
Striking ligand-disproportionative Cl/aryl scrambling in a simple Au(III) system. Solvent role, driving forces and mechanisms†
Sara
Fernández-Moyano
 ,
Marconi N.
Peñas-Defrutos
,
Camino
Bartolomé
* and
Pablo
Espinet
*
,
Marconi N.
Peñas-Defrutos
,
Camino
Bartolomé
* and
Pablo
Espinet
*
IU CINQUIMA/Química Inorgánica, Facultad de Ciencias, Universidad de Valladolid, Valladolid-47071, Spain. E-mail: espinet@qi.uva.es
First published on 20th November 2020
Abstract
Aryl rearrangements triggered by Cl− extraction from trans-[AuIII(Rf)2Cl2]− (Rf = C6F3Cl2-3,5), led quickly to a mixture of [Au(Rf)3(solv)], cis-[Au(Rf)2Cl(solv)] and [Au(Rf)Cl2(solv)] (solv = OEt2, OH2). 19F NMR and X-ray diffraction studies led us to identify the species present in solution and the role of the solvent in their formation, while DFT calculations confirm the thermodynamic basis of their evolution. Very different Rf–Rf coupling rates are found from (μ-Cl)2[cis-Au(Rf)2]2 or cis-[Au(Rf)2ClL] species (L = OEt2, NCMe, Cl−) depending on the coordination strength of the ligand or solvent in the fourth position.
Organometallic complexes bearing pentafluorophenyl (Pf) or other fluorinated aryls have been used in chemistry for decades because of the extra stability of the M–(fluoroaryl) bonds. Their use in gold chemistry in the 1970s was decisive for an explosive development of different families of gold complexes, which later have made accessible the study of properties such as aurophilicity,1a mesomorphism,1b and luminescence.2
A few years ago, Toste et al. reported an extremely fast aryl–aryl coupling based in the AuIII/AuI redox couple, occurring at very low temperatures and not needing the use of ligands specially designed for such a purpose.3 This finding boosted the interest in a topic, C–C coupling from AuIII, started forty years before.4 Two excellent reviews of recent advances in gold(III),5 the first aryl–aryl coupling reported,6 and some recent reductive elimination studies are worth mentioning.7
The work presented here started because of a casual discovery when we wanted to prepare stable gold(III) complexes containing the cis-AuIII(Rf)2 fragment (Rf = C6F3Cl2-3,5). The synthesis of complexes trans-[AuIII(Pf)2Cl2]−,8cis-[AuIII(Pf)2Cl2]−,8 and (μ-Cl)2[Au(Pf)2]2,9 was reported long ago, but we prefer to use Rf derivatives because they greatly simplify the 19F NMR spectra (there is no intra-ring 19F–19F coupling in C6F3Cl2-3,5).10 This NMR simplification is useful to discern, in mixtures, signals that with C6F5 can overlap due to their multiplicity. For this reason, we chose to synthesize the cis- and trans-AuIII(Rf)2 homologues of the complexes mentioned above.
In our large experience, Rf and Pf behave identically as far as chemical reactivity of their complexes is concerned. We were surprised when, applying with Rf the same synthetic procedures reported for Pf-complexes,8,9 we observed the formation of tris-Rf and mono-Rf complexes. This complication was not mentioned for Pf complexes in the original papers or, more likely, it went unnoticed in their syntheses. Its formation reveals the occurrence of some unexpected and unreported aryl scrambling processes. These complications deserve attention because they can be a serious problem in the context of cross-coupling catalysis. In Pd, they are a frequent source of loss of selectivity, giving rise to homocoupling products and opening the door to other undesired reaction pathways. The scrambling mechanisms operating in gold(III) are poorly known and understood. In fact, to the best of our knowledge, only three aryl rearrangement reactions, incidentally involving AuIII Pf derivatives, have been reported in recent times.11–13 They occur in very different conditions but, in all of them, either strong oxidants (nitrosyl, selectfluor, or PhI(OAc)2) or mixtures of gold compounds with different oxidation states are involved. In the study that follows, we take advantage of the highly informative observation of Rf complexes to uncover the origin and mechanistic details of scrambling processes in gold chemistry.
Following the procedures reported in the literature for the Pf complexes, the oxidation of (NBu4)[Au(Rf)2],14 using a CCl4 solution of Cl2, led to the expected gold(III) complex (NBu4)(trans-[Au(Rf)2Cl2]) (1), which was isolated and fully characterized. Also, as expected, the cis isomer (NBu4)(cis-[Au(Rf)2Cl2]) (2) was obtained upon heating a solution of 1 in toluene, and was fully characterized (Scheme 1). The X-ray structures of 1 and 2 are shown in Fig. S5 and S6, ESI,† respectively. The cis isomer is thermodynamically preferred, obeying the antisymbiotic effect that avoids strongly donor ligands in mutually trans positions,15 but the trans isomer did not isomerize unless it was submitted to heating.
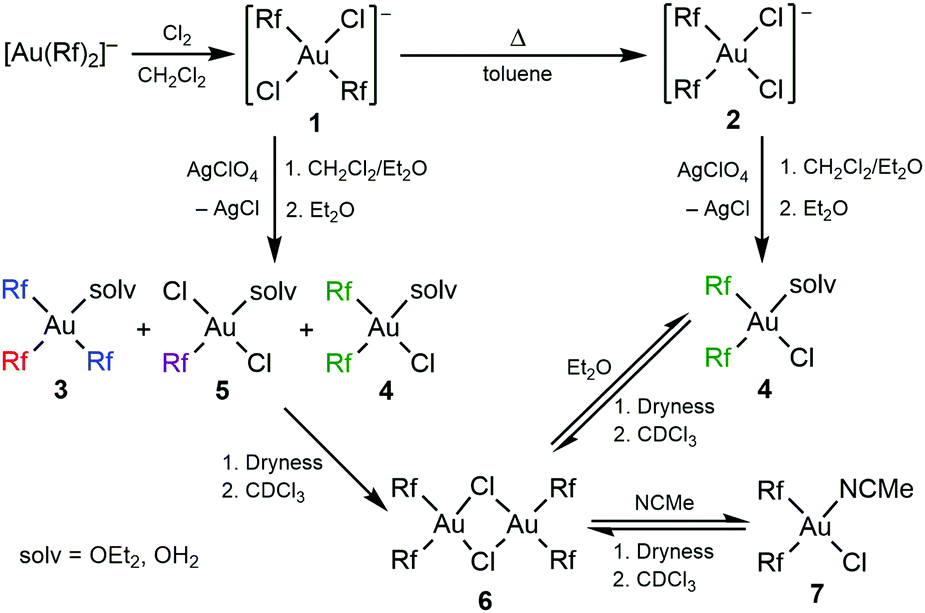 | ||
Scheme 1 Synthesis of Rf dichlorodiaryl gold (III) anionic complexes and evolution with AgClO4 (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1). The cation is (NBu4)+. :1). The cation is (NBu4)+. | ||
In the literature, the Pf analogues of 1 and 2 are indistinct precursors for the synthesis of the Pf analogue of (μ-Cl)2[Au(Rf)2]2 (6) by reaction with 1 equivalent of AgClO4.9 However, the reaction of 1 carried out in CH2Cl2/Et2O, leads to a mixture of compounds 3–5, which requires aryl rearrangement (Scheme 1). Fig. 1 shows the 19F NMR spectrum of an aliquot of this reaction after solvent evaporation and subsequent extraction with Et2O in order to remove NBu4ClO4 (see experimental details in ESI†). The Fpara region (upper right corner) shows five signals corresponding to the five non-equivalent Rf groups of the three species. The multiplicity of the Fortho signals, due to through-space Fortho–Fortho coupling, is perfectly studied for square planar complexes with this particular aryl,10 and allows for unambiguous assignment of a tris-aryl fragment (1:2 quintuplet:triplet, labelled in red and blue, respectively), and a cis-diaryl fragment with two non-equivalent Rf groups (two 1:1 pseudotriplets labelled in green). The solv ligand (Et2O or adventitious water, both O-donor ligands) fills the coordination vacancies, leading to [Au(Rf)3(solv)] (3), cis-[Au(Rf)2Cl(solv)] (4), and trans-[Au(Rf)Cl2(solv)] (5). This ligand-disproportionative scrambling of aryl and chloro ligands replaces in these conditions the expected formation of (μ-Cl)2[Au(Rf)2]2 (6).
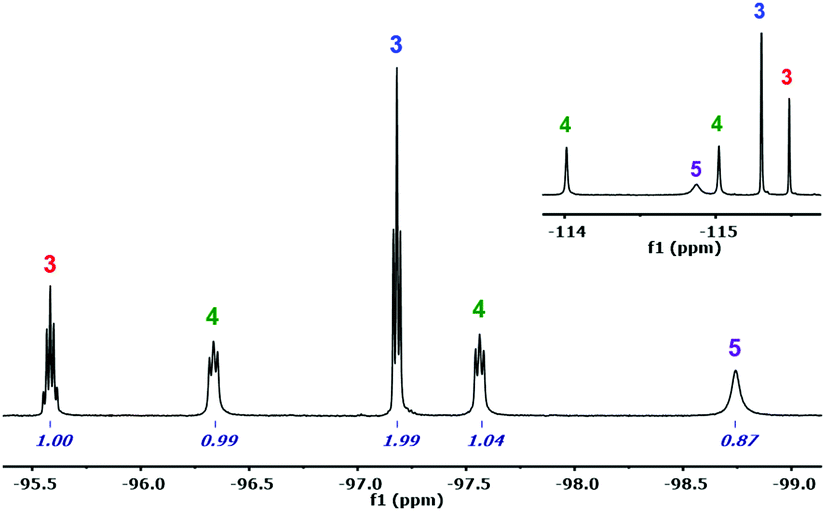 | ||
| Fig. 1 19F NMR spectrum of an aliquot (in Et2O, ref. acetone-d6) of the reaction of 1 with AgClO4: Fortho region with integrations. Fpara region in upper right corner. | ||
If the reaction mixture of Fig. 1 is evaporated to dryness and then dissolved in non-coordinating dry CDCl3, only one species is observed in solution. Its 19F NMR spectrum (Fig. 2, above) displays two singlets (2:1) assigned, respectively, to Fortho and Fpara of the desired dimer (μ-Cl)2[Au(Rf)2]2 (6). Additionally, signals of the coupling product Rf–Rf start to be observed.
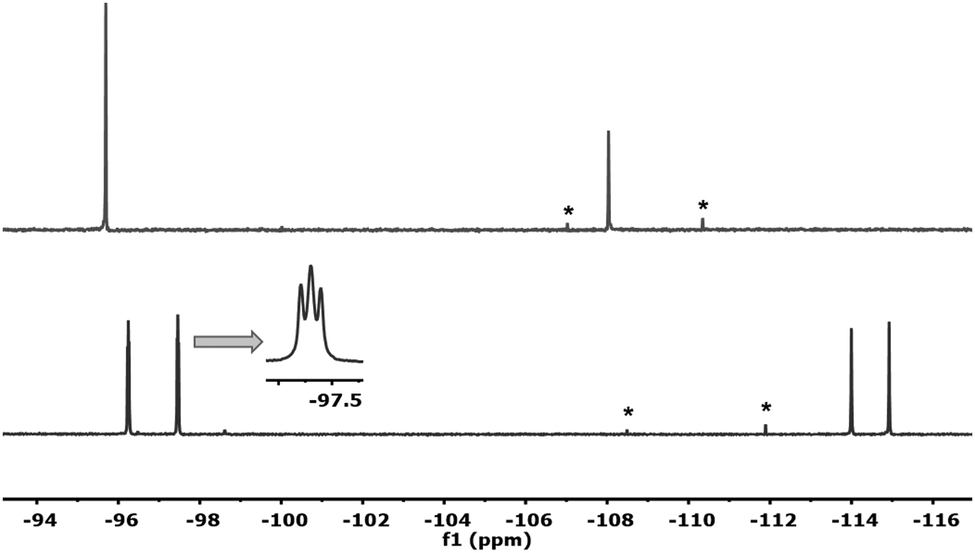 | ||
| Fig. 2 19F NMR spectra of 6 formed in CDCl3 (above), and 4 formed in Et2O (below), ref. acetone-d6 capillary. Asterisks are signals of Rf–Rf formed from 6. | ||
If, once more, the CDCl3 is removed completely and Et2O is added, the coordinating solvent (or adventitious water in it) splits the weak Cl bridges and only cis-[Au(Rf)2Cl(solv)] (4) is formed (Fig. 2, below, see details in ESI†). This explains why, in the absence of NMR monitoring and depending on the use of solvents, often varied in synthesis, the aryl scrambling could go unnoticed in the reported syntheses of the Pf analogue of 6.9 We have confirmed that under our conditions, the same unreported aryl scrambling occurs with Pf.
Slow evaporation of the mixture of products in Fig. 1 led to isolation of colourless crystals of [Au(Rf)3(OH2)]·2Et2O (3·OH2) in up to 20% yield (Fig. 3 left). This is one of the few gold(III) aquo-complexes ever reported.16,17 The fourth coordination position in the crystal is occupied by a water molecule, in this case, H-bonded to two diethyl ether molecules. In solution, fast ligand exchange between Et2O and water is expected. The Au–C distance of the aryl group trans to OH2 is 1.987(12) Å, significantly shorter than those found for the other two mutually trans Rf groups, with longer Au–C distances of 2.056(7) Å. This is expected from the high trans influence of Rf and the low trans influence of OH2.
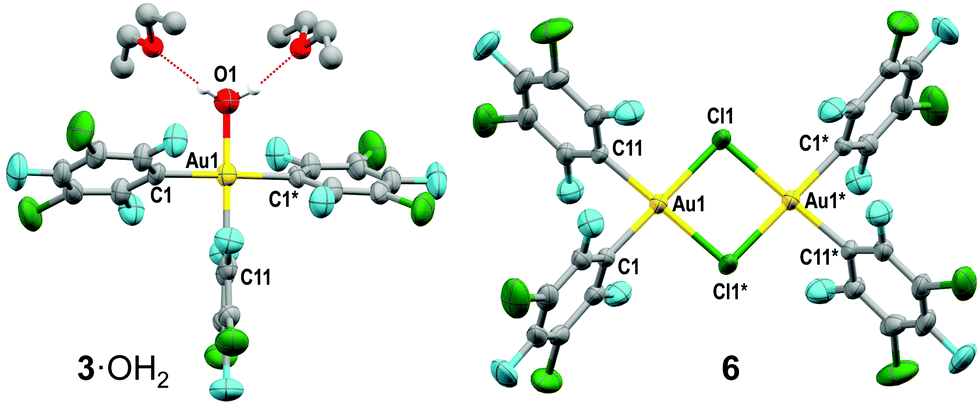 | ||
| Fig. 3 X-ray structure of 3·OH2 (left) and 6 (right). | ||
The dimer (μ-Cl)2[Au(Rf)2]2 (6) could also be crystallized and characterized by X-ray diffraction (Fig. 3 right).
The sequence of reaction from 1 in Scheme 1 cannot be reproduced for solv = MeCN because it complexes the Ag+ cations and prevents quite efficiently the precipitation of AgCl, but solutions of cis-[Au(Rf)2Cl(NCMe)] (7) are reversibly formed easily from 6 (more details in ESI†). The stability of the different species in front of Rf–Rf homocoupling follows the order 2 ≫ 7 > 4 ≫ 6, according to the following observations: (i) complex 2 requires heating in toluene above 373 K to observe reductive elimination; (ii) solutions of 6 in toluene with similar concentrations of MeCN or OEt2 (toluene:solv = 80:20 molar ratio) are stable for days at room temperature, but the solutions with OEt2 decompose much faster at 333 K (see details in ESI†); (iii) in the absence of any coordinating solvent or ligand the solutions of 6 in CDCl3 produce fast Rf–Rf coupling at 293 K. This rate sequence supports a very plausible sequence of the coordinating ability of the fourth ligand: CDCl3 ≪ bridging Cl < OEt2/OH2 < MeCN < terminal Cl−. The RE rates observed strongly suggest coupling via dissociation to a tricoordinate intermediate. This interpretation is also in line with the extremely fast Ar–CF3 coupling from [Au(Ar)(CF3)I(PR3)] in CH2Cl2 by treatment with AgSbF6,18 and the high-energy barrier for Ar–Ar′ coupling in four-coordinated [Au(Ar)(Ar′)Cl(PR3)].19 Dissociative coupling mechanisms are common in isoelectronic and isostructural PdII complexes.20
Stoichiometric abstraction of one halide from (NBu4)(cis-[Au(Rf)2Cl2]) (2) in CH2Cl2/Et2O gives selective access to fairly stable solutions of cis-[Au(Rf)2Cl(solv)] (4), preventing the formation of 3 and 5. In non-coordinating CDCl3, 6 is formed but it has only limited stability at room temperature. Other Au/Ag proportions, not illustrative for the discussion, are commented on in the ESI.† In order to understand the effect of changing these proportions, it is important to be aware that, as most of the initial AgClO4 is not dissolved, its reaction with 1 is heterogeneous. Hence, the change of solid AgClO4 proportion is not directly reflected in a proportional change of Ag+ concentration.
The previous results allow for some mechanistic analysis of the processes sketched there. First of all, a concerted mechanism for the oxidative addition of Cl2 to [AuIRf2]− can be excluded: it should produce cis-[AuIII(Rf)2Cl2]− (2), which is the most stable isomer, and 1 would never be observed. Consequently, an electrophilic attack to gold by Cl–Cl, followed by Cl− release and coordination must operate (Scheme 2).
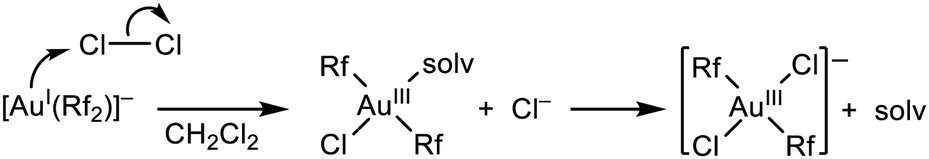 | ||
| Scheme 2 The most plausible mechanism for the oxidative addition of Cl2 to the nucleophilic anionic gold(I) complex. | ||
The thermodynamically driven 1 to 2 isomerization must follow the mechanism proposed much earlier by Kochi and Hoffmann from studies combining experiments and calculations.21 This isomerization consists of ligand dissociation (Cl− dissociation in this case), followed by topomerization from trans-R2 to the thermodynamically favourable cis-R2 coordination, and ligand (Cl−) recoordination (Scheme 3).
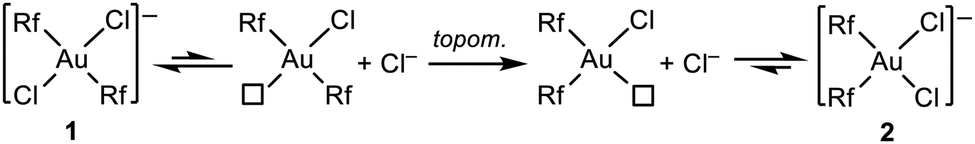 | ||
| Scheme 3 Isomerization via ligand dissociation plus topomerization. | ||
If the first easy leaving chloride is precipitated with a silver salt of a non-coordinating anion, AgClO4, the remaining chloride coordinates additionally to the empty coordination position created, to form a bridged dimer 6 in non-coordinating chloroform. As mentioned, these chloro bridges are not very strong. In fact, they are easily split by coordinating molecules such as Et2O or OH2, forming cis-[Au(Rf)2Cl(solv)] (4) (Scheme 1).
At this point it is clear why the presence of a coordinating solvent can be determining for the different fate of 1. In CH2Cl2, CHCl3 or toluene, and in the absence of AgClO4 the relatively high dissociation energy of a Cl−, required for topomerization, makes the isomerization of 1 to 2 very slow at room temperature: heating in toluene is required for fast isomerization. When the presence of OEt2 helps for progressive partial solubilization of AgClO4, irreversible precipitation of AgCl produces a non-observed intermediate trans-[Au(Rf)2Cl(solv)] (I1). Since the dissociation of the OEt2 ligand is easier than for Cl−, the topomerization at room temperature to the cis isomer 4 becomes much faster (Scheme 4).
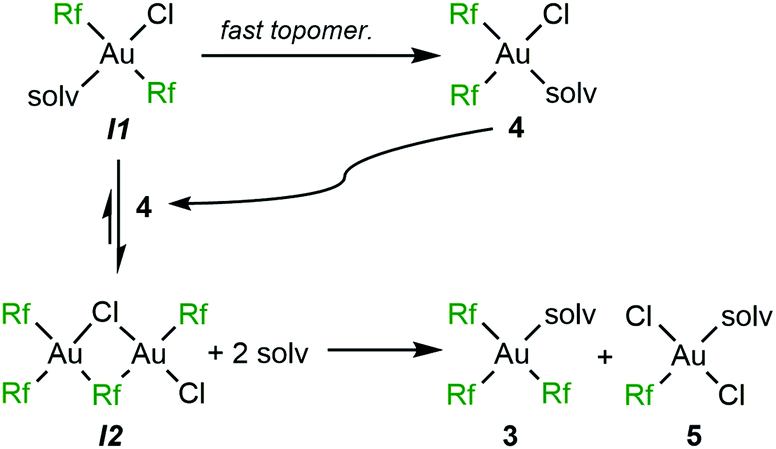 | ||
| Scheme 4 Ag+-fuelled mechanism of the ligand-disproportionative Rf/Cl rearrangement from trans-[Au(Rf)2Cl(solv)] in the presence of a coordinating solvent (OEt2). | ||
The coexistence of I1 and 4 opens a new 1:1 reaction pathway that involves disproportionative Cl-for-Rf exchange affording compounds 3 and 5 (Scheme 4). For this reason, they are in equimolecular proportion in the mixture (Fig. 1). The rate competition between the formation of 4, that of I1, and their consumption through the Rf/Cl mixed bridged dimer I2 (see Fig. S8, ESI†) determines the final proportion of 4vs.3 and 5 in the products. It is worth remarking that the three aryl products 3, 4 and 5 are not in equilibrium, since the reaction of 6 with Et2O (solv) only produces 4 (Scheme 1).
Density functional theory (DFT) calculations at the wb97x-D level were carried out in order to establish the thermodynamics of the reactions in Scheme 4 (see computational details in the ESI†). The results confirm our guess that trans-[Au(Rf)2Cl(OEt2)] (I1) is the highest energy form.15 It is however accessible because the formation of insoluble AgCl compensates the thermodynamic balance. Its cis isomer 4 is 17.3 kcal mol−1 more stable. The rearrangement of trans-[Au(Rf)2Cl(OEt2)] (2 equivalents) into [Au(Rf)3(OEt2)] (3) + trans-[Au(Rf)Cl2(OEt2)] (5) (more stable than its cis isomer) viaI2 is favourable by 27.5 kcal mol−1, and the splitting of I2 with 2OEt2 is also favourable by 11.5 kcal mol−1. These values support that isomerization and Rf/Cl rearrangements on OEt2 complexes are irreversible processes with low activation barriers that make the proposed intermediates unobservable. Only the fact that in the end a considerable amount of 4 survives needs to be explained by kinetic reasons: its formation is faster than its consumption rate to produce I2, and this rate difference produces neat 4 observed when the process is finished. Most probably, the rate of consumption of 4 is limited by the rate of production of I1, which depends on the solubilization of AgClO4 in OEt2. This is supported by the observation that adding preformed 4 does not alter the rate of formation of 3 and 5 significantly.
In summary, the initially striking variety of complexes produced from a single precursor finds a fully satisfactory explanation. This ligand-disproportionative rearrangement, arising simply depending on the choice of solvent or other often disregarded details, is a warning on possible complexities in catalysis under oxidative conditions where complexes of this kind are formed (e.g., in Ar–Ar coupling via gold(I)/gold(III) cycles), or when using gold(III) complexes for materials. For synthetic applications, (NBu4)(cis-[Au(Ar)2Cl2]) (Ar = Pf, Rf) is a stable and storable most convenient starting precursor of cis-Au(Ar)2 species.
We thank the Spanish MINECO (Projects CTQ2017-89217-P and CTQ2016-80913-P) for their financial support. M. N. P-D. is grateful to the Spanish MECD for a FPU grant.
Conflicts of interest
There are no conflicts to declare.Notes and references
- (a) A. Luquin, E. Cerrada and M. Laguna, in Gold Chemistry: Applications and Future Directions in the Life Sciences, ed. Mohr, F., Wiley-VCH, Weinheim, 2009, ch. 3, pp 93–181 Search PubMed; (b) S. Coco and P. Espinet, Gold Chemistry, ch. 8, pp 357–396 Search PubMed.
- (a) V. R. Bojan, J. M. L. de-Luzuriaga, E. Manso, M. Monge and M. E. Olmos, Organometallics, 2011, 30, 4486–4489 CrossRef CAS; (b) M. Bachmann, O. Blacque and K. Venkatesan, Chem. – Eur. J., 2017, 23, 9451–9456 CrossRef CAS; (c) T. von Arx, A. Szentkuti, T. N. Zehnder, O. Blacque and K. Venkatesan, J. Mater. Chem. C, 2017, 5, 3765–3769 RSC.
- W. J. Wolf, M. S. Winston and F. D. Toste, Nat. Chem., 2014, 6, 159–164 CrossRef CAS.
- A. Tamaki, S. A. Magennis and J. K. Kochi, J. Am. Chem. Soc., 1974, 96, 6140–6148 CrossRef CAS.
- (a) L. Rocchigiani and M. Bochmann, Chem. Rev., 2020 DOI:10.1021/acs.chemrev.0c00552; (b) A. Nijamudheen and A. Datta, Chem. – Eur. J., 2020, 26, 1442–1487 CrossRef CAS.
- J. Vicente, M. D. Bermúdez and J. Escribano, Organometallics, 1991, 10, 3380–3384 CrossRef CAS.
- (a) A. Genoux, J. A. González, E. Merino and C. Nevado, Angew. Chem., Int. Ed., 2020, 59, 17881–17886 CrossRef CAS; (b) L. Rocchigiani, J. Fernandez-Cestau, P. H. M. Budzelaar and M. Bochmann, Chem. – Eur. J., 2018, 24, 8893–8903 CrossRef CAS.
- R. Usón, A. Laguna, J. García and M. Laguna, Inorg. Chim. Acta, 1979, 37, 201–207 CrossRef.
- R. Usón, A. Laguna, M. Laguna and M. Abad, J. Organomet. Chem., 1983, 249, 437–443 CrossRef.
- P. Espinet, A. C. Albéniz, J. A. Casares and J. M. Martínez-Ilarduya, Coord. Chem. Rev., 2008, 252, 2180–2208 CrossRef CAS.
- For the rearrangement leading to [Au(Pf)2(tht)2][Au(Pf)4] as a by-product, from mixtures of [AuIII(Pf)3(tht)] and [AuI(Pf)(tht)] see: J. Coetzee, W. F. Gabrielli, K. Coetzee, O. Schuster, S. D. Nogai, S. Cronje and H. G. Raubenheimer, Angew. Chem., Int. Ed., 2007, 46, 2497–2500 CrossRef CAS.
- For the formation of [AuIII(Pf)3(NCMe)] during the oxidation of NBu4[Au(Pf)2] with [NO]PF6 (not observed with its C6Cl5 analogue) see: A. T. Overton, J. M. L. de-Luzuriaga, M. E. Olmos and A. A. Mohamed, Organometallics, 2012, 31, 3460–3462 CrossRef CAS.
- Oxidation of [Au(Pf)(PPh3)] with selectfluor or PhI(OAc)2 led to the rearrangement products [Au(PPh3)2][Au(Pf)4] and [Au(Pf)3(PPh3)] respectively: M. Hofer and C. Nevado, Eur. J. Inorg. Chem., 2012, 1338–1341 CrossRef CAS.
- E. J. Fernández, A. Laguna, J. M. L. de-Luzuriaga, M. Monge, M. Montiel, M. E. Olmos, J. Pérez, R. C. Puelles and J. C. Sáenz, Dalton Trans., 2005, 1162–1164 RSC.
- The well-known antisymbiotic effect or transphobia, states that the trans arrangement of two strong σ-donor groups (in our case the Rf groups), is a destabilizing contribution compared to their cis arrangement: (a) R. G. Pearson, Inorg. Chem., 1973, 12, 712–713 CrossRef CAS; (b) J. Vicente, A. Arcas, D. Bautista and P. G. Jones, Organometallics, 1997, 16, 2127–2138 CrossRef CAS.
- For neutral [AuMe2(OTf)(OH2)] see: S. Komiya, J. C. Huffman and J. K. Kochi, Inorg. Chem., 1977, 16, 2138–2140 CrossRef CAS.
- For examples of cationic gold aquo complexes see: (a) J. Serra, P. Font, E. D. Sosa Carrizo, S. Mallet-Ladeira, S. Massou, T. Parella, K. Miqueu, A. Amgoune, X. Ribas and D. Bourissou, Chem. Sci., 2018, 9, 3932–3940 RSC; (b) M. S. Messina, J. M. Stauber, M. A. Waddington, A. L. Rheingold, H. D. Maynard and A. M. Spokoyny, J. Am. Chem. Soc., 2018, 140, 7065–7069 CrossRef CAS.
- M. S. Winston, W. J. Wolf and F. D. Toste, J. Am. Chem. Soc., 2014, 136, 7777–7782 CrossRef CAS.
- K. Kang, S. Lui, T. Xu, D. Wang, X. Leng, R. Bai, Y. Lan and Q. Shen, Organometallics, 2017, 36, 4727–4740 CrossRef CAS.
- M. Pérez-Rodríguez, A. A. C. Braga, M. García-Melchor, M. H. Pérez-Temprano, J. A. Casares, G. Ujaque, A. R. de Lera, R. Álvarez, F. Maseras and P. Espinet, J. Am. Chem. Soc., 2009, 131, 3650–3657 CrossRef.
- S. Komiya, T. A. Albright, R. Hoffmann and J. K. Kochi, J. Am. Chem. Soc., 1976, 98, 7255–7265 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Synthesis and full characterisation of the complexes, X-ray and computational details; 27 pages. CCDC 2025754–2025757 and 2043412. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d0cc06450e |
| This journal is © The Royal Society of Chemistry 2021 |