became a postdoctoral associate with Dr J. Legros and Pr. I. Chataigner, working on (3+2) cycloaddition reactions in the frame of the Interreg V European project “LabFact” at the same university. |
 Antoine Bigot | Antoine Bigot received an associate degree in chemistry at the University of Rouen Normandy (France) in 2016. He then went on to study chemical engineering at the National Institute of Applied Sciences (INSA). Between 2018 and 2019, he worked as a research scholar at the Broad Institute of MIT and Harvard, under the supervision of Dr Florence Wagner and Michel Weïwer. He earned his engineering degree in Chemistry in 2020, and started an industrial PhD with the PSA Group and the Claude Bernard University of Lyon 1, under the guidance of Prof. Emmanuel Lacôte and Jean Raynaud. |
 Léo Birbaum | Léo Birbaum received his MSc degree in 2018 from the University of Rouen Normandie (France). In 2019, he began his PhD at the same university, under the supervision of Prof. I. Chataigner and Dr M. De Paolis, on the dearomatization of nitroarenes. |
 Maxime Manneveau | Maxime Manneveau was educated at the University of Rouen Normandy (France), where he received his MSc degree in 2016. He performed his master project under the guidance of Prof. Hélène Lebel at the Université de Montréal (Canada) on a continuous flow process for the synthesis of trifluoromethylthioethers. In 2017, he joined the team of Prof Isabelle Chataigner and Dr Julien Legros at the CNRS (Rouen) in the frame of the Interreg V European project “LabFact”, where he is currently working on dearomatization cycloaddition reactions using innovative technologies such as flow chemistry and high pressure system. |
 Michaël De Paolis | Michaël De Paolis received his PhD degree in 2003 under the guidance of Dr Jieping Zhu (ICSN, Gif sur Yvette) on the total synthesis of ecteinascidin 743. After postdoctoral stays with Professors Istvan Marko (UCL, Belgium) and Léon Ghosez (IECB, Pessac) in the fields of organocatalysis and total synthesis (with Rhodia and Syngenta fellowships), he joined the CNRS as Chargé de Recherches (Research Associate) in 2007 at the University of Rouen Normandy in the group of Dr Jacques Maddaluno (COBRA), beginning his research on the synthesis of aureothin. His interests encompass asymmetric synthesis using carbanions, organocatalysis, domino reactions and total synthesis of natural products. |
 Julien Legros | Julien Legros was educated at the University of Paris-South, where he received his PhD under the guidance of Dr D. Bonnet-Delpon and J.-P. Bégué (2002). After an Alexander von Humboldt postdoctoral position with Prof. C. Bolm at the RWTH Aachen University (Germany), he was appointed as CNRS Research Associate in 2004 (Faculty of Pharmacy Paris South), and then moved to the University of Rouen Normandy in 2011 (promoted as CNRS Research Director in 2018). In 2019, he founded the ‘national French network for flow chemistry’ (GdR Synth_Flux). Julien Legros’ research is oriented toward the use of non-conventional media in organic chemistry (flow microreactors, hyperbaric conditions, and fluorous solvents). |
 Isabelle Chataigner | Isabelle Chataigner received her PhD degree in 1997 under the guidance of Dr Jean Villiéras (University of Nantes, France) on stereoselective allylboration reactions. After a postdoctoral period (1998–99) in Milano (Italy), under the supervision of Prof. Cesare Gennari, she joined the University of Rouen Normandy in 1999 as a lecturer. Her research is oriented toward synthetic organic chemistry, dealing with dearomatization reactions, cycloadditions and high pressure organic chemistry. She received her habilitation in 2009 and turned a full professor in 2014. Since 2017, she also works at the LCT at UPMC/Sorbonne University on DFT calculations. |
1. Introduction
Indoles and their derivatives are important scaffolds in organic chemistry.1 Notably, the indoline motif has found major applications as a biologically relevant compound, for instance in the indole alkaloid family with aspidospermine or vindoline molecules as salient examples (Scheme 1). These patterns are also found in useful ligands and organocatalysts. For many years, indole dearomatization strategies have led to elegant alkaloid syntheses mainly by taking advantage of the well-recognized C3-nucleophilicity of this electron-rich heterocycle.2 Complementarily, indoles substituted by electron-withdrawing groups, in particular 3-nitroindole derivatives 1, have been reported to exhibit an unexpectedly remarkable reactivity toward different electron-rich species, particularly in cycloaddition or annulation reactions.3 This has led to the expedient synthesis of functionalized indolines, which often retain a tetrasubstituted centre at the cis 5-membered cycle junction. The C2C3 double bond of nitroindoles can indeed behave either as a dienophile, a dipolarophile or a Michael acceptor (Scheme 1). Complementarily, the C2C3–NO system of 3-nitroindoles has been shown to exhibit a heterodienic character. Herein, we present an overview of the reactivities of 3-nitroindoles towards different electron-rich species, mainly in (formal) cycloaddition reactions, including catalytic and stoichiometric methodologies. Reductive transformations of 3-nitroindoles for the generation of 3-aminoindole derivatives are not discussed here.
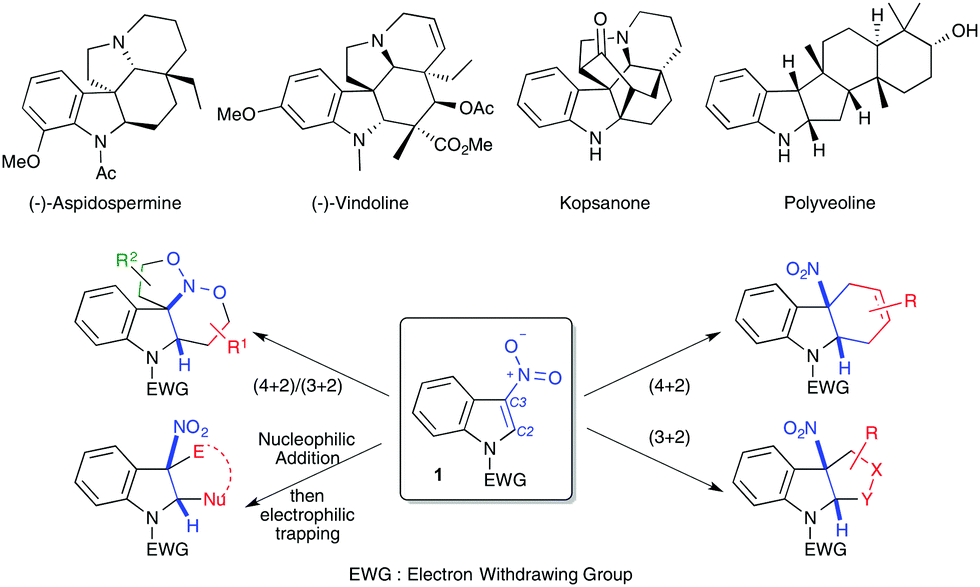 | ||
| Scheme 1 Selected indoline containing compounds and general reactivity of 3-nitroindoles 1 with electron-rich species. | ||
2. (3+2) (Formal) cycloadditions
2.1. With azomethine ylides and related dipoles
(3+2) Cycloaddition reactions involving electron-poor indoles were first reported by Gribble et al., in 1998, in the presence of mesoionic münchnones (1,3-oxazolium-5-olates) 2a–b, bearing the same substituents in positions 2 and 4 (Table 1).4,5 The reactive species were generated in situ, using the appropriate N-acylamino acid derivative in the presence of N,N′-diisopropylcarbodiimide (DIPC) as a dehydrating agent. These electron-rich 1,3-dipoles were thus reacted with 3-nitroindoles 1, protected/activated as N-carbamates or -sulfonamides, in refluxing THF. At this temperature, the (3+2) cycloadditions could be achieved and the bridged tetracyclic cycloadduct intermediates 3 instantaneously lost carbon dioxide and nitrous acid to form the re-aromatized pyrrolo[3,4-b]indoles 4, isolated in moderate to good yields (Table 1, entries 1–4).4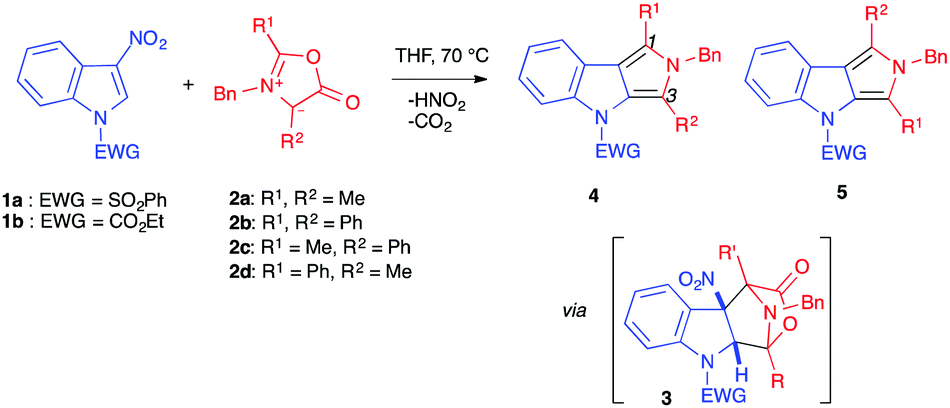
Two years later, the same group used unsymmetrical münchnones 2c–d in the same transformation (Table 1, entries 5–8).5 Surprisingly, the regioselectivity displayed in the formation of the dihydropyrrolo[3,4-b]indoles 4 and/or 5 did not systematically follow the frontier molecular orbital (FMO) theory rules. The major or exclusive product featured the phenyl group in position 1 of the isolated dihydropyrrolo[3,4-b]indole. Although the exact reasons for this regioselectivity were not clear, the authors proposed that either favourable π-interactions between the indole nitro group and the phenyl ring of the münchnone in the transition state could induce the observed regioselectivity, or that it may be a consequence of a highly asynchronous transition state where the bond forming between the methylated carbon atom of the münchnone and C2 of the nitroindole precedes the bond forming involving the phenyl substituted carbon atom of the münchnone, for steric reasons. Note, the indolic nitrogen atom has to bear an electron-withdrawing group for the reaction to proceed, as N-methyl-3-nitroindole was completely recovered after 24 h of reflux in the presence of 2d. This requirement will be found in many other transformations involving 3-nitroindoles as electrophilic species.
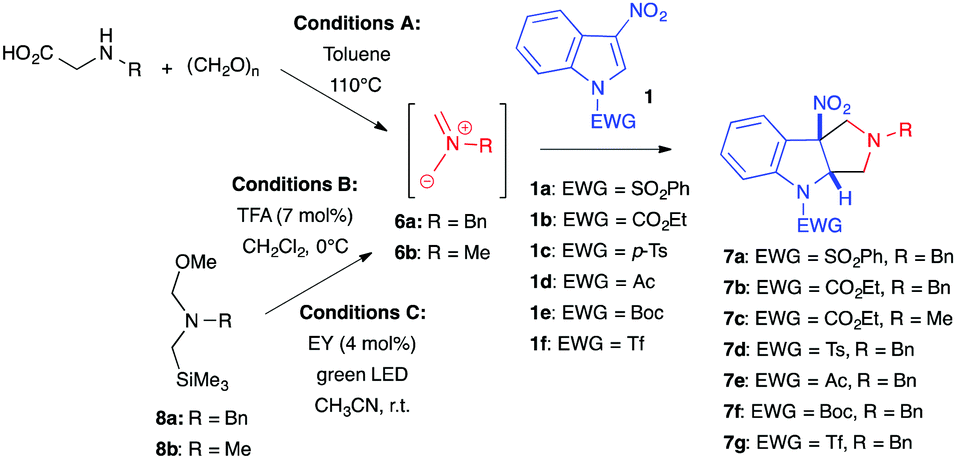
In 2007, Gribble et al. described non-stabilized azomethine ylides as suitable 1,3-dipoles for the (3+2) cycloaddition of 3-nitroindoles (Table 2, conditions A).6 Azomethine ylides 6 were classically generated in situ from the corresponding α-amino acids and paraformaldehyde in refluxing toluene. In this case, treatment of 3-nitroindoles 1a–b with an excess of dipole precursor cleanly afforded the dearomatized hexahydropyrrolo[3,4-b]indoles 7a–c, in good to excellent yields, without the loss of nitrous acid (Table 2, entries 1–3). 2-Nitroindoles also proved reactive toward the same dipole under these conditions. In contrast, however, attempting the cycloaddition with 1,2-bis(phenylsulfonyl)indole failed, underlying the importance of the nitro group for this transformation. In 2011, our group revisited these (3+2) dearomative cycloadditions with the same non-stabilized azomethine ylide 6a, generated this time from hemiaminal 8a at 0 °C under TFA catalysis (Conditions B).7 These conditions were compatible with different nitroarenes, including nitrobenzenes, and were applied to 3-nitroindoles to deliver the corresponding dearomatized cycloadduct in high yields (Table 2, entries 4–7).8 The lability of some protecting groups such as tert-butoxycarbonyl (Boc) or trifluoromethylsulfonyl (Tf) limited their use in this case but these milder thermal conditions allowed the quantity of dipole to be significantly decreased from 5 to 1.2 equiv. Note, this (3+2) cycloaddition with azomethine ylides generated under acidic conditions not only applies to 3-nitroindoles but could also be extended to indoles bearing keto, ester, or cyano groups at position 3.8,9 More recently, Lequeux et al. developed a photocatalyzed reaction for the formation of azomethine ylide 6a, at room temperature, under non-acidic conditions (Conditions C).10 To this end, they used eosin Y (EY), an organic dye, to decompose hemiaminal 8a and generate the dipole under green light irradiation. Although the article focuses mainly on the preparation of fluorinated pyrrolidines, the authors included one example of a (3+2) dearomative cycloaddition with N-tosyl-3-nitroindole 1c, which showed excellent conversion of the heteroaromatic substrate under these photocatalysis conditions (Table 2, entry 8).
|
|
|||||
|---|---|---|---|---|---|
| Entry | Conditionsa | Indole | Dipole | Cycloadduct | Yield (%) |
| a Conditions A: 5 equiv. of amino acid and 12 equiv. of paraformaldehyde in toluene at 110 °C. Conditions B: 1.2 to 2 equiv. of 8a and TFA (7 mol%) in CH2Cl2, at 0 °C to r.t. Conditions C: 2 equiv. of 8a and eosin Y disodium salt (4 mol%) under a 5 W green LED in CH3CN at r.t. | |||||
| 1 | A | 1a | 6a | 7a | 95 |
| 2 | A | 1b | 6a | 7b | 96 |
| 3 | A | 1b | 6b | 7c | 69 |
| 4 | B | 1c | 6a | 7d | 85 |
| 5 | B | 1d | 6a | 7e | 86 |
| 6 | B | 1e | 6a | 7f | 59 |
| 7 | B | 1f | 6a | 7g | 14 |
| 8 | C | 1c | 6a | 7d | 92 |
In 2014, Arai et al. reported the first example of an enantioselective (3+2) dearomative formal cycloaddition involving 3-nitroindoles 1 and stabilized azomethine ylide derivatives (Scheme 2).11 The authors used glycine-derived imino esters 9 in the presence of Cs2CO3 as a base and Cu(OTf)2 associated with the chiral PyBidine ligand 10 as a catalyst to form the reactive chiral enolate 11 acting as a surrogate of a stabilized azomethine ylide dipole. The proposed mechanism involves a stepwise transformation, via two anti-selective Michael/Mannich reactions. The PyBidine-Cu(OTf)2 chiral complex would first drive the anti-attack on the 3-nitroindole. After C–N single bond rotation, the least sterically hindered nitronate intermediate would cyclize, leading to the formation of pyrroloindolines 12 in good to excellent yields, with nearly perfect exo′-selectivity.
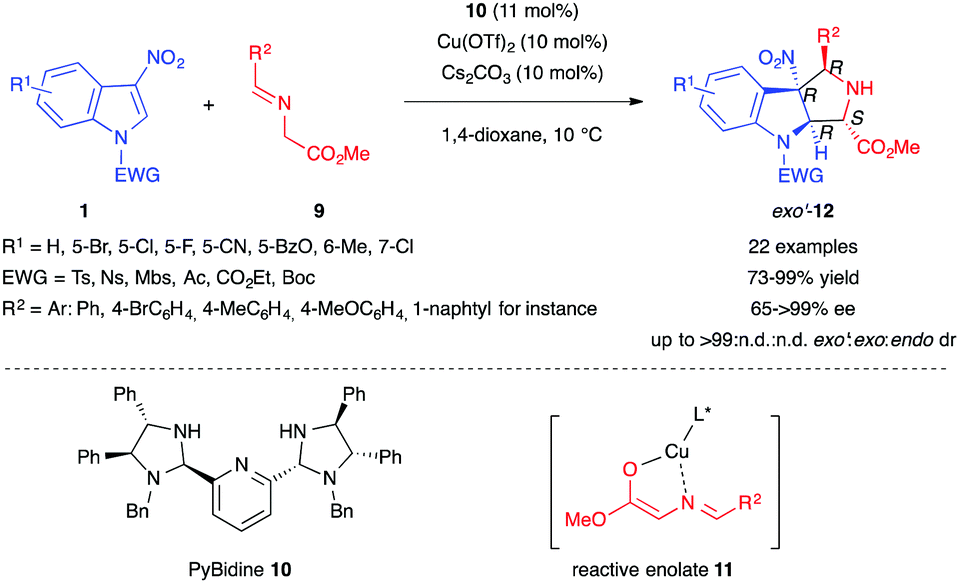 | ||
| Scheme 2 Enantioselective (3+2) formal cycloaddition of 3-nitroindoles with glycine-derived iminoesters. | ||
Two years later, Gerten and Stanley extended this type of reactivity to enolates derived from alanine imino esters 13, generating a cycloadduct bearing a quaternary center at position 3 of the pyrrolidinoindoline 14 (Scheme 3).12 The authors used a catalytic system based on Cu(OTf)2 and bisphosphine ligand (R)-difluorphos to obtain good diastereo- and enantio-selectivity. They also demonstrated the existence of a competitive non-catalyzed endo-selective reaction, but showed that the copper complex also catalyzed the epimerization of the endo-14 cycloadduct to the exo′-14 one.
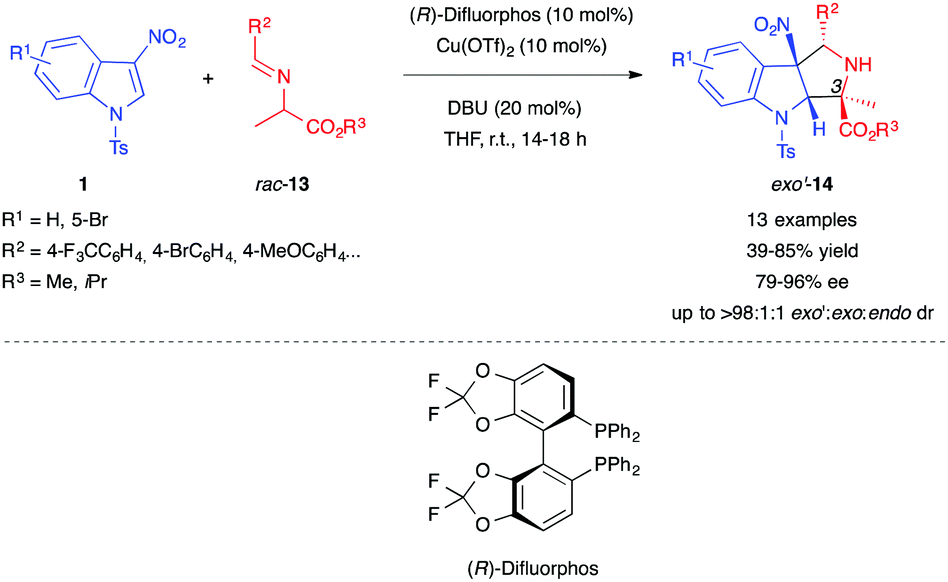 | ||
| Scheme 3 Enantioselective (3+2) formal cycloaddition of 3-nitroindoles with alanine-derived iminoesters 13. | ||
In 2017, Wang et al. reported the reactivity of 3-nitroindoles 1 as dipolarophiles toward a different kind of nucleophilic 1,3-dipole, cyclic azomethine imines 15, in a catalyst-free transformation.13 The reaction proceeded smoothly in most organic solvents, with ethyl acetate giving the best results, to afford pyrazolidinic cycloadducts 16 in a diastereoselective fashion, with a diastereomeric ratio above 20![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 in all the reported examples (Scheme 4). According to the authors, this selectivity comes from the favourable π–π interaction between the 6-membered aromatic rings of the indole and the azomethine imine.
:1 in all the reported examples (Scheme 4). According to the authors, this selectivity comes from the favourable π–π interaction between the 6-membered aromatic rings of the indole and the azomethine imine.
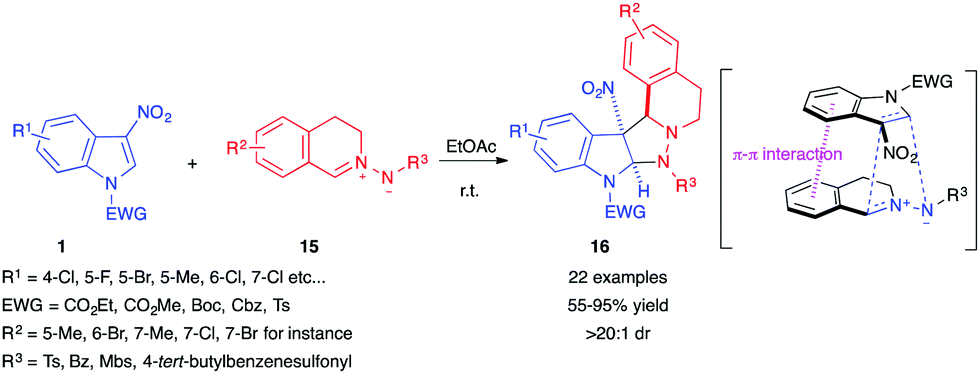 | ||
| Scheme 4 (3+2) Dearomative cycloaddition of 3-nitroindoles 1 with cyclic azomethine imines 15. | ||
2.2. Dipoles generated under palladium or copper catalysis
Different types of nucleophilic 1,3-dipoles or surrogates can be formed using transition metal catalysts, such as palladium. In 2014, Trost et al. first used the palladium-catalyzed trimethylenemethane (TMM) cycloaddition methodology14 to explore the dearomatization of nitroarenes. By using the TMM precursor 17, Pd(dba)2 and the bisdiamidophosphite ligand 18, the authors were able to dearomatize diverse nitroarenes, including nitroindoles, in quantitative yields (Scheme 5).15 This process was the first in which an all-carbon 1,3-dipole generates methylidenecyclopentane containing structures in such dearomatizing (3+2) formal cycloadditions. Applied to phenylsulfonyl- and Boc-protected 3-nitroindoles, the racemic version of the reaction, performed in the presence of ligand (±)-18, led to the corresponding cycloadducts 19 in quantitative yields, while with the chiral ligand, a 66% ee was reached. A stepwise pathway was suggested for this transformation. The TMM dipole 20, generated in situ, adds to the C2 electrophilic position of the nitroindole. Then, in the second step of this formal cycloaddition process, the zwitterionic nitronate intermediate undergoes cyclization via an intramolecular attack of the soft carbon nucleophile onto the π-allylpalladium.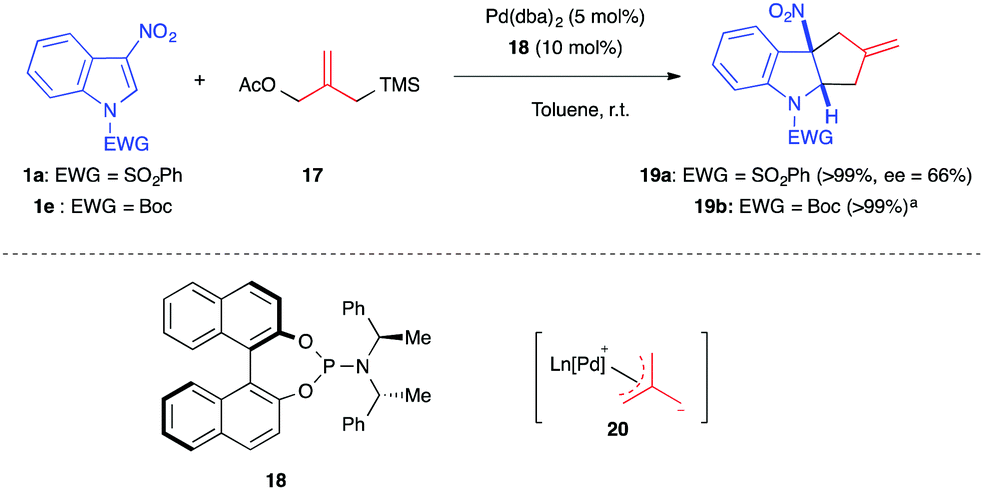 | ||
| Scheme 5 Pd-catalyzed (3+2) annulation with TMM. aPerformed in the presence of the racemic ligand. | ||
Three-membered rings bearing a vinylic appendage constitute relevant 1,3-dipole precursors in the presence of a transition metal such as palladium.16 In late 2016, the Hyland group reported the Pd-catalyzed diastereoselective synthesis of pyrrolo[2,3-b]indoline using vinylaziridines 21 as dipole precursors.17 In the presence of the phenanthroline (BPhen) ligand and Pd(dba)3·CHCl3, the authors were able to access pyrroloindolines 23 in high yields with good diastereoselectivity (Scheme 6). They proposed that the stereocontrol of this two-step formal cycloaddition reaction is linked to the nature of the C4-substituent of the indole. With a proton at this position, the nitro-enolate intermediate would preferentially adopt the TS1 conformation, which may be stabilized by cation–π interactions between the cationic π-allyl Pd complex and the electron-rich indole. This transition state would then lead to the trans-product. Alternatively, if the indole C4-position is substituted, the resulting steric repulsions could disrupt these interactions, leading to the less hindered TS2. Additionally, an ester at C4 would be able to interact with the π-allyl Pd complex, stabilizing TS2 even more, ultimately leading to the formation of cis-23.
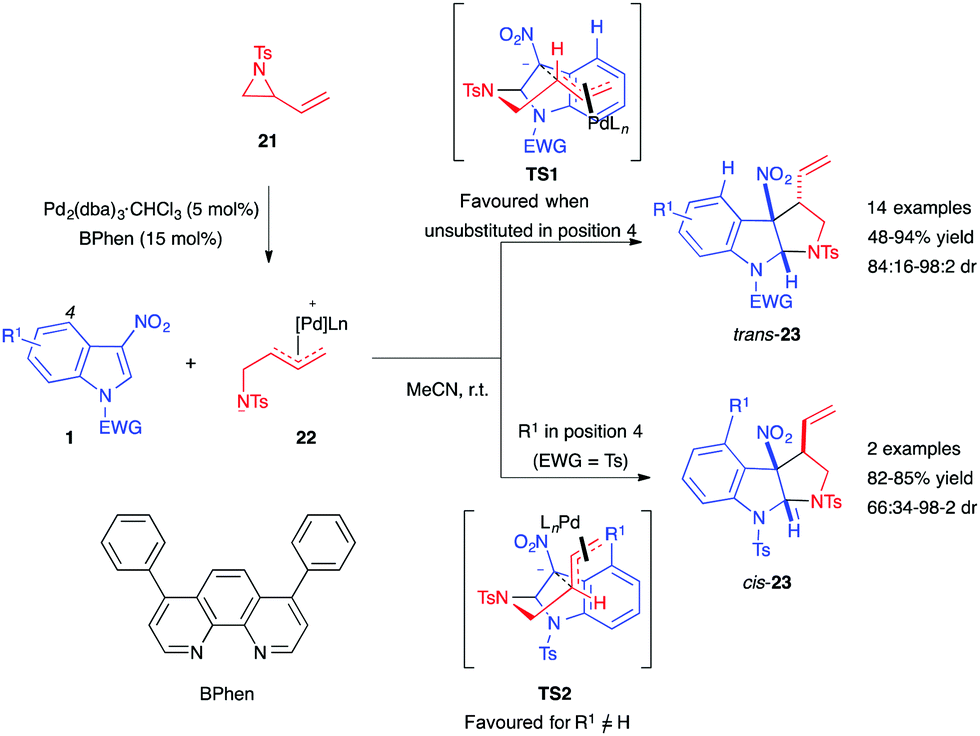 | ||
| Scheme 6 Pd-catalyzed (3+2) annulation with vinylaziridines 21. | ||
In 2018, Hou et al. reported the enantioselective dearomative (3+2) version of this reaction.18 The authors used cryogenic temperatures and the chiral ferrocenyl phosphine ligand 24 to obtain high enantio- and diastereocontrol over the reaction between substituted 3-nitroindoles 1 and N-tosyl vinyl aziridines 21 (Scheme 7). In this case, cis-pyrroloindolines 23 were obtained in excellent yields, diastereomeric ratios and enantiomeric excesses. The reaction could be scaled up to a gram scale and the reaction products could be easily converted to amino pyrroloindolines for instance, by reduction of the nitro group using the TMSCl/Zn system.
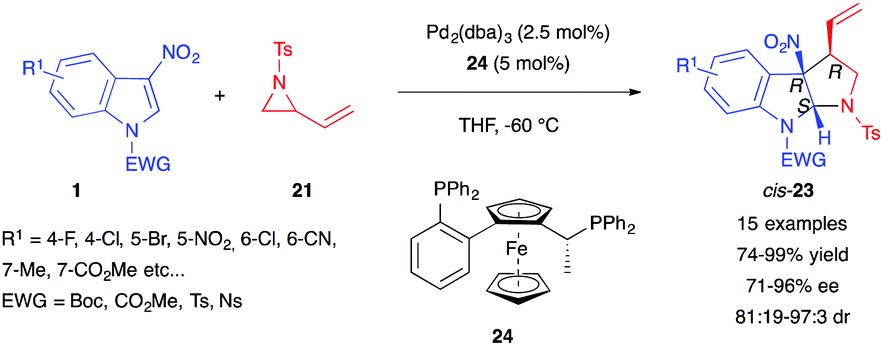 | ||
| Scheme 7 Enantioselective (3+2) annulation of 3-nitroindoles with vinyl aziridines. | ||
Almost simultaneously to Hyland's work, in early 2017, Vitale et al. reported a Pd-catalyzed (3+2) cyclopentannulation of 3-nitroindoles with various vinylcyclopropanes (VCPs) 25, bearing cyano or ester electron-withdrawing groups.19 Their method, involving all-carbon dipoles, allowed the diastereoselective dearomatization of a wide range of nitroindoles, furnishing the corresponding cyclopenta[b]indolines 27 in good to excellent yields and diastereoselectivities (Scheme 8). The method selectively produced the cis-isomer of 27. In order to account for the divergent diastereoselectivity compared to Hyland's work, the authors proposed that, after Michael addition of the Pd-VCP 1,3-dipole 26 to the electrophilic 3-nitroindole 1, subsequent cyclization would occur through either transition state TS1 or TS2, in which the forming cycle is no longer on the indole side. The latter would be favoured as the π-allyl moiety would adopt a pseudo-equatorial conformation, minimizing 1,3-diaxial interactions.
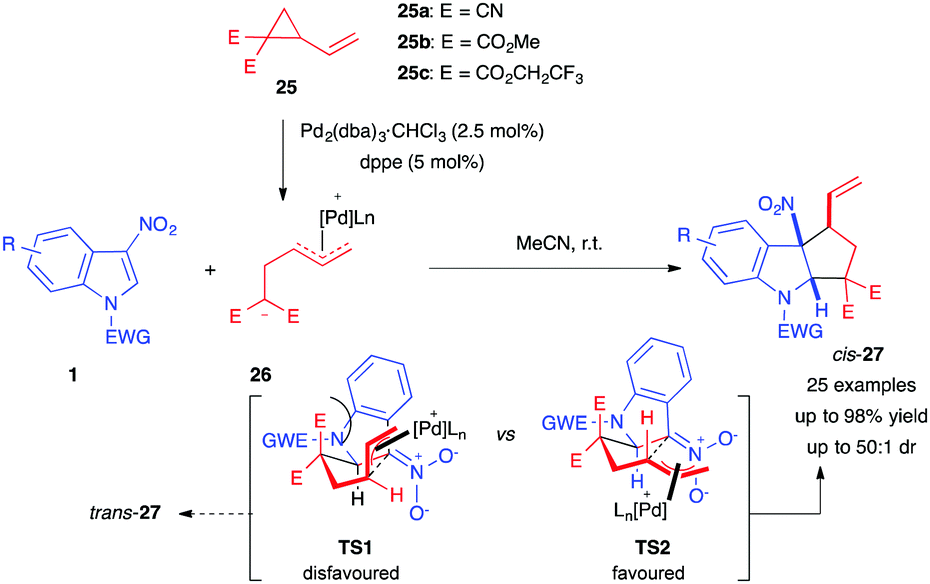 | ||
| Scheme 8 Pd-catalyzed dearomative (3+2) annulation with VCPs 25 and proposed transition states. | ||
Soon after, Hyland also disclosed the use of 2-vinylcyclopropane-1,1-dicarboxylates as all-carbon dipole precursors for the synthesis of cyclopenta[b]indolines 27.20 The authors used the previous catalytic system, composed of Pd and BPhen, to achieve efficient dearomative (3+2) annulation of 3-nitroindoles 1 (Scheme 9). They were able to control the diastereoselectivity of the annulation process by adding nBu4NI as a source of halide. The authors proposed that this could enhance the π–σ–π interconversion rate between the different transition states, leading, in most cases, to the probably more thermodynamically stable trans-isomers. As in their previous report, an ester substituent at the indole C4-position caused an inversion of the diastereoselectivity, and cis-27 ended up being the major product. Only in this case, the halide additive was not required, as it was detrimental to both the yield and the dr.
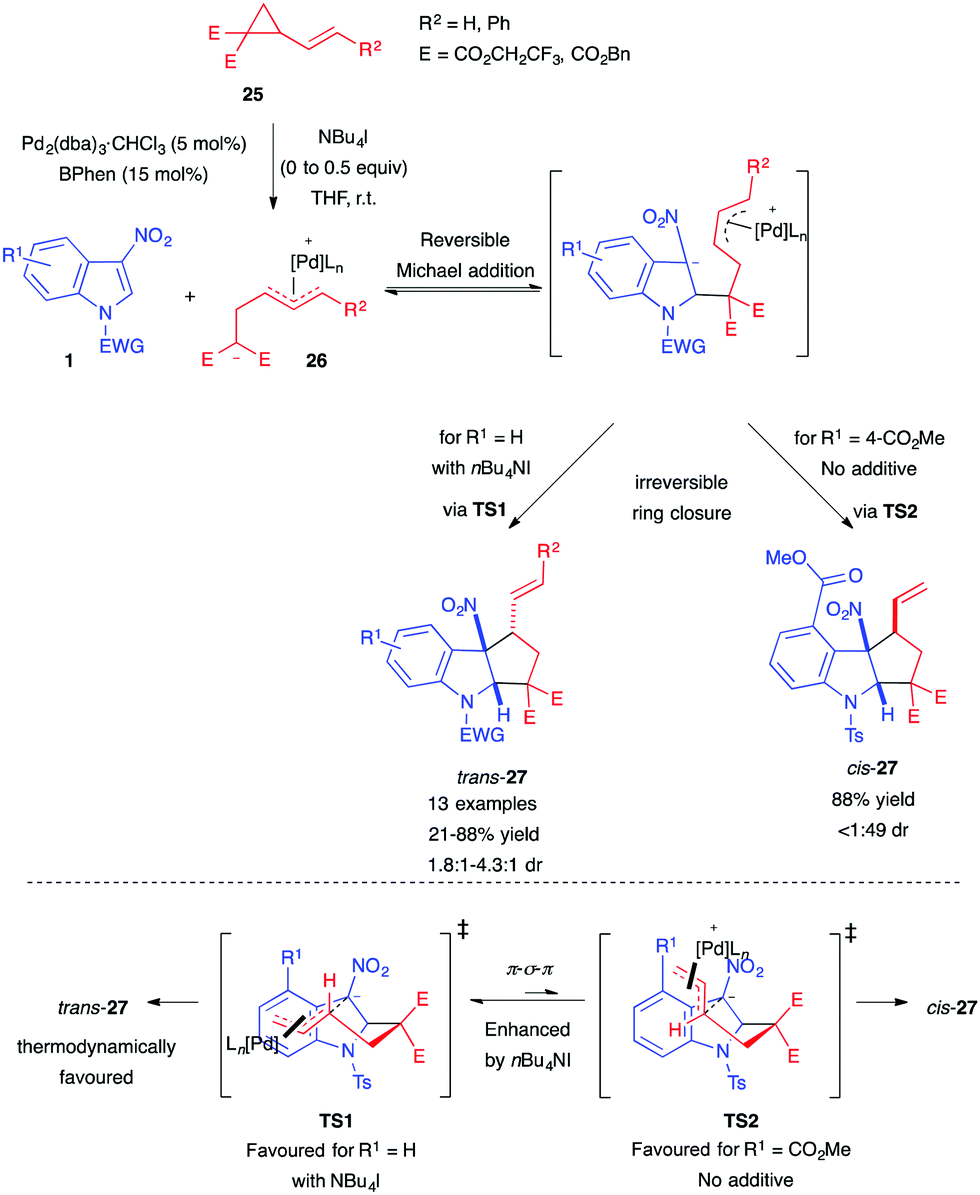 | ||
| Scheme 9 Pd-catalyzed diastereoselective (3+2) annulation with VCPs. | ||
In 2018, Shi et al. used the dicyano VCP 25a to develop an enantioselective version of this dearomative (3+2) cyclopentannulation of 3-nitroindoles.21 The authors found that their optimal catalytic system, composed of Pd(dba)2 and the chiral phosphoramidite ligand 28, was able to convert a range of 3-nitroindoles to the corresponding polycyclic adduct 27 in good yields with high enantioselectivity but still with a moderate cis diastereoselectivity (Scheme 10).
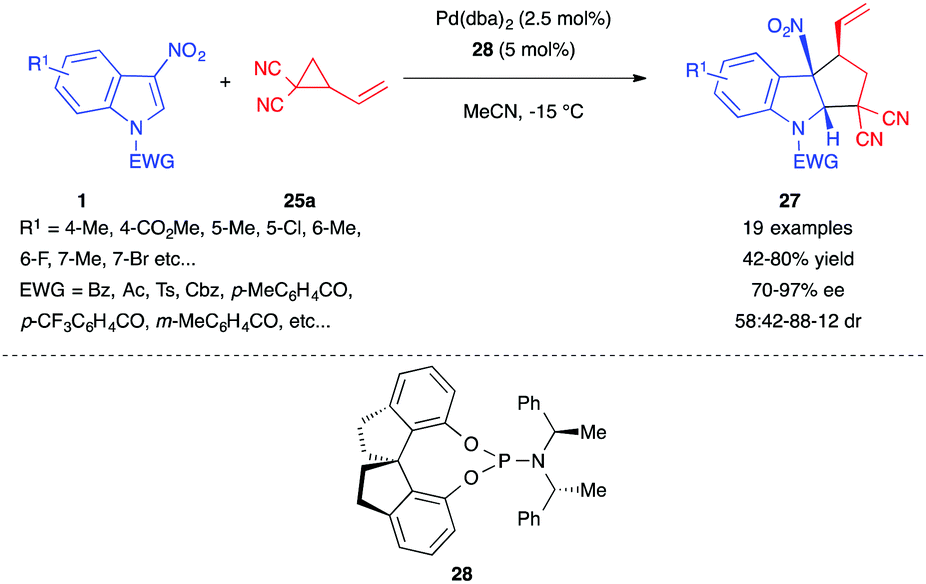 | ||
| Scheme 10 Enantioselective (3+2) annulation of 3-nitroindoles with dicyano VCP 25a. | ||
Simultaneously, Wang et al. developed strategies for the asymmetric (3+2) annulation of 3-nitroindoles with both vinyl aziridines 21 and VCPs 25.22 The authors used chiral bis(oxazoline) (Box) ligands 29a–b to convert 3-nitroindoles 1 into the corresponding highly diastereo- and enantioenriched pyrrolo[2,3-b]indolines trans-23 and cyclopenta[b]indolines trans-27, albeit with lower diastereoselectivity (Scheme 11).
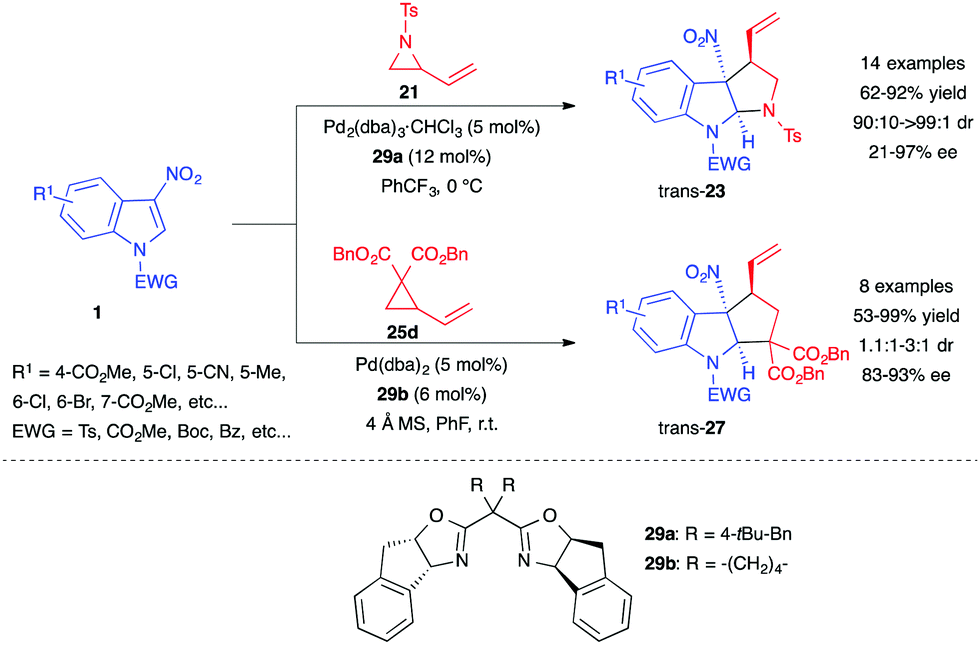 | ||
| Scheme 11 [PdBox]-promoted stereoselective (3+2) annulation with vinyl aziridines 21 and VCP 25d. | ||
Guo, You et al. then reported a stereodivergent method for the synthesis of tetrahydrofuroindoles by Pd-catalyzed asymmetric (3+2) annulation of 3-nitroindoles with vinyl oxiranes 30.23 By using the chiral phosphinooxazoline ligand 31, a large scope of substituted 3-nitroindoles 1 could be reacted with epoxybutenes 30 in good to excellent yields and stereoselectivities (Scheme 12). Unlike many dearomatizing annulations of this type, indoles bearing a 5- or 6-methoxy moiety were found to be suitable substrates. Additionally, by choosing either toluene or a much more polar solvent such as acetonitrile, the authors were able to direct the product diastereoselectivity toward its cis- or trans-isomer. To explain this stereodivergency, they performed a number of mechanistic experiments, ultimately proposing a mechanism (Scheme 12). Oxidative addition of the Pd(0) catalyst to the oxirane would lead to the ring-opened 1,3-dipole 32. In a nonpolar solvent such as toluene (path a), this intermediate would form a tight ion pair, decreasing its nucleophilicity and making the first dearomative Michael addition step rate-determining. Fast cyclization of the resulting intermediate 33 would then occur, giving preferentially cis-34. In contrast, a more polar solvent like acetonitrile (path b) would drive the rapid formation of the intermediate, while stabilization of the zwitterion 33 would slow the cyclization step, thus making it rate limiting. This would give enough time for π–σ–π inversion of the Pd-allyl complex, leading to the formation of diastereoisomer trans-34.
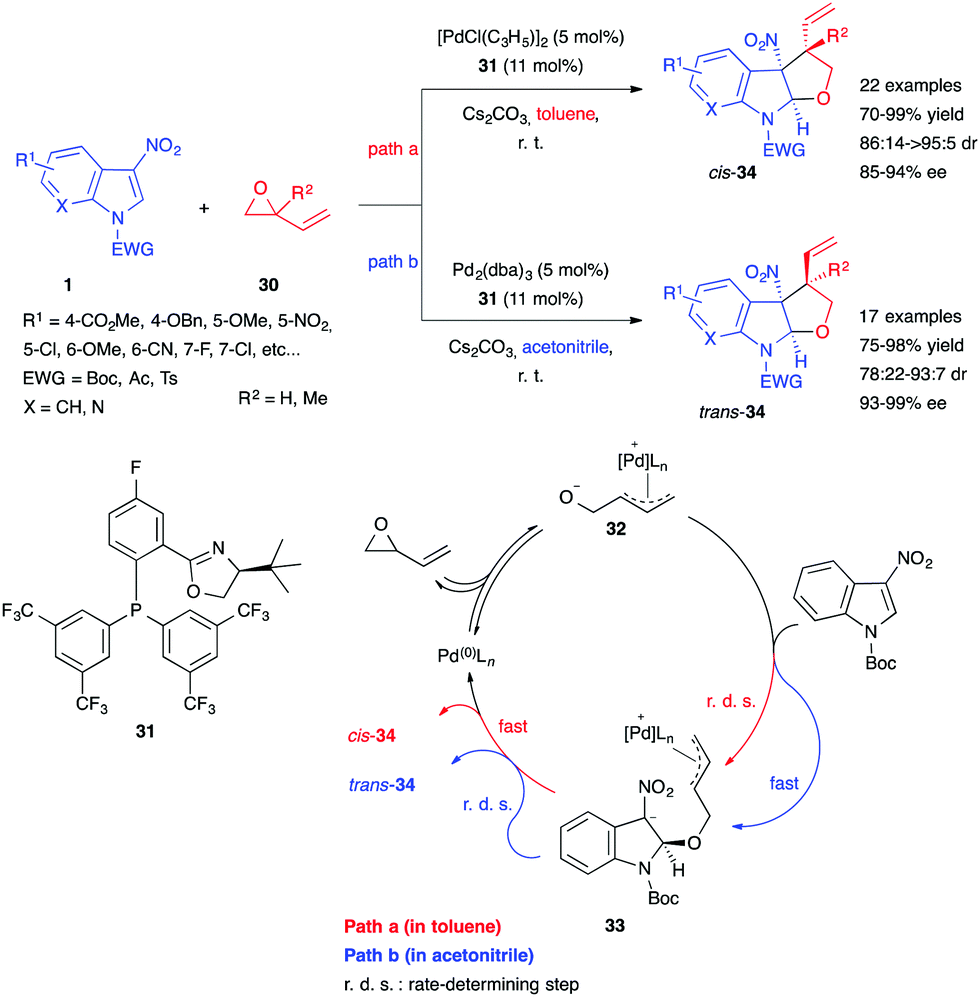 | ||
| Scheme 12 Pd-catalyzed enantioselective (3+2) annulation with vinyl oxiranes 30. | ||
More recently, Vitale et al. reported a different type of dearomative (3+2) annulation of 3-nitroindoles, involving this time propargylic nucleophiles and copper catalysis.24 Using copper(II) triflate, triphenylphosphine and cesium carbonate, the authors were able to convert a wide range of 3-nitroindoles 1 to the corresponding cyclopenta[b]indolines, pyrrolo[2,3-b]indolines and furo[2,3-b]indolines 35 with propargylic partners 33 (Scheme 13). They also demonstrated the possibility of an asymmetric version of their reaction, using (R)-difluorphos (Scheme 3) as a chiral ligand, albeit with a modest ee (51%). The authors proposed the mechanism shown in Scheme 13. Reduction of Cu(OTf)2 to the copper(I) catalyst, followed by complexation to the propargylic nucleophile 35, may form the π-alkyne complex A, possibly in equilibrium with the alkynylcopper(I) derivative A′. A would undergo deprotonation to give 1,3-dipole B, which would then react with nitroindole 1 to form a nitronate intermediate adduct. It would then cyclize in a pseudo-chair-like transition state, either through an anti-carbocupration (C) or a syn-carbocupration (C′) pathway, forming intermediate D. Subsequent protodecupration with either hydrogenocarbonate 35 or A would then afford cycloadduct 36, as well as regenerating copper(I).
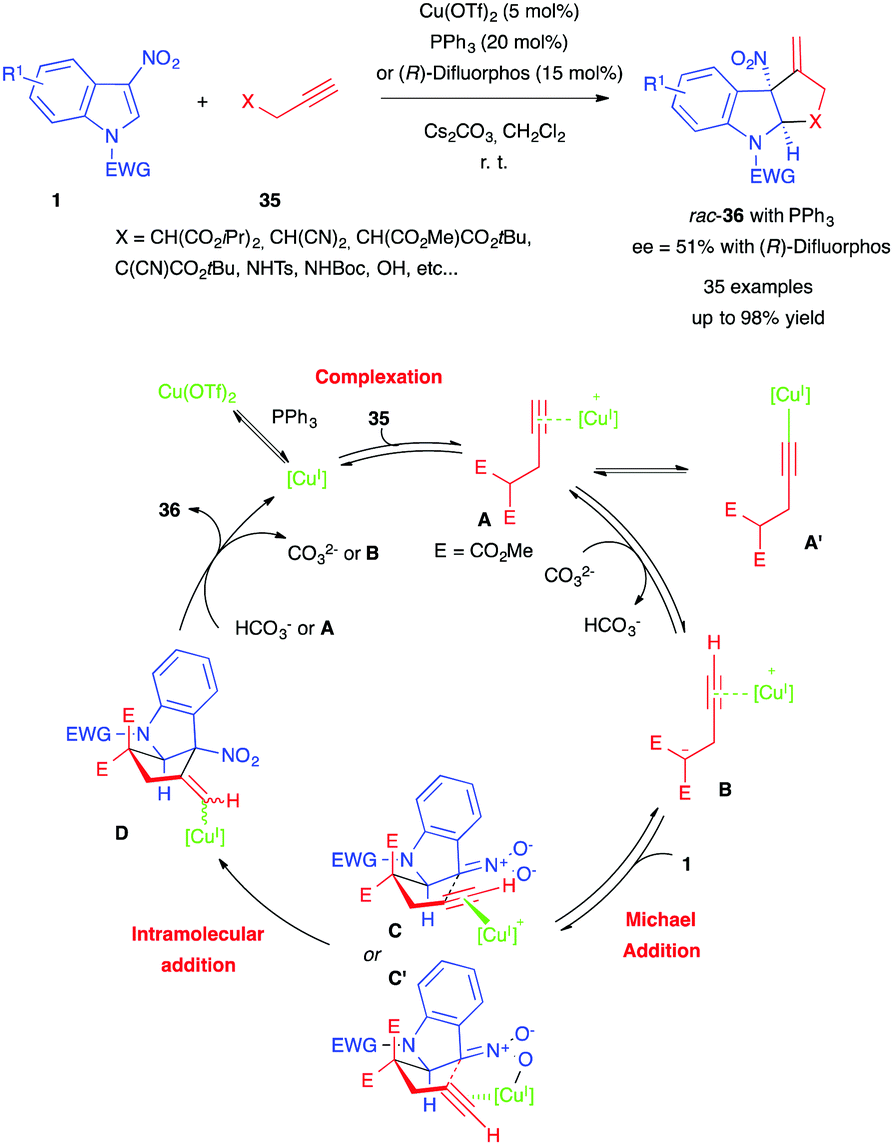 | ||
| Scheme 13 Cu-catalyzed racemic dearomative annulation with propargylic nucleophiles. | ||
2.3. 1,3-Dipoles (or surrogates) generated in the presence of organic promoters
3-Nitroindoles are prone to react with enolates in nucleophilic addition reactions (vide infra). If the enolate anion bears an additional reactive site, in position 3 with respect to the enolate carbon atom, this can lead to an intramolecular cyclization and an overall formal (3+2) cycloaddition process. Different enolates of this kind have been used throughout the years in such annulation reactions.In 1997, Gribble et al. first showed that 3-nitroindoles 1 protected/activated as N-benzyl, N-pyridyl or N-ethoxycarbonyl compounds could react with ethyl isocyanoacetate 37a or tosylmethyl isocyanide 37b in the presence of an organic base such as DBU (Scheme 14).25 Compounds 35 were formed, according to the Barton–Zard reaction, by conjugate nucleophilic addition of the enolate anion on the nitroindole substrate, followed by intramolecular 5-endo-dig cyclisation of the intermediate nitronate on the isocyanide. After reprotonation of the formal cycloadduct, base-catalysed elimination of the nitro group and tautomerization leading to the formation of a pyrrole ring, compounds 38 were obtained in moderate to high yields, depending mainly on the electron-withdrawing character of the nitrogen substituent of 1 (CO2Et > pyridyl > benzyl). The more attracting groups probably facilitated the key initial nucleophilic addition. When even more electron-withdrawing groups such as phenylsulfonyl were used, however, an abnormal regioselectivity was observed for this reaction, leading to a ring opening/rearrangement and a regioisomeric cycloadduct (vide infra, Section 5.1.).26
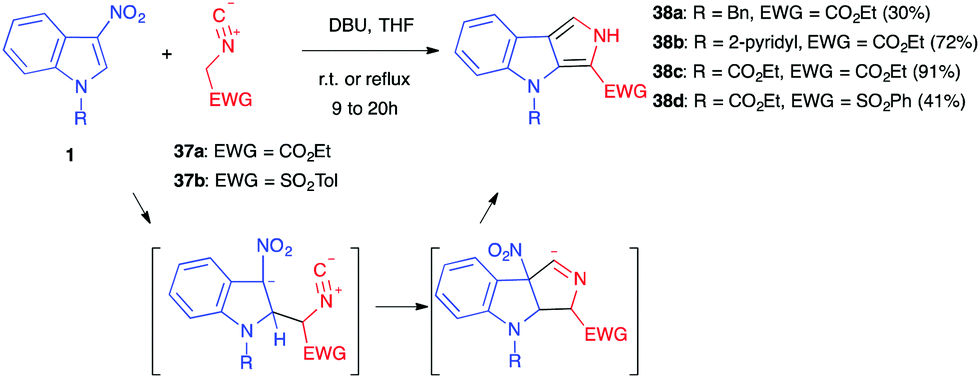 | ||
| Scheme 14 Barton–Zard reactions involving 3-nitroindoles. | ||
In 2015, Yuan et al. reported the organocatalyzed asymmetric Michael/cyclization cascade reaction between 3-nitroindoles 1 and 3-isothiocyanato oxindoles 39.27 Using the chiral bifunctional catalyst 40, based on the quinine structure, the authors were able to access a wide range of spirocyclic oxindoles 41 in excellent yields with great stereoselectivity (Scheme 15). The first step of the process was the Michael addition of the enolate derived from 39 to the electrophilic C2 position of the activated 3-nitroindole and this was followed by an intramolecular cyclization on the electrophilic central carbon atom of the isothiocyanide. The same year, this group updated the transformation by switching the organocatalyst to a Zn(OTf)2/diphenylamine-linked bis(oxazoline) complex (Scheme 15).28 The use of the Lewis acid catalyst with ligand 42 improved the results. The proposed mechanism of this latter process suggested a transition state where the catalytic complex plays a dual role. Indeed, the Zn(II) core would activate the 3-nitroindole, while the NH link would serve as a Lewis base, directing the 3-isothiocyanato oxindole for the key Michael addition step.
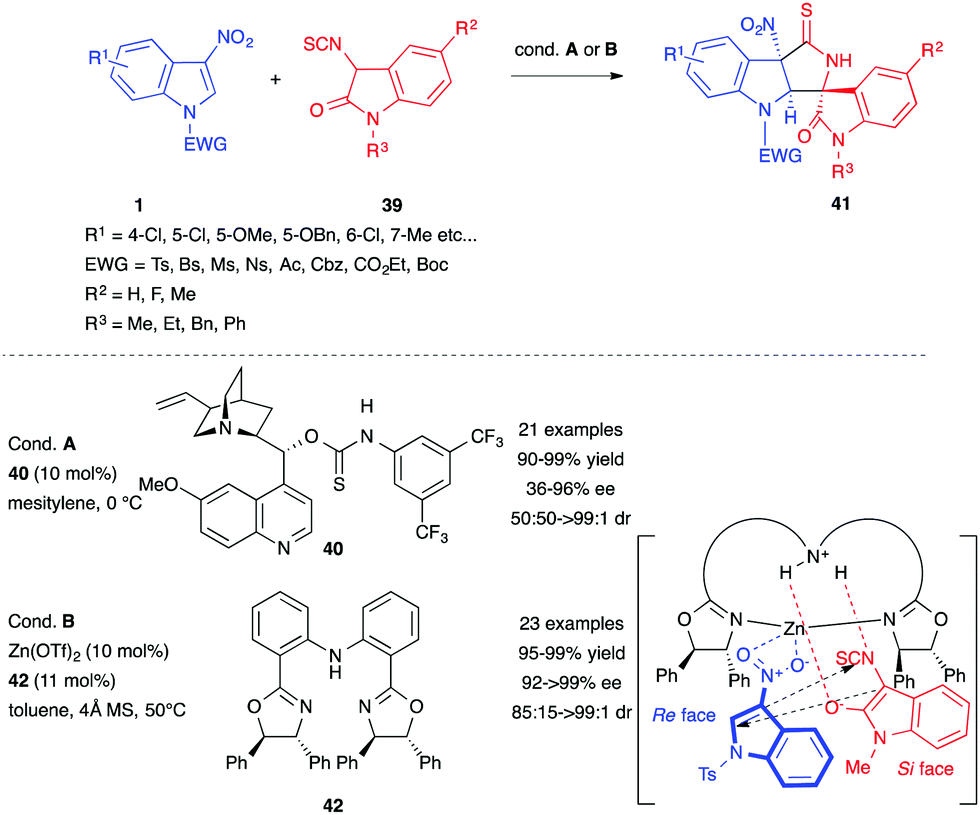 | ||
| Scheme 15 Organocatalyzed vs. metal-catalyzed asymmetric spirocyclic oxindole synthesis. | ||
In 2019, Yuan et al. reported a different type of asymmetric dearomative (3+2) annulation of 3-nitroindoles, triggered by a thiol 43 bearing a Michael acceptor moiety.29 They used a chiral tertiary amine-squaramide bifunctional organocatalyst 44 to control the enantioselective, stepwise double Michael addition between ethyl 4-mercapto-2-butenoate 43 and 3-nitroindoles 1 (Scheme 16). This reaction tolerated a wide range of substitution on the electrophilic 3-nitroindoles and furnished tetrahydrothiopheindolines 45 in good to excellent yields with great enantio- and diastereo-selectivity.
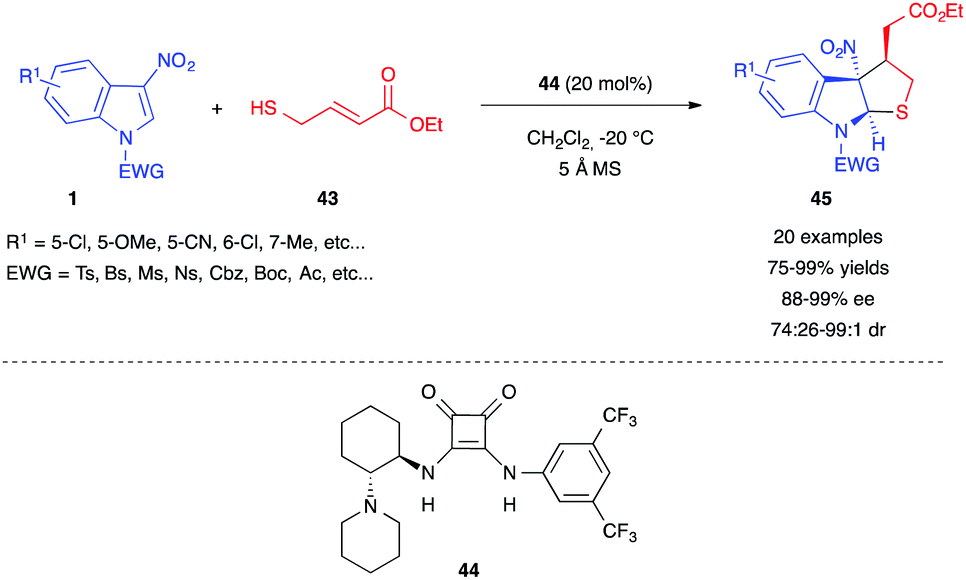 | ||
| Scheme 16 Organocatalyzed thiol-triggered (3+2) annulation of 3-nitroindoles. | ||
In 2019, almost simultaneously, different publications reported the reactivity of allenoates 46 toward 3-nitro-indole derivatives 1, in the presence of phosphine organocatalysts, to generate fused cyclopentannulated indolines.30–35 In the literature, Lu (3+2)-annulation processes have been shown to lead to two possible alpha and gamma regioisomers, the result of the two possible additions of the mesomeric 1,3-dipole A or B. Thus, the nucleophilic addition of the dipole to the 2 position of the electrophilic nitroindole can potentially lead to the formation of two regioisomers, 47 and 48. In all these publications, the unique formation of the α-cycloadduct 47 was observed (Scheme 17).
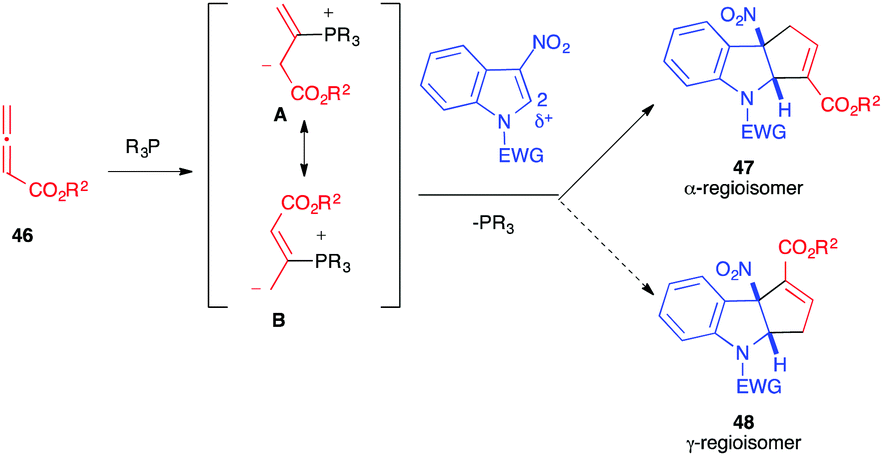 | ||
| Scheme 17 Regioselectivity of the dearomative (3+2) Lu annulation with 3-nitroindoles. | ||
The mechanism proposed for this reaction and supported by DFT calculations34,35 involves, first, the addition of the phosphine catalyst to the allenoate leading to the reactive 1,3-dipole A, stabilized by the positive P+⋯O− interaction.35 The electron-rich dipole would then add regioselectively to the nitroindole dipolarophile to furnish nitronate C that would cyclize intramolecularly generating the formal α-cycloadduct D. A proton shift is then required to produce E. The presence of water molecules (or other proton relays) in the reaction medium would facilitate this otherwise highly energetic reaction. The formal cycloadduct 47 would then be formed by expulsion of the catalyst (Scheme 18). Lu et al. showed, through intermolecular competition experiments, that the nucleophilic addition of the dipole A onto 1 was the rate determining step in this case, involving γ-unsubstituted allenoates.31
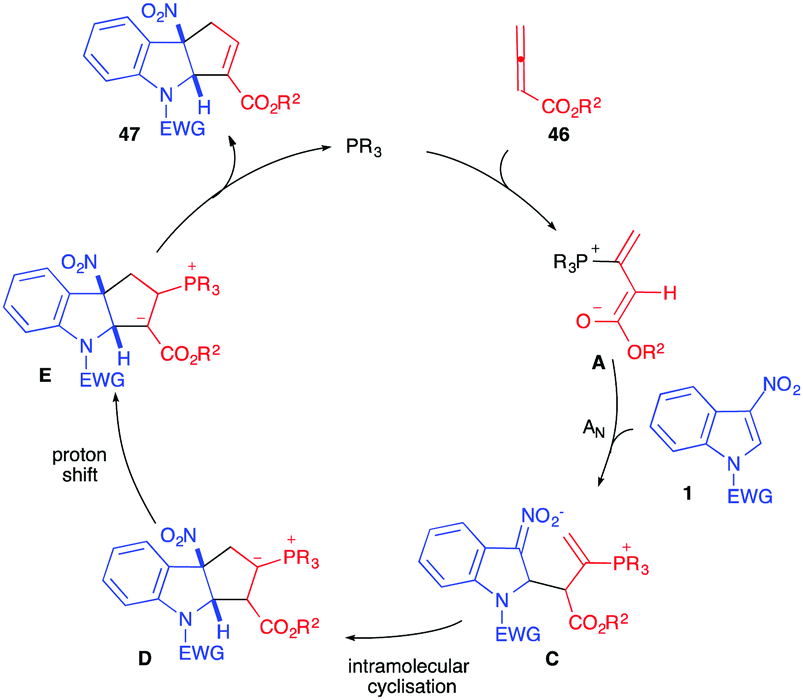 | ||
| Scheme 18 Catalytic cycle proposed for the dearomatizing (3+2) annulation reaction. | ||
These (3+2) dearomative cycloadditions were first reported by the groups of Zhang and Lu, with unsubstituted allenoates 46 in the presence of dipeptidic phosphine catalysts 46 and 47. Different indoles 1 were dearomatized in a highly enantioselective fashion with these chiral bifunctional organocatalysts (Scheme 19).30,31 These catalysts play a dual role here, allowing the formation of the reactive dipole while activating the electrophilic nitroindole by hydrogen bonds in the meantime, thus bringing the reactive parts into close proximity. The presence of the amido protons probably also helped the proton shift required within the catalytic cycle. The SPINOL-derived phoshine and amido-phosphine derived from cyclohexyl-1,2-diamine could also be used as chiral organocatalysts in these reactions albeit with lower enantioselectivity.32,33
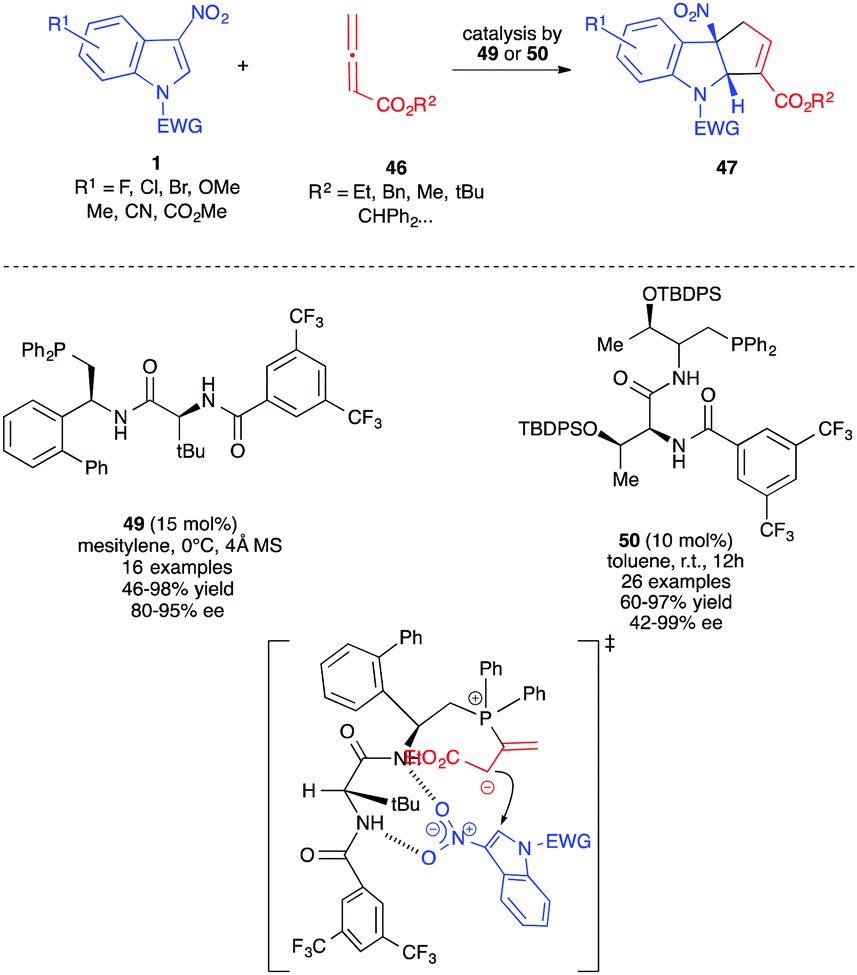 | ||
| Scheme 19 Enantioselective Lu (3+2) annulations with 3-nitroindoles in the presence of bifunctional dipeptidic phosphine organocatalysts. | ||
Zhang et al. showed that indoles 1 protected as carbamates were more reactive than their sulfonamidic analogues.30 This was correlated by the authors to the unexpected lower aromaticity of the former, a feature already observed for different electrophilic indole derivatives in (4+2) cycloaddition processes.36
Bandini et al. reported the formal (3+2) cycloaddition in the presence of γ-alkyl allenoates 51 bearing methyl or n-propyl substituents.34 In the presence of the commercially available tri(p-methoxyphenyl)phosphine, the corresponding α-cycloadducts 52 were formed in good to high yields and diastereoselective ratios ranging from 2:1 to 3.5:1, with a preferentially trans relative stereochemistry (Scheme 20).
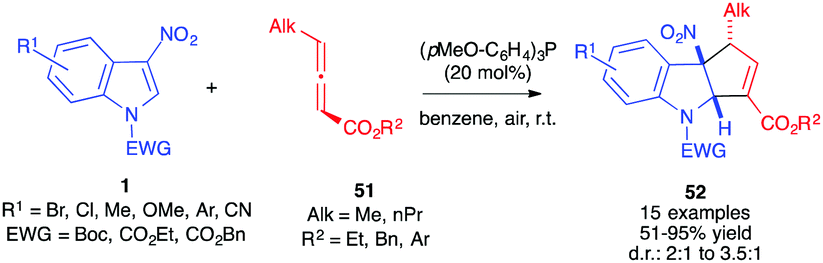 | ||
| Scheme 20 Dearomative (3+2) annulations with 3-nitroindoles 1 in the presence of γ-alkyl allenoates. | ||
Soon after, we reported the dearomative (3+2) annulation of 3-nitroindoles 1 with other γ-substituted allenyl esters 53 (Scheme 21).35 γ-Allenoates bearing more hindered alkyl groups such as iPr or Cy, with diverse functionalities such as alkenes, alkynes, protected alcohols or amines, or substituents derived from biomolecules such as amino and deoxycholic acids proved to react smoothly with 3-nitro-N-Ts indoles 1c in the presence of a commercially available phosphine catalyst (PPh3 or PBu3). The reaction also led to the exclusive formation of the functionalized α-regioisomer 54, in moderate to good yields and with diastereoselectivities up to 3:1, in favour of the trans diastereomer. The reaction was best performed in the presence of water to facilitate the [1,2]-H shift.
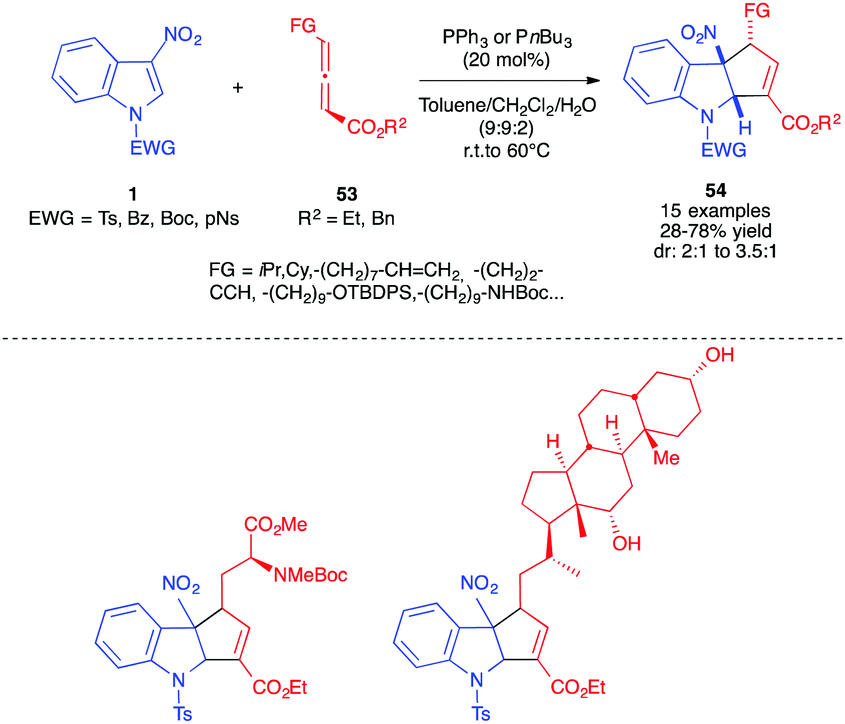 | ||
| Scheme 21 (3+2) Formal cycloadditions in the presence of γ-functionalized allenoates 53. | ||
Starting from a different substrate that is an isatin-derived Morita–Baylis–Hillman (MBH) carbonate 55, Gao, Ge, Tian et al. recently showed that a related enantioselective (3+2) annulation reaction could be performed on electrophilic 3-nitroindoles 1, in the presence of a bifunctional DMAP-thiourea organocatalyst 56.37 Spirooxindoles 57, bearing a cyclopentannulated indoline and three consecutive stereocenters, were thus obtained in high yields and enantioselectivities (Scheme 22).
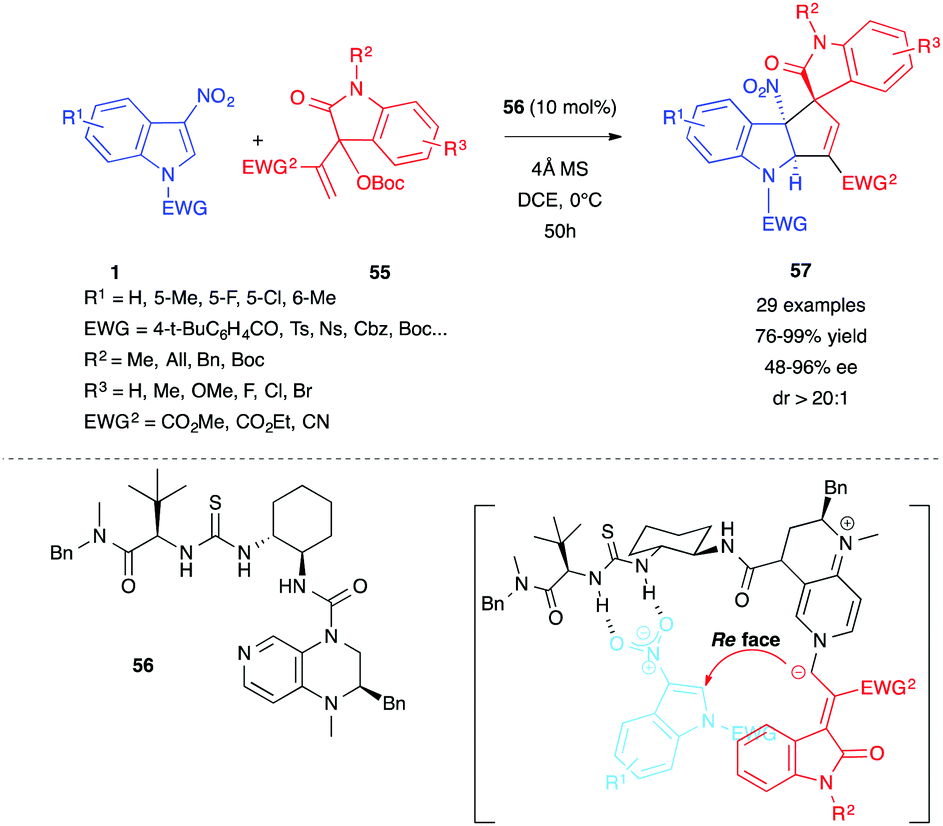 | ||
| Scheme 22 (3+2) Annulation between 3-nitroindoles 1 and isatin-derived MBH carbonate 55 in the presence of the bifunctional organocatalyst 56. | ||
3. (4+2) (Formal) cycloadditions
As a complement to the (3+2) cycloadditions/annulations, the (4+2) transformations involving 3-nitroindoles 1 generate tricyclic indolines combining 6-5-6 rings, encountered in (polyhydro)carbazole derived structures. In this case, the C2C3 double bond is involved as an electron-poor dienophile with electron-rich 1,3-dienes or 1,4-dipoles.
3.1. (4+2) cycloadditions
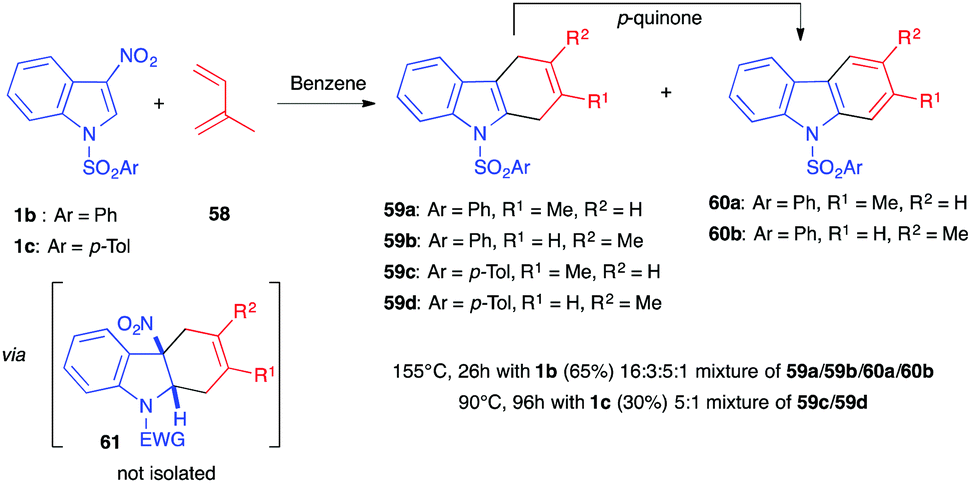
Before the (3+2) reactions mentioned above were described, Wenkert et al. first reported in 1988 the normal electron demand (NED) Diels–Alder reaction involving different electron-poor indole derivatives.38 3-Nitroindoles were the most reactive ones and 1-benzenesulfonyl-3-nitroindole 1b reacted with a 12-fold excess of isoprene (58) to generate a 16:3:5:1 mixture of dihydrocarbazoles (59a and 59b) and carbazoles (60a and 60b) in an overall 65% yield (Scheme 23). With this neutral apolar electron-rich diene, more drastic conditions were needed compared to those of the (3+2) cycloadditions/annulations described previously, and the reaction was performed at high temperature (155 °C) for 26 h. This led to the re-aromatization of the primary cycloadducts by extrusion of nitrous acid and aromatization, by oxidation, into the corresponding carbazoles. The authors showed that the cycloaddition required the presence of an electron-withdrawing group on the nitrogen atom of the indole dienophile, a feature shared with the (3+2) reactions described above. Later, Mancini et al. showed that using the similar N-tosylated nitroindole 1c would lead to a 3:1 mixture of regioisomeric dihydrocarbazoles 59c and 59d, along with unreacted nitroindole, and that no carbazole formation was observed when decreasing the temperature to 90 °C for 96 h,39 revealing that complete aromatization is a process favoured by high temperature. The primary nitroindoline cycloadduct 61 could not be observed under these thermal conditions. This feature contrasts with the reactivity observed with other electrophilic indoles (substituted at position 3 by an acetyl, ketoester or ketoamide group, for example), for which the cycloaddition reaction led to the formation of a dearomatized indoline bearing a quaternary center at the ring junction.39,40
 | ||
| Scheme 23 (4+2) Diels–Alder reaction between 3-nitroindoles 1 and isoprene under thermal activation. | ||
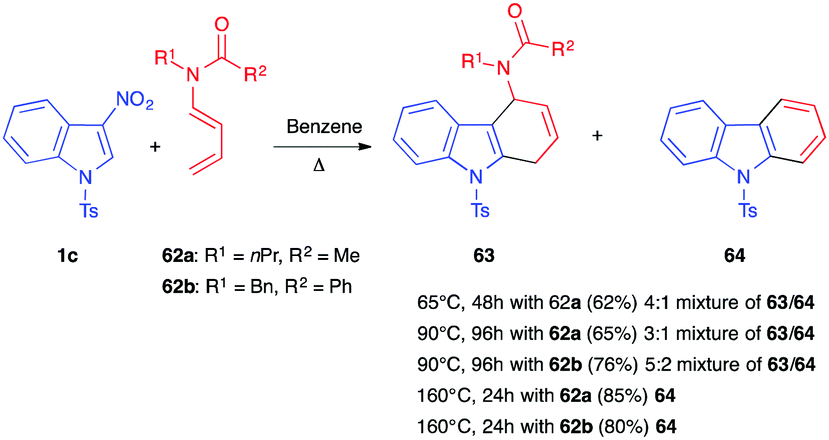
In 1999, Mancini et al. described the reactivity of 3-nitroindole 1c toward dienamides 62.41 Heating the mixture at 90 °C for 4 days afforded a mixture of the dihydrocarbazole 63 and carbazole 64 in an overall 65% and 76% yield, respectively (Scheme 24). The presence of a unique regioisomer 63 suggested a total control of the regiochemistry in the presence of this more polarized diene. Increasing the temperature to 160 °C led, here again, to the aromatization, and carbazole 64 was the only product isolated in this case. In 2001, the same team reported that lowering the temperature to 65 °C did not allow isolation of the primary nitroindoline cycloadduct, and a similar 4:1 mixture of 63/64, along with unreacted nitroindole 1c, was obtained.39
 | ||
| Scheme 24 (4+2) Diels–Alder reaction between 3-nitroindoles 1c and dienamides 62 under thermal activation. | ||
When the diene bears a trimethylsilyloxy substituent at position 1, the thermal reaction was shown later to lead to the hydroxycarbazole after 48 h at 90 °C in benzene.42
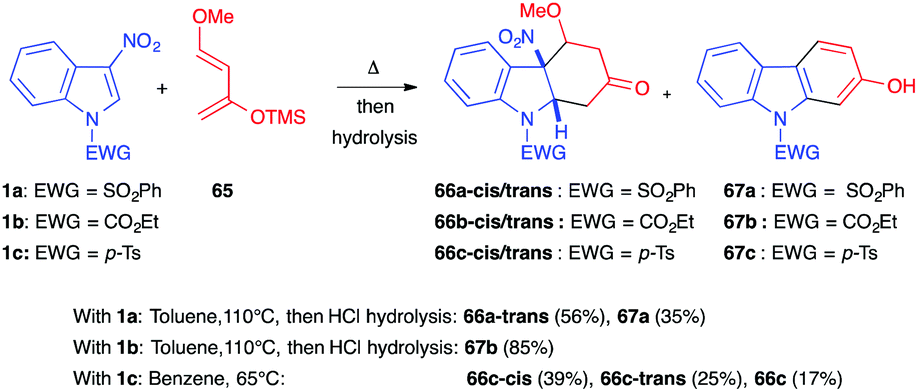
In the presence of the 1,3-dioxygenated Danishefsky diene 65, Gribble et al. showed in 2001 that differently protected 3-nitroindole derivatives could react under thermal conditions (Scheme 25).43 Reacting the N-phenylsulfonylindole 1a with 65 in refluxing toluene afforded a mixture of the nitroindoline 66a-trans (56%) along with the re-aromatized carbazol-2-ol 67a (35%). Thus, with this more reactive diene, the (4+2) dearomatized cycloadduct was stable and could be isolated, even with a leaving group such as nitro. With the carbamate protected indole 1b, the cycloaddition led to the isolation of carbazolol 67b in a good yield of 85%. The same year, Mancini et al. showed that, in the presence of sulfonylindole 1c, decreasing the temperature to 65 °C allowed isolation of both the cis and trans diastereomers in an overall 64% yield (39% cis-diastereomer, 25% trans).39 A smaller amount of the re-aromatized carbazol-2-ol 67 was also isolated in this case (17%). This suggested that the cis isomer, which was not isolated in the reaction with 1a, was more sensitive to heating.
 | ||
| Scheme 25 (4+2) Diels–Alder reaction between 3-nitroindoles 1c and Danishefsky diene 65 under thermal activation. | ||
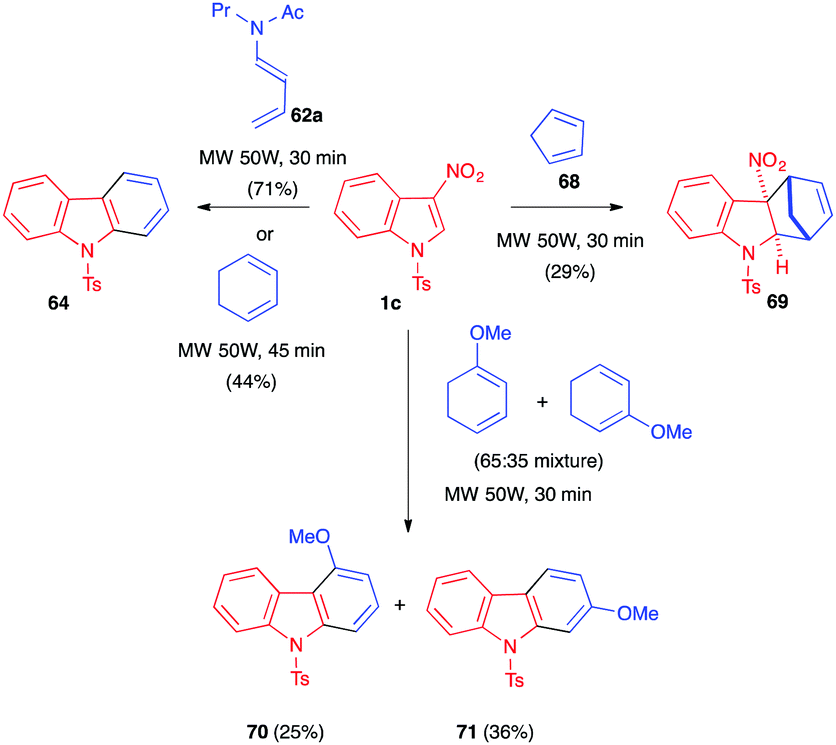
These reactions were also performed under microwave irradiation as an alternative to conventional heating.44 Gómez et al. showed, in 2009, that 3-nitroindole 1c reacted with a variety of dienes under MW irradiation (50 W, 100 °C) and solvent-free conditions. As shown in Scheme 26, this activation mode allowed the reaction time to be significantly decreased to 30–45 minutes and furnished the aromatized cycloadducts. These were obtained via (4+2) cycloaddition/elimination/retro-Diels–Alder or (4+2) cycloaddition/elimination(s) sequences except in the case of cyclopentadiene (68). With this latter reactive diene, the dearomatized nitroindoline 69 was isolated as a single diastereomer, without loss of nitrous acid, in a modest 29% yield. Computational studies suggested that this stereochemical outcome would arise from an unexpected hetero-Diels–Alder cycloaddition involving the CC–NO electron-poor dienic moiety of the indole and one of the electron-rich CC bonds of the cyclopentadiene and would be followed by a [3,3]-sigmatropic shift.
 | ||
| Scheme 26 Reactivity of 3-nitroindole 1c toward different dienes under microwave irradiation. | ||
In 2012, Mancini and co-workers showed that these Diels–Alder reactions could also be performed in protic ionic liquids such as [HMIM][BF4].41 Thus, nitroindole 1c reacted with dienes such as isoprene 55, 1-trimethylsilyloxybutadiene 72 or Danishefsky diene 65, for 24–48 h at 60 °C in this neoteric solvent (Scheme 27). After the polar cycloaddition, the carbazole adducts were isolated in good yields. These conditions seemed to favour the elimination reactions occurring after cycloaddition leading to the completely aromatized adducts as the sole or main compounds in all cases.
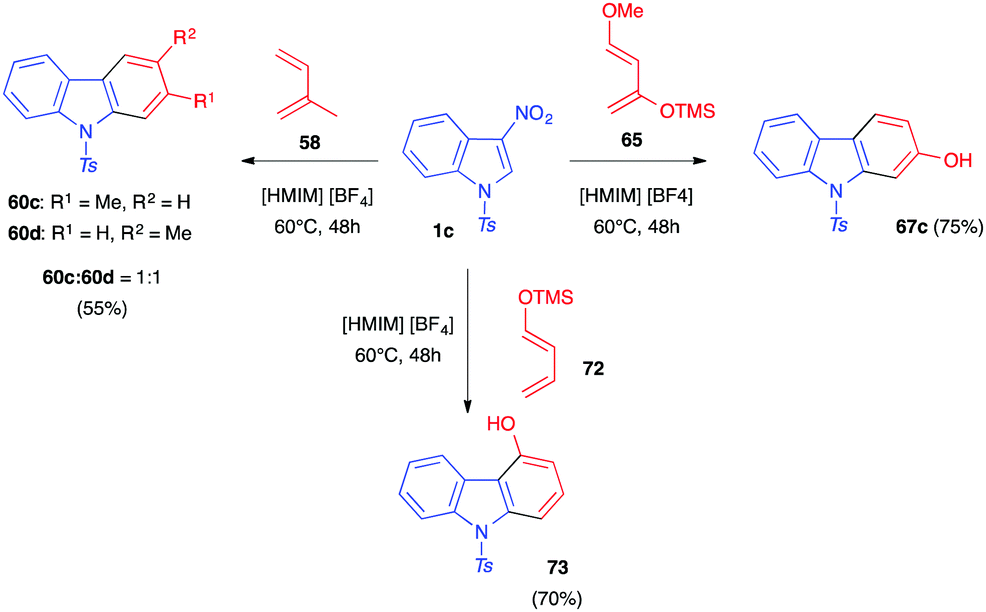 | ||
| Scheme 27 Reactivity of 3-nitroindole 1c toward different dienes in the protic ionic liquid [HMIM][BF4]. | ||
Compared to thermal conditions, the reactions are favoured in this solvent due to the activation of the dienophilic nitroindole by hydrogen bonding between the oxygen atoms of the nitro group and the acidic hydrogen atom of [HMIM][BF4]. Evaluation of the electrophilicity of the nitroindole by DFT computations indeed showed that if nitroindole 1c is a “strong electrophile” on the electrophilicity scale developed by Domingo and co-workers (global electrophilicity, ω = 2.25 eV),45 the activated dienophile 1c-[HMIM][BF4] is even more electrophilic (ω = 3.48 eV in the gas phase, and ω = 2.98 eV when an implicit solvent is considered in the calculations). The regioselectivity observed for these processes was also in line with the calculated local electrophilicities that showed that the C2 carbon atom of the nitroindole was much more electrophilic than the C3 carbon atom (0.48 vs 0.07 eV for 1c and 0.75 vs 0.04 eV for 1c-[HMIM][BF4]).
Diels–Alder reactions are generally characterized by largely negative volumes of activation (−25 to −45 cm3 mol−1). Thus, these transformations are often highly accelerated under high pressure. In 2001, taking advantage of this phenomenon, electron-poor indoles bearing electron-withdrawing groups at position 3 were reported to react at room temperature under 12–16 kbar pressures with different electron-rich dienes to yield the corresponding dearomatized primary indolines.46 Under these conditions, the cycloaddition reaction was promoted while, in contrast, the re-aromatization process, which involves elimination reaction(s), generally characterized by positive activation volumes, was disfavoured.
Soon after, Mancini et al. elegantly demonstrated the reactivity of N-sulfonyl-3-nitroindoles 1c toward various 1,3-dienes under hyperbaric conditions. Interaction of 1c and isoprene 58 under an 11.5 kbar pressure, at room temperature, for 24 h led to the formation of the expected dearomatized cycloadduct 61a, obtained as a single regioisomer, showing the positive effect of high pressure on the transformation (Scheme 28).39 The isolated yield, however, remained low (22%). Increasing both the temperature to 40 °C and the reaction time to 3 days enhanced the yield to 75%. Under these conditions, traces of the corresponding re-aromatized carbazole were observed (5%), together with some unreacted indole (20%). Indole 1c could also react with the dienamide 62a under the same pressure. The cycloaddition led, this time, to a mixture of the desired dearomatized cycloadduct 74 and the corresponding denitrated compound 63, obtained in a 1:2 ratio, along with unreacted indole (24%). Satisfyingly, the cycloaddition was also performed with Danishefsky diene 65. With this more reactive diene, a total conversion of the indole 1c was observed, leading to the nitrocycloadduct 66c (1:1 d.r.) with no traces of the re-aromatized cycloadducts.
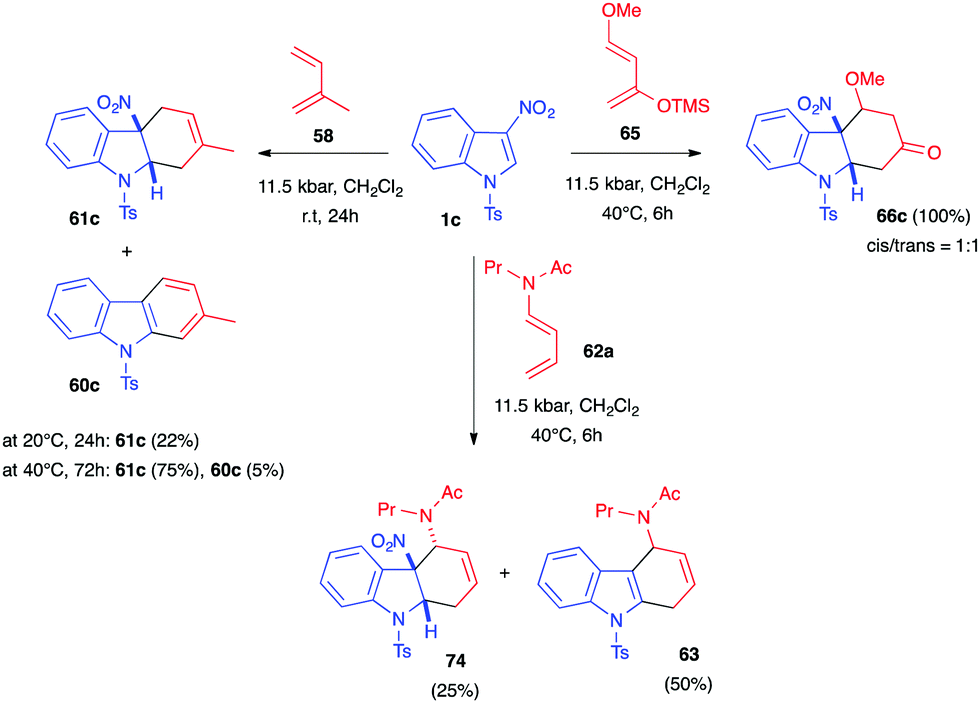 | ||
| Scheme 28 Dearomative (4+2) cycloadditions of 3-nitroindole 1c with different dienes under high pressure activation. | ||
In 2015, our group reported the dearomative (4+2) cycloaddition of 3-nitroindole 1c in the presence of Danishefsky diene 65 at room temperature in the presence of a thiourea organocatalyst.47 The achiral Schreiner thiourea catalyst 75 indeed promoted the Diels–Alder reaction at room temperature. Under these mild thermal conditions, no elimination reaction occurred after cycloaddition and the corresponding adducts 66c (63:37 of trans/cis diastereomers) were isolated in 95% yield (Scheme 29). Attempts to perform this reaction in an enantioselective manner in the presence of chiral thiourea or squaramide catalysts, however, led to low enantioselectivities.
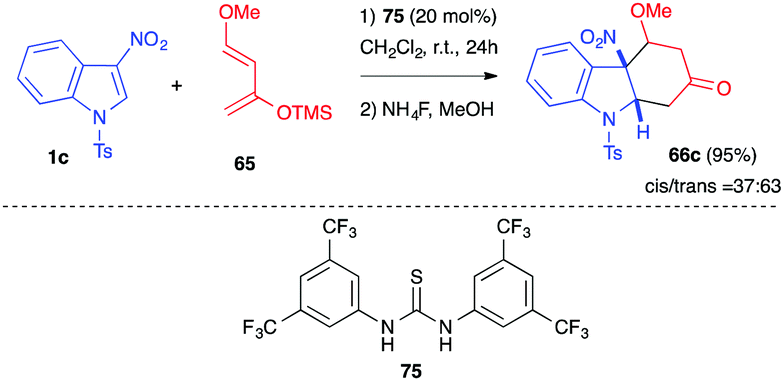 | ||
| Scheme 29 Dearomative (4+2) cycloadditions of 3-nitroindole 1c with Danishefsky diene 65 under thiourea organocatalysis. | ||
An achievement in the field of enantioselective (4+2) cycloadditions with 3-nitroindoles was the organocatalytic approach reported by Jørgensen and co-workers in 2016 (Scheme 30).48 The cycloaddition of 3-nitroindole derivatives 1 with 2,4-dienals 76 was performed in a highly enantioselective fashion in the presence of the proline-urea organocatalyst 77. This trienamine pathway was found to be efficient at low temperature and furnished, under the basic conditions of the reaction, the chiral dihydrocarbazole 78 in good yields (up to 87%) and enantioselectivies (up to 97% ee) after nitrous acid elimination. The bifunctionnal organocatalyst brings closer the diene/dienophile substrates, by allowing the formation of the electron-rich trienamine while activating the nitroindole by hydrogen bonding, thus inducing an efficient facial discrimination.
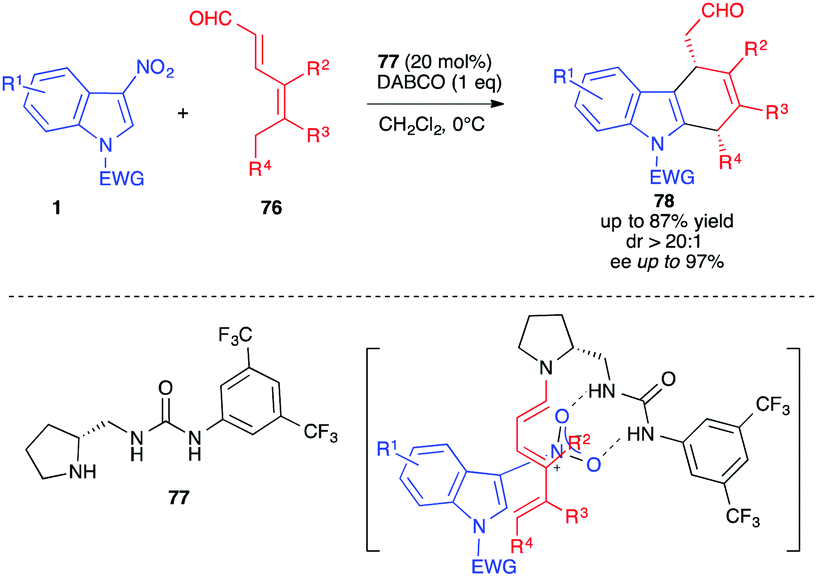 | ||
| Scheme 30 Organocatalyzed enantioselective (4+2) cycloadditions of 3-nitroindoles with 2,4-dienals 72. | ||
3.2. (4+2) annulations
(4+2) Dearomative annulations involving 3-nitroindoles as electrophiles have emerged more recently, taking advantage of the possible addition of nucleophiles at the C2 position of 3-nitroindoles (Scheme 31). If the nucleophile is a 1,4-zwitterionic dipole, the nitronate intermediate can subsequently cyclize intramolecularly in a (4+2) formal cycloaddition process. The first addition step is the dearomatizing one, often reversible, and the second cyclization step thus shifts the equilibrium toward the formation of the dearomatized cycloadduct. This strategy is compatible with the use of relatively mild nucleophiles for the first determining step.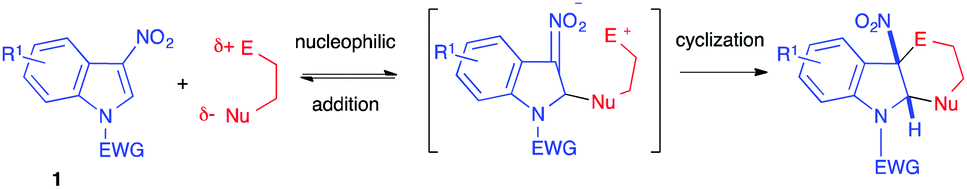 | ||
| Scheme 31 General dearomative (4+2) annulations of 3-nitroindoles. | ||
In 2017, Xu, Yuan et al. reported the first organocatalyzed reaction of 3-nitroindoles 1 with Nazarov reagents 79, which possess a nucleophilic α-carbon and an electronically deficient CC double bond, in the presence of the thiourea organocatalyst 80 (Scheme 32).49 Its bifunctional nature allowed, here again, activation of the 3-nitroindole 1 by the formation of multiple hydrogen bonds with the amide/thiourea moiety and the simultaneous activation of 79 through the tertiary amine function of the catalyst. This dual activation was crucial for the reactivity and the stereocontrol. After acylation, the chiral hydrocarbazole skeletons 81 were obtained in good to high yields with excellent diastereo- and enantio-selectivities.
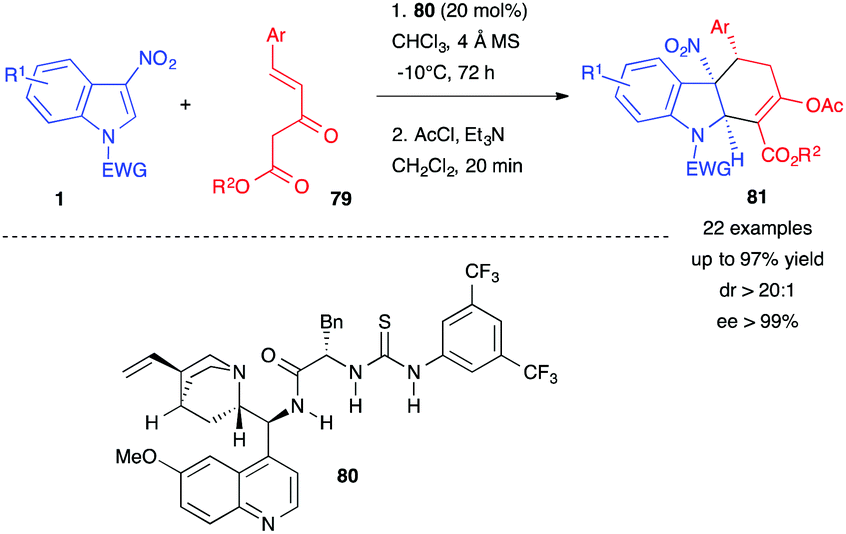 | ||
| Scheme 32 Enantioselective dearomative (4+2) annulations of 3-nitroindoles with Nazarov reagents 79. | ||
In 2018, Yang et al. reported the (4+2) annulation of 3-nitroindoles 1 with alkylidene malononitriles 82 (Scheme 33).50 In the presence of triethylamine, carbazol-4-amine derivatives 83 were obtained via a four-step sequence involving (i) vinylogous Michael addition, (ii) cyclization, (iii) isomerization, and (iv) nitrous acid elimination. This reaction was recently extended to different α,α-dicyanoalkenes 82.51
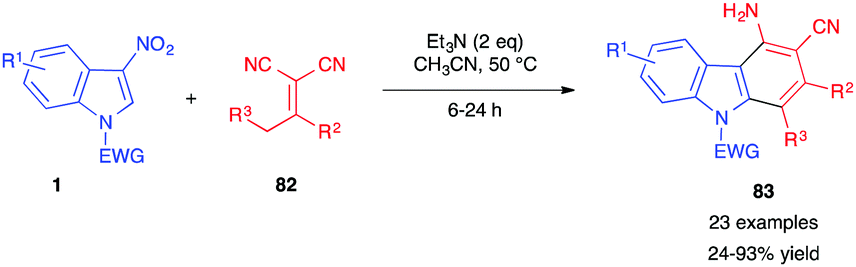 | ||
| Scheme 33 (4+2) annulations of 3-nitroindoles with alkylidene malononitriles 82. | ||
In 2019, Ding, Hou et al. developed a palladium-catalyzed asymmetric decarboxylative (4+2) annulation of 3-nitroindoles 1 with benzoxazinanones 84 (Scheme 34).52 In the presence of Pd2dba3·CHCl3 (2.5 mol%) and a chiral SIOCPhox ligand 85 (5 mol%), a series of chiral dearomatized tetrahydro-5H-indolo[2,3-b]quinolines 86 was obtained in high yields, up to 97%, and good to excellent stereoselectivities (dr > 95:5, and ee = 79–95%). N-Tosyl protected vinyl benzoxazinanones 84 were required and no reaction took place when replacing the tosyl group with a proton, a nosyl or an acetyl group. The authors proposed a transition state involving a dual role of the catalyst, allowing the formation of the π-allylic 1,4-dipole and activating the nitro group of the indole with the phenol hydroxyl group of the ligand to account for the observed stereoselectivities.
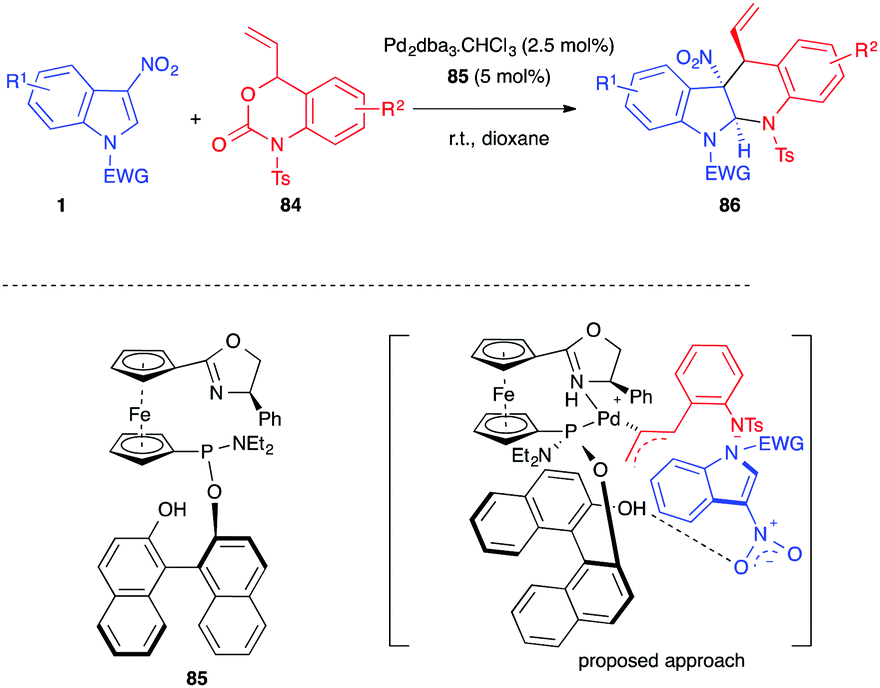 | ||
| Scheme 34 Enantioselective palladium catalyzed decarboxylative (4+2) annulations of 3-nitroindoles with N-tosyl vinyl benzoxazinanones 84. | ||
Hyland and co-workers demonstrated recently that the same reaction was also possible in the presence of unprotected vinyl benzoxazinanones 87, using a higher catalyst loading of Pd2dba3·CHCl3 (7.5 mol%) and ligand (15 mol%) (Scheme 35).53 In a diastereoselective version of the process, the reaction could be promoted by the simple and air-stable phenanthroline ligand. Excellent yields and trans diastereoselectivities were reported with this catalytic system, even in the absence of any electrophilic activation of the nitro group. To account for this trans preference, the authors proposed a reversible nucleophilic attack at the C2 position of the indole to generate a nitronate, which would undergo a ring-closure via a favoured boat-like transition state. Note, N-sulfonylated nitroindoles 1 were required for this process as N-carbonylated substrates proved inert or led to decomposition. In addition, C3-substituted acetyl, cyano, and methyl ester indoles failed in the reaction, showing, here again, the importance of the nitro group at position 3 for enhancing the electrophilicity of the indole ring. When performed in the presence of a chiral phosphinooxazoline ligand 88, a high yield and ee for the trans diastereomer (>99% ee) were reported. However, the dr was significantly impacted (Scheme 35).
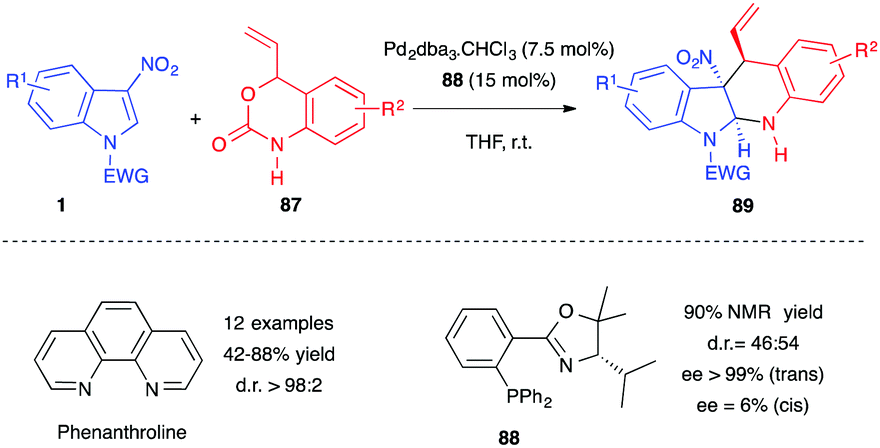 | ||
| Scheme 35 Palladium catalyzed decarboxylative (4+2) annulations of 3-nitroindoles with unprotected vinyl benzoxazinanones 87. | ||
In 2019, the dearomative (4+2) annulation of 3-nitroindoles 1 in the presence of enolizable vinylketones 90 was reported under basic conditions and at low temperatures (Scheme 36).54 The in situ generated lithium enolates add to the C2-position of the nitroindoles 1. Then, depending on the substrates, trans 1,4-adducts 91 or, most of the time, in situ cyclized adducts 92 were isolated in good yields. This method constitutes an alternative to the classical Diels–Alder cycloaddition performed in the presence of 2-silyloxy-1,3-dienes, such as Danishefsky diene, for which the re-aromatized carbazolols 64 were often isolated (vide supra).
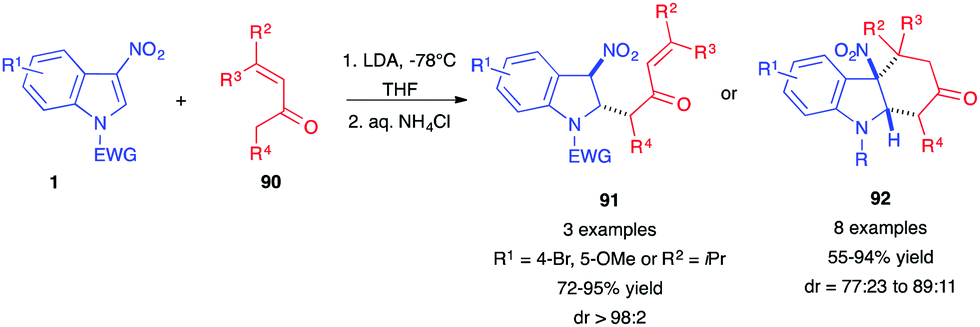 | ||
| Scheme 36 Dearomative (4+2) annulations of 3-nitroindoles with enolizable vinyl ketones 90. | ||
Guo et al. recently reported the phosphine-catalyzed enantioselective (4+2) annulation of 3-nitroindoles 1 with allenoates 93 at room temperature, in the presence of a chiral phosphine catalyst 94 (Scheme 37).55 Such a process generally leads to the generation of tetrahydrocyclopenta[b]indoline 47, according to a (3+2) Lu annulation (vide supra, Scheme 17). With allenoates 93, bearing an alpha substituent, benzylic most of the time, a different and complementary pathway was observed. The dipole A generated after nucleophilic addition of the phosphine on the allenoate reacted with the electrophilic indole at the less sterically hindered γ-position, this time, leading to a nitronate zwitterion B. Migration of the double bond followed by intramolecular cyclization would lead to D, which would release the catalyst and furnish the formal (4+2) cycloadduct E. Under the reported reaction conditions, re-aromatization of the 5-membered ring by loss of nitrous acid would then afford the isolated dihydrocarbazole 95 in good yields and enantioselectivities.
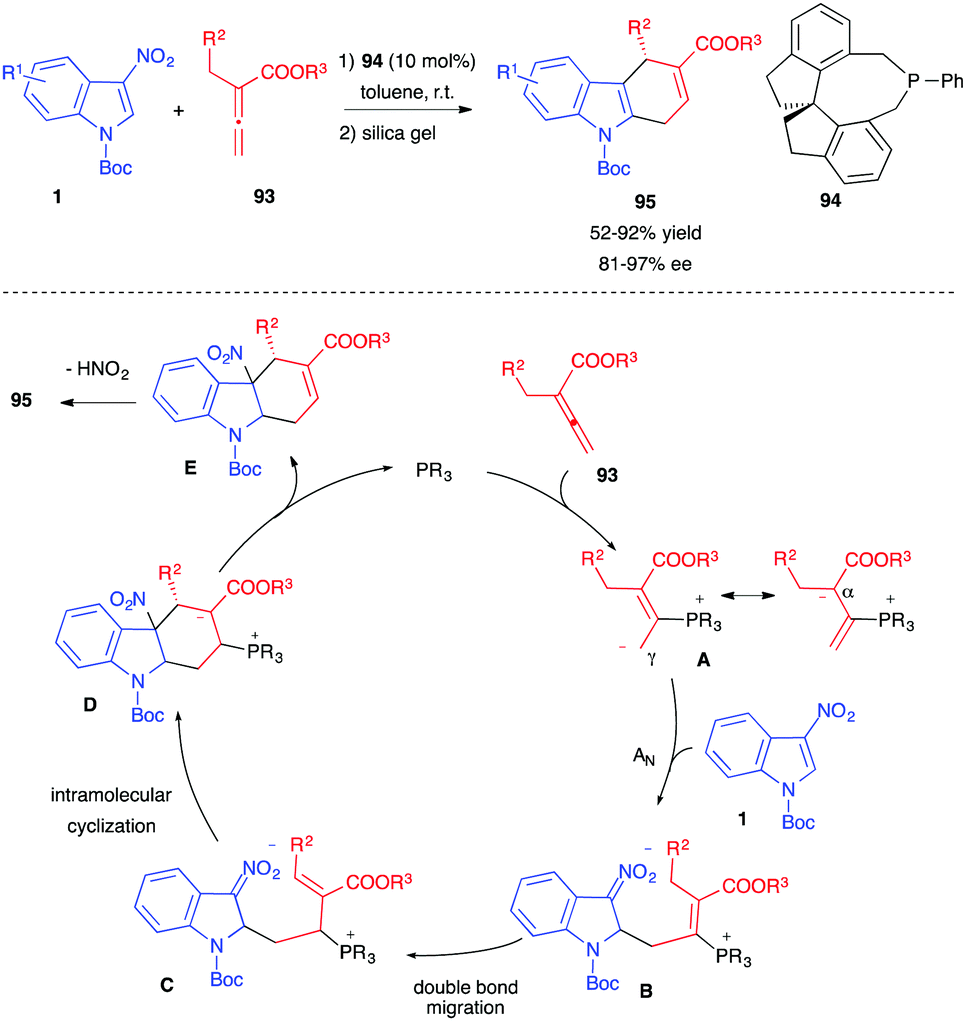 | ||
| Scheme 37 Enantioselective phosphine-catalyzed (4+2) annulations of 3-nitroindoles with allenoates 93. | ||
In 2020, Huang, Li et al. reported a novel type of dearomative (4+2)-annulation of 3-nitroindoles 1 with β-substituted crotonaldehydes 96 (Scheme 38).56 Under an oxidative N-heterocyclic carbene (NHC) catalyzed process, dearomatized hydrocarbazolones 97 were obtained in good yields. The use of a chiral NHC catalyst allowed an enantioselective version of this reaction to be performed, with ee up to 74%. This was, however, detrimental to the isolated yield (18%).
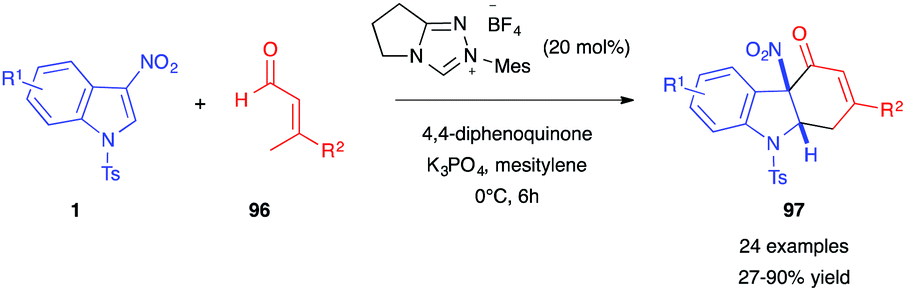 | ||
| Scheme 38 Oxidative NHC-catalyzed (4+2) annulation of 3-nitroindoles 1 with β-substituted crotonaldehydes 96. | ||
4. (4+2)/(3+2) cycloaddition reactions
The cycloadditions/annulations described so far rely on the electrophilic character of the C2C3 double bond of the 3-nitroindole derivatives. Based on the reactivity of the C2C3–NO system as an electron-poor hetero-diene,57 in 2007, the first multicomponent domino (4+2)/(3+2) cycloaddition process was reported in the presence of N-tosyl-3-nitroindoles 1c. Performed in the presence of an electron-rich vinyl ether dienophile 98 and an electron-poor acrylate dipolarophile 99, the reaction led to the formation of a nitrosoketal 100 in one operation (Scheme 39).58 Under reductive conditions, 100 could be converted into 101 in high yields, through (i) N–O hydrogenolyses, (ii) formation of an imine and its reduction into pyrrolidine, and (iii) lactamization.
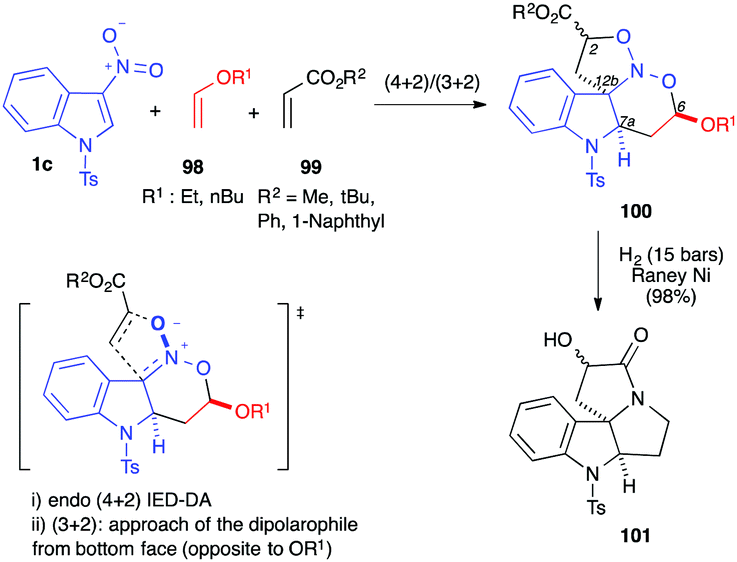 | ||
| Scheme 39 Domino (4+2)/(3+2) cycloaddition of 3-nitroindole 1c with vinylethers 98 and acrylates 99. | ||
In an attempt to develop an enantioselective version of this reaction, it was performed in the presence of different chiral thioureas or sulfonamides, but the enantioselectivities remained low (up to 30% ee).47
In this reaction, 4 stereocenters were created and 8 couples of diastereomers were potentially obtained. High pressure conditions gave the best diastereoselectivity (Table 3, compare entries 1–4). The two diastereomers obtained under these conditions were observed to differ in the relative positions of the centre alpha to the ester group, the result of an endo (4+2) inverse electron demand hetero-Diels–Alder reaction (relative stereochemistry of C7a and C6), and a face selective (3+2) cycloaddition (dipolarophile from the face opposite to OR1, and relative stereochemistry of C12b), while the endo vs. exo approach of the dipolarophile is poorly controlled (two diastereomers differing in the relative stereochemistry of C2). Switching to an acrylate bearing a more hindered aromatic moiety led to a higher diastereoselectivity (Table 3, entry 7).
| Entry | Conditions | R2 | Yield (100) (%) | dr |
|---|---|---|---|---|
| 1 | CH2Cl2, r.t., 168 h | Me | 20 | 50/50/0/0 |
| 2 | Tol., 110 °C, 60 h | Me | 83 | 45/45/5/5 |
| 3 | MW (120 W), 130 °C, 1 h | Me | 75 | 35/35/15/15 |
| 4 | HP (12 kbar), r.t., 24 h | Me | 83 | 55/45/0/0 |
| 5 | HP (12 kbar), r.t., 24 h | tBu | 83 | 60/40/0/0 |
| 6 | HP (12 kbar), r.t., 24 h | Ph | 92 | 66/34/0/0 |
| 7 | HP (12 kbar), r.t., 24 h | 1-Naphthyl | 99 | 78/22/0/0 |
DFT calculations reported on these multicomponent domino (4+2)/(3+2) cycloadditions of 3-nitroindoles confirmed that the endo approach with the regiochemistry observed experimentally was indeed the most favourable pathway for the primary (4+2) cycloaddition reaction.59 The 3-nitroindole reacted through its LUMO as an electron-poor heterodiene in a concerted though asynchronous inverse demand hetero-Diels–Alder manner. The electronic rearrangements observed during this step were in line with the expected pathway and, for this first cycloaddition process, the FMO model provided perfectly reliable predictions (Scheme 40).
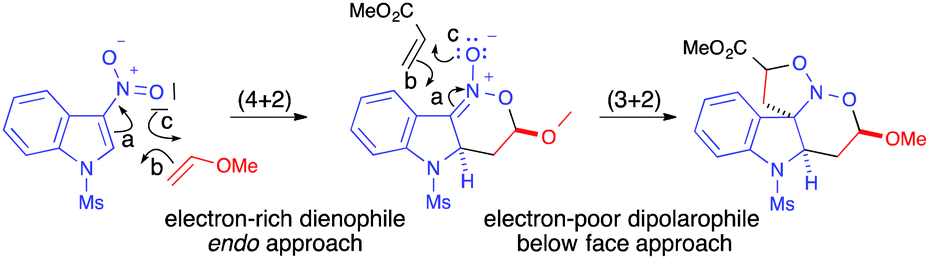 | ||
| Scheme 40 Electronic rearrangements computed for the most favoured pathway in the two step (4+2)/(3+2) sequence. | ||
The results computed for the subsequent (3+2) cycloaddition reaction were also in line with the experimental results, with a more favoured approach of the dipolarophile from the face opposite to the alkoxy group of the intermediate. A small difference between endo and exo approaches also accounted for the poor diastereoselectivity experimentally observed. During the second (3+2) cycloaddition step, however, unexpected electronic rearrangements were observed. Even if the electron-deficient acrylate appeared to react in preference to the electron-rich vinyl ether with the intermediate nitronate, the calculations demonstrated that the electron-poor dipolarophile provided the electrons to the electron-rich dipole (Scheme 40, arrow b for the (3+2) step).
5. Reactions with other nucleophiles
5.1. Nucleophilic addition reactions
The electrophilicity of 3-nitroindole derivatives allows the addition of different types of nucleophiles to the C2 position of the heterocycle, leading to a nitronate intermediate. The addition can be reversible and leads to different types of adducts, depending on the quenching of the intermediate. When reacted with ethyl isocyanoacetate 37a in the presence of an organic base such as DBU, it has been shown above that 3-nitroindoles 1 protected as N-benzyl, N-pyridyl or N-ethoxycarbonyl compounds underwent the Barton–Zard reaction, by conjugate nucleophilic addition of the enolate anion on the nitroindole substrate, followed by intramolecular 5-endo-dig cyclization of the intermediate nitronate on the isocyanide (vide supra, Scheme 14).25 When more electron-withdrawing groups such as phenylsulfonyl were activating the indole ring, an abnormal regioselectivity was observed for this reaction, leading to a ring opening/rearrangement and a regioisomeric cycloadduct 102 (Scheme 41).26 The mechanism proposed by Gribble et al. to account for this unexpected regioselectivity involved first the expected Michael addition of the enolate anion resulting from the deprotonation of the isocyanide on the C2 position of the nitroindole. Then, an expected ring opening would occur. After prototropy, the nitronate anion would add to the isocyanide to form the 5 membered ring. After prototropy, ring closure of the central ring and concomitant elimination of nitrite, a final isomerisation would lead to the isolated compound 102.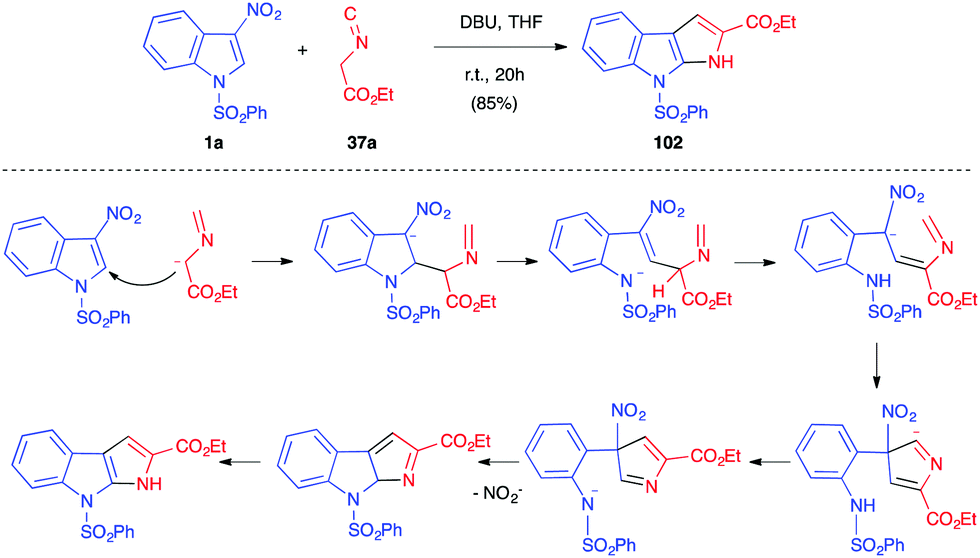 | ||
| Scheme 41 Abnormal Barton–Zard reaction between indole 1a and isocyanide 37a. | ||
Later in 2003, Gribble disclosed several nucleophilic additions of enolates to 3-nitroindole 1a. Thus, the “simple” addition of the anion at the C2 position of the indole results, after hydrolysis, in the dearomatized indolines 103 (Scheme 42).60 Thus, the use of deprotonated diethyl malonate or cyclohexanone led to the formation of the Michael-type nitronate adduct, which furnished selectively the trans indoline after hydrolysis. Similarly, addition of a vinyl Grignard reagent led to the trans adduct 103c.
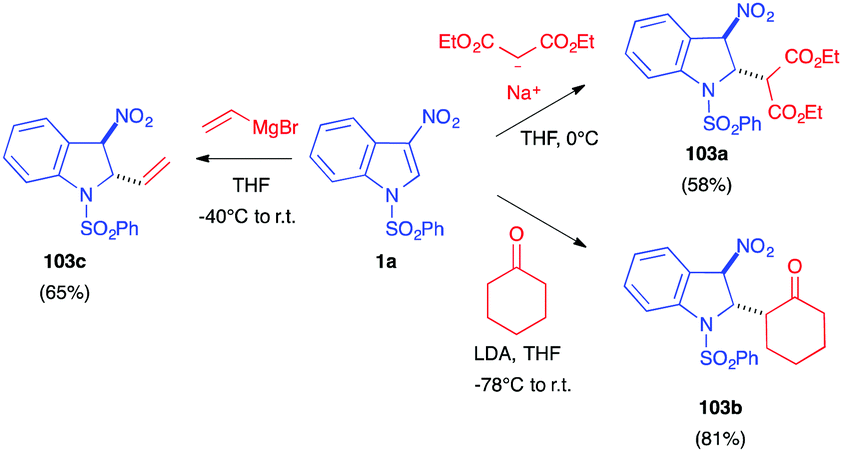 | ||
| Scheme 42 Michael addition of enolates and vinyl Grignard reagents to nitroindole 1a. | ||
The use of alkyl Grignard reagents led to the same Michael adducts 103, however with a small amount of the ketones 104, resulting from the Nef reaction (Scheme 43). Re-aromatization of 103 to 105 could easily be performed in the presence of Pd/C.
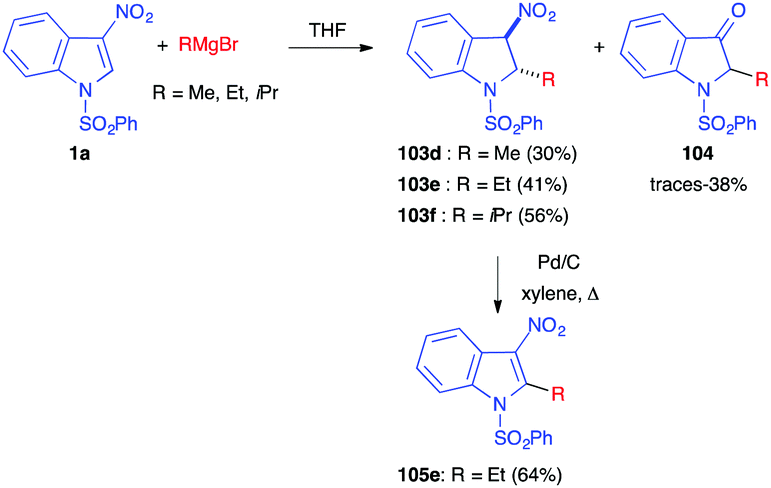 | ||
| Scheme 43 Nucleophilic addition of alkyl Grignard reagents to nitroindole 1a. | ||
In 2010, the same group reported the nucleophilic addition of (hetero)aryllithiated compounds to 3-nitroindole 1a. In most cases, the reaction led to the N-deprotected adduct 107 in good yields (Scheme 44).61 The authors proposed a mechanism involving the nucleophilic addition of the lithiated compound to the C2 position of the indole, followed by an elimination of the sulfinate anion and prototropy. In one case only, involving a 2-lithioindolic nucleophile, the corresponding trans indoline 103 was isolated.
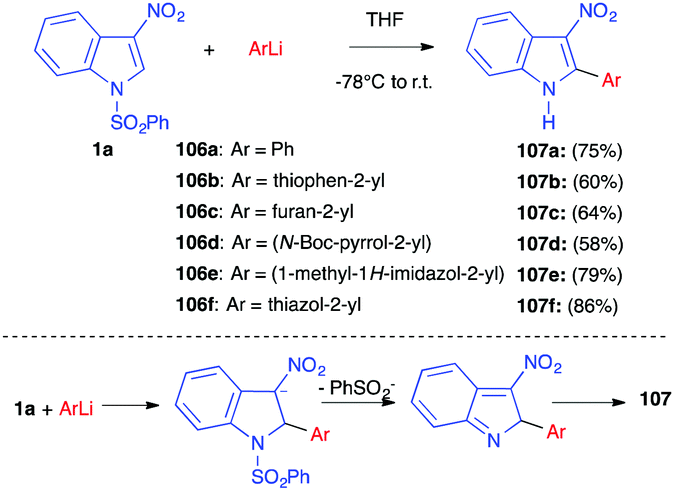 | ||
| Scheme 44 Nucleophilic addition of organolithium nucleophiles 106 to nitroindole 1a. | ||
5.2. Nucleophilic additions: the special case of enamines
More recently, the addition of enamines to nitroindoles 1 has been studied. In 2016, the stoichiometric addition of 4-(cyclohex-1-en-1-yl)morpholine 108 to 1c, under mild anhydrous conditions, was reported to lead to the formation of the unexpected dearomatized indoline 109, as a single regioisomer and diastereomer (Scheme 45).62 It was isolated in an excellent 95% yield, even on a 10 g scale. This formal ene adduct 109 is formed by nucleophilic addition of the enamine at the C2 position of the nitroindole followed by selective proton abstraction under the conditions of the reaction. The reaction proved to be reversible in solution and the ene adduct 109 was sensitive to water. It was efficiently isolated in this case thanks to its highly crystalline character. When compared to the addition of vinylethers that required high temperatures or high pressure conditions, the addition of enamines is much easier, and reflects the much higher nucleophilicity of enamines. DFT computations in this field suggested that the greatest difference between these oxygenated and nitrogenated nucleophiles would come from the much higher stability of the zwitterionic nitronate intermediates resulting from the addition (iminium vs oxonium).63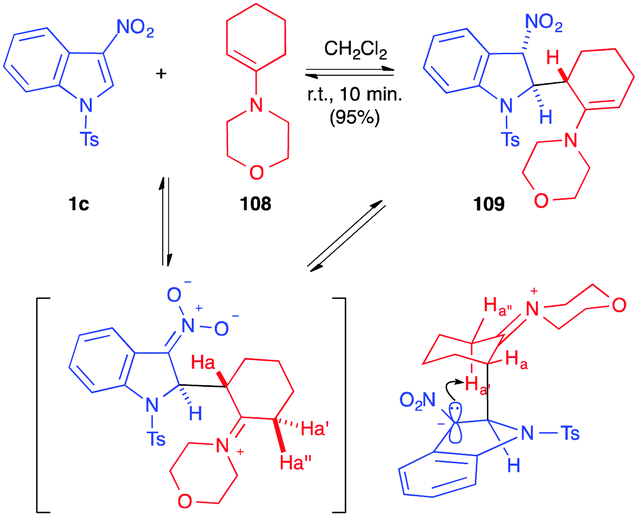 | ||
| Scheme 45 Dearomatizing formal ene reaction on nitroindole 1c. | ||
In 2017, John et al. studied the addition of in situ generated enamines toward nitroindoles.64 They showed that reacting the nitroindole 1c with an enolizable ketone 110 and a primary amine 111, in toluene at 60–110 °C in the presence of 4 Å MS, resulted in the aromatized pyrrolo[3,2b]indoles 112, isolated in good yields (Scheme 46). In this case, the involvement of a primary amine would imply the formation of an imine, in equilibrium with the enamine 113. The authors proposed a mechanism involving first the nucleophilic addition of the enamine to the C2 position of the indole derivative, as previously observed. In this case, however, the iminium proton could be trapped by one of the nitronate oxygen atoms. A second enamine addition would then allow for the five-membered ring closure. This latter state would then aromatize by elimination of hyponitrous acid and water, generating 112.
 | ||
| Scheme 46 Multicomponent reaction involving nitroindole 1c to generate pyrrolo-indoles 112. | ||
Soon after, the same authors reported an extended version of the methodology toward the synthesis of indolo[3,2b] indoles 114 (Scheme 47).65 In this case, the intermediate pyrrolo[3,2b]indoles, formed after 12 h in toluene at 60 °C in the presence of cyclic ketones, were oxidized in situ by addition of chloranil as the oxidant. The corresponding fully aromatized compounds 114 were isolated in good overall yields.
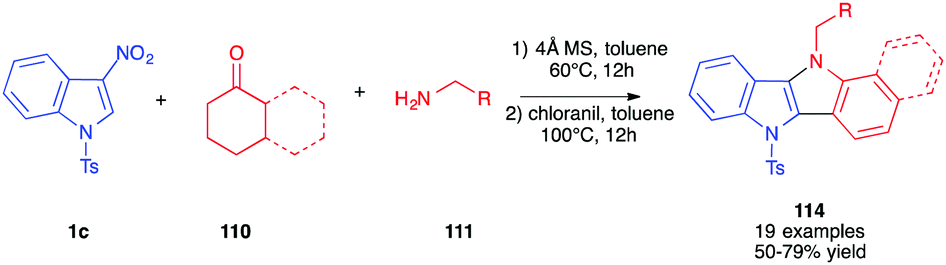 | ||
| Scheme 47 One-pot generation of indolo-indoles 111 from nitroindole 1c. | ||
5.3. Nucleophilic aromatic substitution reactions
In 2006, Gribble and Roy described an efficient SNAr reaction involving secondary amines and an activated 2-iodo-3-nitroindole 115 (Scheme 48).66 The reaction was performed under mild conditions, with a range of secondary amines 116, affording the corresponding 2-amino-3-nitroindoles 117 in very good yields (76–96%), while the yield dropped with the less nucleophilic primary cyclohexylamine.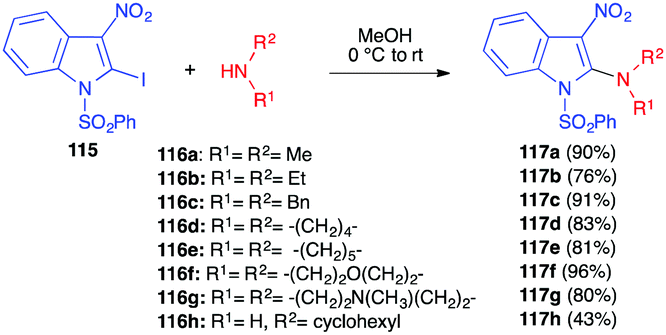 | ||
| Scheme 48 Nucleophilic aromatic substitution between 2-iodo-3-nitroindole 99 and amines 100. | ||
6. Conclusions
3-Nitroindoles 1 exhibit an unexpected, albeit remarkable, electrophilic reactivity toward different electron-rich species, notably in dearomative cycloaddition and annulation reactions. This peculiarity is directly related to the C2C3 double bond of nitroindoles that can behave either as a dipolarophile, a dienophile or a Michael acceptor in dearomatizing (3+2) and (4+2) cycloadditions or nucleophilic addition reactions. In a complementary manner, the heterodienic character of the C2C3–NO system of 3-nitroindoles explains their reactivity in multicomponent tandem (4+2)/(3+2) cycloadditions. Dearomatizing nucleophilic additions at the C2 center have also been reported.
The reported cycloadditions can be concerted; the corresponding transformations generally involve a highly asynchronous transition state with a smaller forming bond at the C2 position. The limit of this concerted asynchronous mechanism is encountered in many other cases with stepwise processes, in which the first step involves a reversible dearomatizing nucleophilic addition, and the nitronate intermediate is then trapped intramolecularly in a formal cycloaddition process.
Recent developments of these reactions include efficient enantioselective strategies, often taking advantage of bifunctional catalysis, activating the electrophilicity of the indole moiety by hydrogen bonding with the nitro group, for instance with thiourea- or squaramide-containing catalysts. Meanwhile, the reacting nucleophilic species are formed/activated, both being linked by a chiral tether.
All these transformations recently reported for the electrophilic 3-nitroindoles underline how this heteroaromatic motif has become a model substrate for electron-poor aromatic compounds in dearomatization strategies.
The reported transformations have led to the expedient synthesis of differently functionalized indolines, which often bear a tetrasubstituted centre at the cis junction of the heterocycle, a feature often retained in natural products of interest. These new structures themselves are potentially relevant compounds for biological or catalytic applications. In addition, after dearomatization, even if the nitro group is still at a benzylic position, it can be simply reduced into the corresponding amino group, using for instance the TMSCl/Zn system, in most cases. Denitration reactions can also be easily performed, notably through radical denitration strategies. Re-aromatization of the 5-membered cycle, by elimination of nitrous acid, is also an easy process. All these transformations and post-functionalizations of the 3-nitroindole motif thus make this compound a good scaffold for diversity-oriented synthesis (Scheme 49).
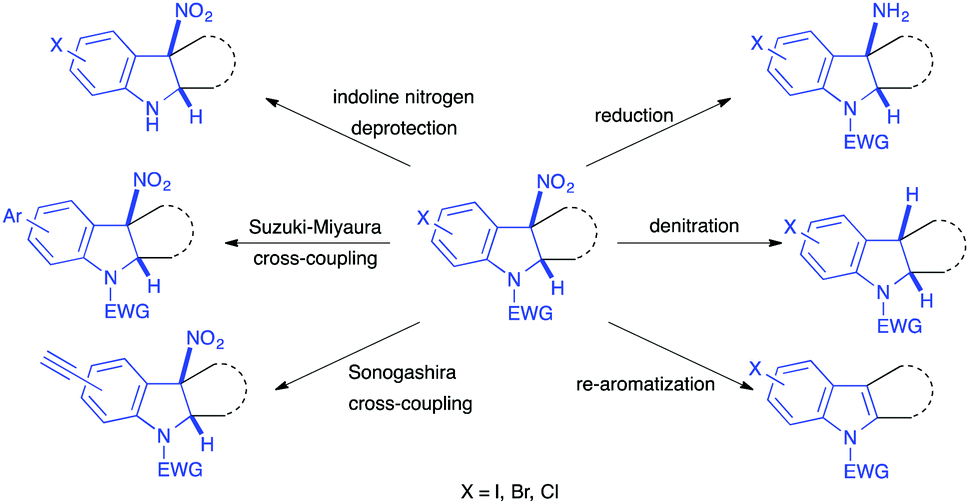 | ||
| Scheme 49 Representative post-functionalizations of the nitroindolines obtained through dearomatizations of 3-nitroindoles. | ||
Conflicts of interest
There are no conflicts to declare.Acknowledgements
Financial support from the Agence Nationale pour la Recherche (“ArDCo”, ANR-17-CE07-0050), the European France-(Manche)-England cross-border cooperation program INTERREG V A “LABFACT” and “SMARTT”, co-financed by ERDF, and Janssen R&D is acknowledged. The authors also thank Normandie Université, CNRS, INSA Rouen, Tremplin Carnot I2C, Labex SYNORG (ANR-11-LABX-0029) and the Région Normandie.Notes and references
- I. G. Sonsona, Synlett, 2015, 2325–2326 Search PubMed.
- J. M. Saya, E. Ruijter and R. V. A. Orru, Chem. – Eur. J., 2019, 25, 8916–8935 CrossRef CAS.
- A. Cerveri and M. Bandini, Chin. J. Chem., 2020, 38, 287–294 CrossRef CAS.
- G. W. Gribble, E. T. Pelkey and F. L. Switzer, Synlett, 1998, 1061–1062 CrossRef CAS.
- G. W. Gribble, E. T. Pelkey, W. M. Simon and H. A. Trujillo, Tetrahedron, 2000, 56, 10133–10140 CrossRef CAS.
- S. Roy, T. L. S. Kishbaugh, J. P. Jasinski and G. W. Gribble, Tetrahedron Lett., 2007, 48, 1313–1316 CrossRef CAS.
- S. Lee, I. Chataigner and S. R. Piettre, Angew. Chem., Int. Ed., 2011, 50, 472–476 CrossRef CAS.
- S. Lee, S. Diab, P. Queval, M. Sebban, I. Chataigner and S. R. Piettre, Chem. – Eur. J., 2013, 19, 7181–7192 CrossRef CAS.
- M. Manneveau, S. Tanii, F. Gens, J. Legros and I. Chataigner, Org. Biomol. Chem., 2020, 18, 3481–3486 RSC.
- T. Thierry, C. Lebargy, E. Pfund and T. Lequeux, J. Org. Chem., 2019, 84, 5877–5885 CrossRef CAS.
- A. Awata and T. Arai, Angew. Chem., Int. Ed., 2014, 53, 10462–10465 CrossRef CAS.
- A. L. Gerten and L. M. Stanley, Org. Chem. Front., 2016, 3, 339–343 RSC.
- X. Liu, D. Yang, K. Wang, J. Zhang and R. Wang, Green Chem., 2017, 19, 82–87 RSC.
- B. M. Trost and G. Mata, Acc. Chem. Res., 2020, 53, 1293–1305 CrossRef CAS.
- B. M. Trost, V. Ehmke, B. M. O’Keefe and D. A. Bringley, J. Am. Chem. Soc., 2014, 136, 8213–8216 CrossRef CAS.
- T. Vivekanand, B. Satpathi, S. K. Bankar and S. S. V. Ramasastry, RSC Adv., 2018, 8, 18576–18588 RSC.
- D. J. Rivinoja, Y. S. Gee, M. G. Gardiner, J. H. Ryan and C. J. T. Hyland, ACS Catal., 2016, 1053–1056 Search PubMed.
- J.-J. Suo, W. Liu, J. Du, C.-H. Ding and X.-L. Hou, Chem. – Asian J., 2018, 13, 959–963 CrossRef CAS.
- M. Laugeois, J. Ling, C. Férard, V. Michelet, V. Ratovelomanana-Vidal and M. R. Vitale, Org. Lett., 2017, 19, 2266–2269 CrossRef CAS.
- Y. S. Gee, D. J. Rivinoja, S. M. Wales, M. G. Gardiner, J. H. Ryan and C. J. T. Hyland, J. Org. Chem., 2017, 82, 13517–13529 CrossRef CAS.
- M. Sun, Z.-Q. Zhu, L. Gu, X. Wan, G.-J. Mei and F. Shi, J. Org. Chem., 2018, 83, 2341–2348 CrossRef CAS.
- J.-Q. Zhang, F. Tong, B.-B. Sun, W.-T. Fan, J.-B. Chen, D. Hu and X.-W. Wang, J. Org. Chem., 2018, 83, 2882–2891 CrossRef CAS.
- Q. Cheng, F. Zhang, Y. Cai, Y.-L. Guo and S.-L. You, Angew. Chem., Int. Ed., 2018, 57, 2134–2138 CrossRef CAS.
- J. Ling, D. Mara, B. Roure, M. Laugeois and M. R. Vitale, J. Org. Chem., 2020, 85, 3838–3848 CrossRef CAS.
- E. T. Pelkey, Chem. Commun., 1997, 1873 RSC.
- E. T. Pelkey, L. Chang and G. W. Gribble, Chem. Commun., 1996, 1909–1910 RSC.
- J.-Q. Zhao, M.-Q. Zhou, Z.-J. Wu, Z.-H. Wang, D.-F. Yue, X.-Y. Xu, X.-M. Zhang and W.-C. Yuan, Org. Lett., 2015, 17, 2238–2241 CrossRef CAS.
- J.-Q. Zhao, Z.-J. Wu, M.-Q. Zhou, X.-Y. Xu, X.-M. Zhang and W.-C. Yuan, Org. Lett., 2015, 17, 5020–5023 CrossRef CAS.
- X.-M. Chen, C.-W. Lei, D.-F. Yue, J.-Q. Zhao, Z.-H. Wang, X.-M. Zhang, X.-Y. Xu and W.-C. Yuan, Org. Lett., 2019, 21, 5452–5456 CrossRef CAS.
- H. Wang, J. Zhang, Y. Tu and J. Zhang, Angew. Chem., Int. Ed., 2019, 58, 5422–5426 CrossRef CAS.
- K. Li, T. P. Gonçalves, K.-W. Huang and Y. Lu, Angew. Chem., Int. Ed., 2019, 58, 5427–5431 CrossRef CAS.
- L.-W. Jin, F. Jiang, K.-W. Chen, B.-X. Du, G.-J. Mei and F. Shi, Org. Biomol. Chem., 2019, 17, 3894–3901 RSC.
- K. Liu, G. Wang, S.-J. Cheng, W.-F. Jiang, C. He and Z.-S. Ye, Tetrahedron Lett., 2019, 60, 1885–1890 CrossRef CAS.
- A. Cerveri, O. N. Faza, C. S. López, S. Grilli, M. Monari and M. Bandini, J. Org. Chem., 2019, 84, 6347–6355 CrossRef CAS.
- L. Birbaum, L. Gillard, H. Gérard, H. Oulyadi, G. Vincent, X. Moreau, M. De Paolis and I. Chataigner, Chem. – Eur. J., 2019, 25, 13688–13693 CrossRef CAS.
- I. Chataigner, C. Panel, H. Gérard and S. R. Piettre, Chem. Commun., 2007, 3288 RSC.
- M.-S. Mei, Y.-H. Wang, Q. Hu, Q.-H. Li, D.-Y. Shi, D. Gao, G. Ge, G.-Q. Lin and P. Tian, Chem. Commun., 2020, 56, 10718–10721 RSC.
- E. Wenkert, P. D. R. Moeller and S. R. Piettre, J. Am. Chem. Soc., 1988, 110, 7188–7194 CrossRef CAS.
- B. Biolatto, M. Kneeteman, E. Paredes and P. M. E. Mancini, J. Org. Chem., 2001, 66, 3906–3912 CrossRef CAS.
- A. Chrétien, I. Chataigner, N. L’Hélia and S. R. Piettre, J. Org. Chem., 2003, 68, 7990–8002 CrossRef.
- B. Biolatto, M. Kneeteman and P. Mancini, Tetrahedron Lett., 1999, 40, 3343–3346 CrossRef CAS.
- C. D. D. Rosa, C. M. Ormachea, A. S. Sonzogni, M. N. Kneeteman, L. R. Domingo and P. M. E. Mancini, Lett. Org. Chem., 2012, 9, 691–695 CrossRef CAS.
- T. L. S. Kishbaugh and G. W. Gribble, Tetrahedron Lett., 2001, 42, 4783–4785 CrossRef CAS.
- M. Victoria Gómez, A. I. Aranda, A. Moreno, F. P. Cossío, A. de Cózar, Á. Díaz-Ortiz, A. de la Hoz and P. Prieto, Tetrahedron, 2009, 65, 5328–5336 CrossRef.
- L. R. Domingo, M. J. Aurell, P. Pérez and R. Contreras, Tetrahedron, 2002, 58, 4417–4423 CrossRef CAS.
- I. Chataigner, E. Hess, L. Toupet and S. R. Piettre, Org. Lett., 2001, 3, 515–518 CrossRef CAS.
- M. Andreini, M. De Paolis and I. Chataigner, Catal. Commun., 2015, 63, 15–20 CrossRef CAS.
- Y. Li, F. Tur, R. P. Nielsen, H. Jiang, F. Jensen and K. A. Jørgensen, Angew. Chem., Int. Ed., 2016, 55, 1020–1024 CrossRef CAS.
- D.-F. Yue, J.-Q. Zhao, X.-Z. Chen, Y. Zhou, X.-M. Zhang, X.-Y. Xu and W.-C. Yuan, Org. Lett., 2017, 19, 4508–4511 CrossRef CAS.
- D. Cao, A. Ying, H. Mo, D. Chen, G. Chen, Z. Wang and J. Yang, J. Org. Chem., 2018, 83, 12568–12574 CrossRef CAS.
- J.-R. Zhuo, B.-X. Quan, J.-Q. Zhao, M.-L. Zhang, Y.-Z. Chen, X.-M. Zhang and W.-C. Yuan, Tetrahedron, 2020, 76, 131115 CrossRef CAS.
- J.-J. Suo, J. Du, Y.-J. Jiang, D. Chen, C.-H. Ding and X.-L. Hou, Chin. Chem. Lett., 2019, 30, 1512–1514 CrossRef CAS.
- M. J. Bird, S. M. Wales, C. Richardson and C. J. T. Hyland, Synlett, 2020, 916–924 Search PubMed.
- K. Pasturaud, B. Rkein, M. Sanselme, M. Sebban, S. Lakhdar, M. Durandetti, J. Legros and I. Chataigner, Chem. Commun., 2019, 55, 7494–7497 RSC.
- H. Wang, Q. Hu, M. Wang and C. Guo, iScience, 2020, 23, 100840 CrossRef CAS.
- H. Huang, Q.-Z. Li, Y.-Q. Liu, H.-J. Leng, P. Xiang, Q.-S. Dai, X.-H. He, W. Huang and J.-L. Li, Org. Chem. Front., 2020, 7, 3862–3867 RSC.
- S. E. Denmark and A. Thorarensen, Chem. Rev., 1996, 96, 137–166 CrossRef CAS.
- I. Chataigner and S. R. Piettre, Org. Lett., 2007, 9, 4159–4162 CrossRef CAS.
- H. Gérard and I. Chataigner, J. Org. Chem., 2013, 78, 9233–9242 CrossRef.
- G. W. Gribble, Curr. Org. Chem., 2005, 9, 1493–1519 CrossRef CAS.
- G. W. Gribble, P. E. Alford and T. L. S. Kishbaugh, Heterocycles, 2010, 80, 831 CrossRef.
- M. Andreini, F. Chapellas, S. Diab, K. Pasturaud, S. R. Piettre, J. Legros and I. Chataigner, Org. Biomol. Chem., 2016, 14, 2833–2839 RSC.
- H. Gérard and I. Chataigner, Chem. – Eur. J., 2017, 23, 13711–13717 CrossRef.
- P. V. Santhini, S. A. Babu, A. Krishnan, R. E. Suresh and J. John, Org. Lett., 2017, 19, 2458–2461 CrossRef CAS.
- P. V. Santhini, A. Krishnan, R. S. A. Babu, B. S. Simethy, G. Das, V. K. Praveen, S. Varughese and J. John, J. Org. Chem., 2017, 82, 10537–10548 CrossRef CAS.
- S. Roy and G. W. Gribble, Tetrahedron Lett., 2007, 48, 1003–1005 CrossRef CAS.
Footnote |
| † Present address: Sorbonne Université, Laboratoire de Chimie Théorique, UMR 7616 CNRS, 75005 Paris, France. |
| This journal is © The Royal Society of Chemistry 2021 |