
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Imidazolium-based catenane host for bromide recognition in aqueous media†
Christopher J.
Serpell
 *ab,
Ah Young
Park
b,
Carol V.
Robinson
b and
Paul D.
Beer
*b
*ab,
Ah Young
Park
b,
Carol V.
Robinson
b and
Paul D.
Beer
*b
aSchool of Physical Sciences, Ingram Building, University of Kent, Canterbury, CT2 7NH, UK. E-mail: C.J.Serpell@kent.ac.uk
bChemistry Research Laboratory, Department of Chemistry, University of Oxford, Mansfield Road, Oxford, OX1 3TA, UK. E-mail: paul.beer@chem.ox.ac.uk
First published on 9th December 2020
Abstract
The synthesis of a novel catenated system which is dense in cationic hydrogen bonding imidazolium units is described. The interlocked host system displays a preference for binding of bromide over other halides, overcoming basicity and Hofmeister trends, under aqueous conditions. This is the first example of an imidazolium-based catenane acting as an anion host through C–H hydrogen bonding.
The achievement of selective anion recognition in aqueous media is challenging due to the competitive nature of the solvent and Hofmeister-biased selectivity preferences.1 Inspired by the creation of solvent-excluded binding pockets in anion-binding proteins,2 we are pursuing the creation of precise cavities within interlocked architectures (catenanes and rotaxanes) as a strategy for selective anion binding.3 The imidazolium unit combines a positive charge and directional hydrogen bonding with chemical stability and synthetic versatility – factors which have led to its use in many anion binding systems,4–6 stretching back over many years.7 These properties make imidazolium a promising candidate for selective anion binding under aqueous conditions, and indeed usage of multiple imidazolium units permits binding of anions in competitive media including water.8–10 It is also possible to obtain a high density of imidazolium units within a molecular structure, separating them by only a methylene unit.11,12 These observations highlight an opportunity for creation of imidazolium-rich interlocked molecules for binding anions, and while a handful of imidazolium rotaxanes have been constructed for anion binding13–15 (and also for other purposes8,16–19), there is currently only one example of an imidazolium-based catenane, and that uses halogen bonding rather than direct C–H hydrogen bonds.20 Because of the additional cyclisation in catenanes as compared to rotaxanes, the binding cavity in a catenane host structure is potentially both better defined (due to loss of conformational freedom), and more shielded from solvent (due to mutual encircling). It is surprising therefore that rotaxane hosts for both cation and anion guest species greatly outnumber their catenane host counterparts.3 We were therefore interested in combining the topological constraints of catenated hosts with a high density of imidazolium C–H hydrogen bond donors. Herein we report the first imidazolium-based catenane 6, which has a dense core of four imidazolium hydrogen bonding units (Fig. 1) and displays a rare unconventional preference for bromide over other halides in aqueous media.
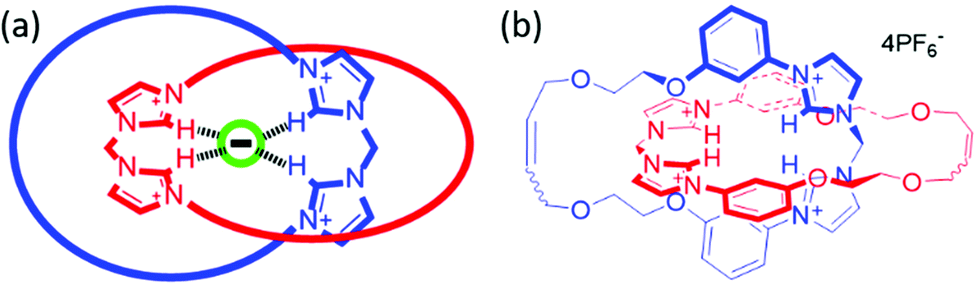 | ||
| Fig. 1 (a) Formation of an anion-binding cavity in an imidazolium-rich catenane. (b) Catenane 6. | ||
To prepare the target catenane 6, an anion-template synthetic strategy21 was adopted (Scheme 1). Tosylated allyloxyethanol 1 was attached to 3-iodophenol in the presence of potassium carbonate and catalytic potassium iodide in DMF with the product 2 being obtained almost quantitatively. Hetero-Ullmann coupling was used to substitute the iodine, giving the imidazole compound 3 in 71% yield. This was then reacted with diiodomethane under microwave irradiation in a sealed vial in acetonitrile to afford, after anion exchange with aqueous NH4PF6, 4 as a pure white crystalline solid in 19% yield.
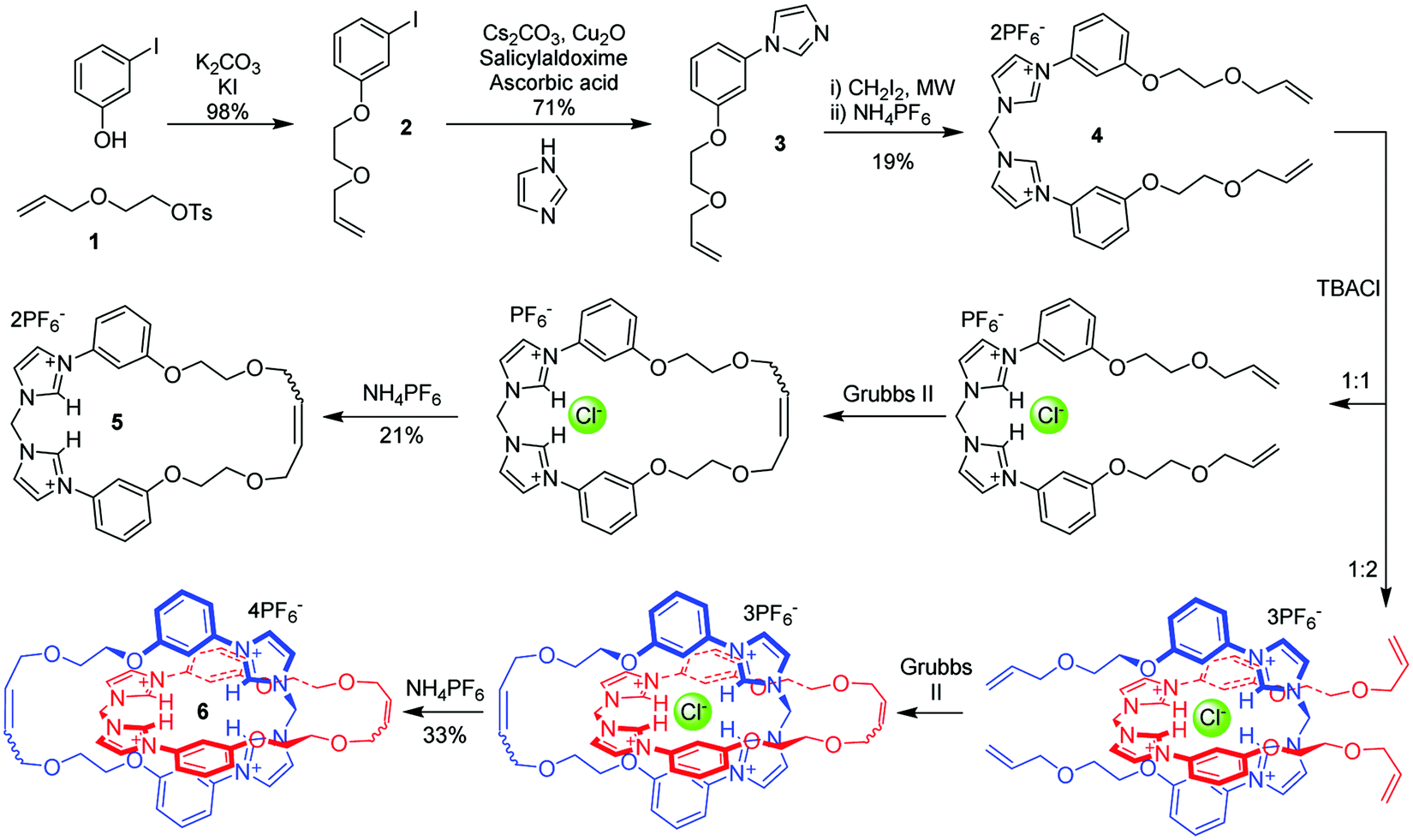 | ||
| Scheme 1 Synthesis of macrocycle 5 and catenane 6. | ||
Crystals of 4 suitable for X-ray diffraction were obtained for both its 2PF6− (see ESI†) and PF6−. Cl− salts by slow diffusion of diisopropyl ether into acetonitrile solutions. In the latter case, chloride was administered as one equivalent of its tetrabutylammonium (TBA) salt. The structure of the monochloride salt of 4 (Fig. 2) reveals the chloride anion bridging two imidazolium molecules, via hydrogen bonds from the 2- and 4-positions, of which the C2 interaction is the shorter. In this structure the PF6− anion sits close to the imidazolium groups and takes part in weak hydrogen-bonding and anion–π interactions. Although the expectation was that the mono-chloride salt would form a chelate with the halide, this structure was not observed. Such a result does not preclude the formation of the chelating structure in solution, since crystallisation requires the formation of networks of supramolecular forces, a condition which promotes divergent hydrogen bonding motifs. The chelating structure has been observed in the solid state by groups using the methylene-bisimidazolium unit for other purposes.22–24
 | ||
| Fig. 2 X-ray crystal structure of the asymmetric unit of 4. Anisotropic displacement parameters shown at 50% probability. | ||
Macrocyclisation and catenation of 4 was performed using 5 wt% Grubbs’ II ring-closing metathesis (RCM) catalyst in dichloromethane. In the absence of any anion other than PF6−, only oligomeric products were formed. The introduction of one equivalent of TBA chloride before the addition of Grubbs’ II resulted in the formation of the macrocycle 5, in an isolated yield of 21% after chromatographic purification, as the most abundant product. Conversely, when the ratio of 4 to chloride was two, the catenane 6 was the major product, isolated in a yield of 33%, together with the macrocycle in 16% yield. (Scheme 1). The difference in product distributions according to presence and stoichiometry of chloride supports the chelating binding shown in Scheme 1. Separation of the cyclic and interlocked products was achieved through preparative thin layer chromatography eluted with 14![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2:1 acetonitrile/water/KNO3 (aq, sat.). After removal of the appropriate bands, the products were extracted using a 98:2 mixture of acetone and saturated aqueous NH4PF6, which gave the PF6− salts as white precipitates after removal of the organic solvent. These results confirm the expected effect of the chloride anion template stoichiometry on the selectivity of the cyclisation reaction to form either the macrocycle or catenane product.
:2:1 acetonitrile/water/KNO3 (aq, sat.). After removal of the appropriate bands, the products were extracted using a 98:2 mixture of acetone and saturated aqueous NH4PF6, which gave the PF6− salts as white precipitates after removal of the organic solvent. These results confirm the expected effect of the chloride anion template stoichiometry on the selectivity of the cyclisation reaction to form either the macrocycle or catenane product.
Both 5 and 6 were characterised by 1H and 13C NMR and HR-ESMS. In the catenane 1H NMR spectrum, the imidazolium and methylene bridge peaks are shifted downfield compared to the macrocycle (Fig. 3a), probably due to their enforced proximity to the polyether oxygen atoms. The alkene peak (H12) is not split, indicating the formation of just one isomer – most likely the trans olefin.
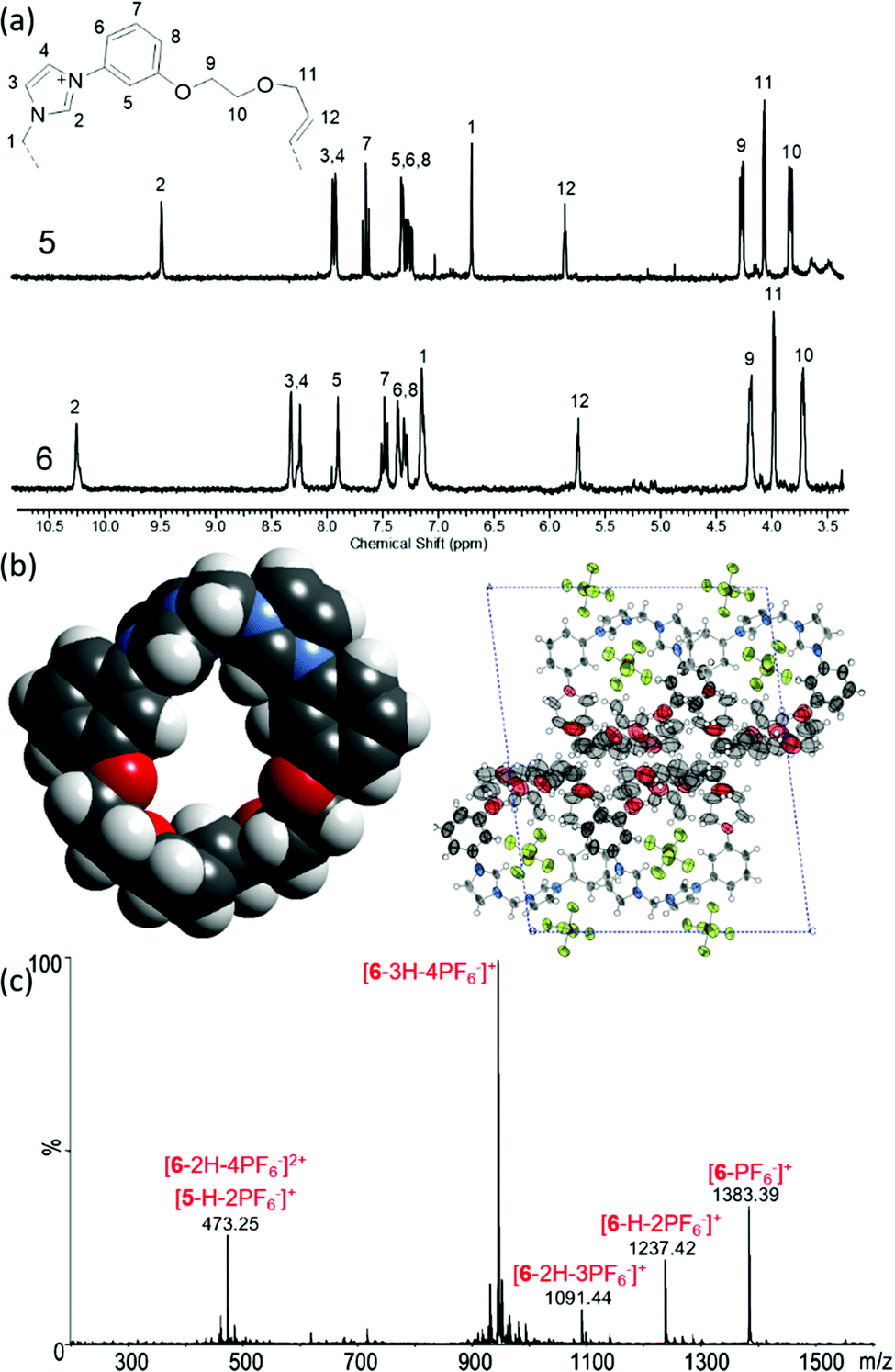 | ||
| Fig. 3 (a) 1H NMR spectra of macrocycle 5 and catenane 6 (MeCN-d3, 293 K) (b) X-ray crystal structure of 5 showing: space-filling representation, omitting anions and disorder; and packing diagram. Thermal ellipsoids shown at 50% probability. (c) Mass spectrum of 6 under high energy conditions. | ||
The two species could also be differentiated by their HR-ESMS spectra. A singly charged peak at m/z 619.1898 (calc. for [M − PF6]+ 619.1903) indicated the macrocycle, whereas the same peak was doubly charged for the catenane, and in addition a new peak at m/z 1383.3466 (calc. for [M − PF6]+ 1383.3465) was observed (Fig. S10 and S14, ESI†).
Single-crystal X-ray diffraction data were obtained for the macrocycle 5. The large hexafluorophosphate anions preclude any inclusion within the macrocyclic annulus. Although the cavity appears small (Fig. 3b), this is due to a contracted conformation controlled by crystal packing requirements. A more open conformation would allow the interpenetration necessary to form 6. One remarkable feature of the crystal structure is the contrast between the ordered ionic portions and the heavily disordered ethylene glycol chains. These are arranged within the crystal giving alternating layers of rigidity and flexibility. Given this level of disorder, it is remarkable that crystallisation occurred at all. Such properties may explain why it has not been possible to crystallise the catenane 6.
In characterising the catenane, it was important to consider the possibility of forming a topologically isomeric [1+1] macrocycle instead of the interlocked product. Such a product could have very similar NMR spectra, and display the same molecular ion peaks in ESMS. To investigate this possibility, mass spectrometric techniques were conducted. Firstly, high energy ionisation conditions were used to induce fragmentation. The [1+1] macrocycle would be expected to produce fragments of various m/z values up to 1383, whereas the catenane should result in no fragments with 619 < m/z < 1383, since cleavage of one bond would result in disassembly into fragments no larger than macrocycle 5.25 Upon raising the energy to the highest instrumental level (Synapt 1, TRAP collision energy: 200 V and bias 200 V), new peaks could be observed at m/z 945.46, 1091.44, and 1237.42 (Fig. 3c). It was found that these corresponded to the sequential loss of HPF6 moieties – presumably due to deprotonation at the imidazolium C2 position, producing a carbene. No other peaks with significant intensity were observed with 619 < m/z < 1383, in keeping with the hypothesised catenane topology. The peak at m/z 473.25 is consistent with a single macrocycle (identical to 5) arising from fragmentation, but is mixed with a double charged peak from the catenane at the same m/z.
Further characterisation was sought using ion mobility mass spectrometry (IM-MS). In this method, ionised molecules enter a drift tube, in which they are drawn by an applied electric field. A carrier buffer gas opposes the motion of the ions, resulting in longer drift times for species with larger cross sections due to collisions with the He atoms. At the end of the drift chamber is a mass spectrometer which can identify the exiting species. Analysis of the time-of-flight through the drift chamber enables calculation of the collision cross section, and thus a measure of the size and shape of the molecules. The collision cross section is rotationally time-averaged and is consequently larger for prolate molecular ions than spherical analogues. There is precedent for use of this technique to distinguish between macrocyclic and catenated isomers.26–29 Energy minimised (MMFF94) anion-free models of the macrocycle 5, and the catenated and macrocyclic topological isomers of 6 were produced (Fig. S16, ESI†). The catenated and macrocyclic topological isomers of 6 should have different levels of extension and hence different collision cross-sections. IM-MS was performed focusing on the peaks corresponding to the imidazolium species flying in the absence of any anions. Collision cross sections of 147.13 and 148.86 Å2 were measured from the [M − H − 2PF6]+ and [M − 2PF6]2+ peaks of the mass spectrum of macrocycle 5 respectively, matching the modelled value of 147.08 Å2. Examination of the [M − 3H − 4PF6]+ peak of 6 gave a collision cross section of 226.18 Å2, slightly closer to the modelled value for the catenane (204.93 Å2) than that of the large [1+1] macrocycle (248.26 Å2). The technique is capable of differentiating different components in the same peak, and therefore the possibility of a mixture of the topological isomers was eliminated. In addition to the results of the anion templating experiments, these mass spectrometric experiments add weight to the proposed catenated topology, and in anion binding experiments discussed below we continued to seek evidence for or against this model.
 | ||
| Fig. 4
1H NMR titration binding isotherms with halides derived from monitoring the imidazolium C2 proton for catenane 6 (9:1 MeCN-d3/H2O, 293 K). Black lines represent the calculated fit (1:1 host:guest model). | ||
1H NMR anion binding experiments were conducted on 5 and 6 in 9:1 MeCN-d3/H2O (Fig. S17–S26, ESI†), to test the capacity of the charged systems to compete with water. Although the water present may not be acting as ‘bulk water’ in this medium, it will compete for coordination of anions (both the intended guest and the hexafluorophosphate). In both cases, significant downfield perturbations of the acidic imidazolium C2 proton peak (up to +0.64 ppm) were observed (Fig. 4 and Fig. S27, ESI†), and the methylene spacer proton resonances were also sensitive to addition of coordinating anions, displaying shifts of up to +0.28 ppm. Further evidence in favour of the catenane structure over the macrocyclic topological isomer can be seen here – it would be expected that the larger macrocycle would possess chirality when twisted around an anion, resulting in diastereotopic splitting of 1H NMR resonances. No such splitting was observed. Analysis using WinEQNMR30 of the titration data obtained by monitoring the imidazolium C2 proton determined 1:1 stoichiometric association constants summarised in Table 1. Amongst the halides, both the macrocycle 5 and the catenane 6 display the greatest preference for bromide, followed by chloride. Although these two values are close, their standard deviations do not overlap, indicating a genuine difference. This preference is contrary to both halide basicity and the Hofmeister series, and is demonstrative of a size selective binding domain. The iodide ion is too large, while water competes more efficiently for the small and more basic fluoride. It is possible that water remains in the coordination sphere of the anion when bound, at least for the macrocycle 5. Notably, the catenane binds anions more strongly than the macrocycle by factors of up to 3.5. Simplistically, a factor of two would be expected if the increase in binding was purely electrostatic due to the doubling of the positive charge. The topology of the cavity can therefore be said to contribute to the enhanced anion binding, again consistent with a catenane structure. Geometrically controlled selectivity is illustrated by the basic, trigonal planar acetate being the weakest bound by catenane 6.
:1 MeCN-d3/H2O (293 K). Errors are given in brackets. Stacked spectra of these titrations can be found in the ESI, Fig. S17–S26
| Anion | Macrocycle 5 | Catenane 6 | 6/5 ratio |
|---|---|---|---|
| F− | 82 (2) | 291 (20) | 3.5 |
| Cl− | 163 (2) | 479 (17) | 2.9 |
| Br− | 171 (6) | 524 (23) | 3.1 |
| I− | 145 (5) | 271 (14) | 1.9 |
| AcO− | 119 (10) | 242 (4) | 2.0 |
In conclusion, the first hydrogen bonding imidazolium catenane has been prepared via a chloride anion template Grubbs’ catalysed RCM double cyclisation. Evidence for the catenated topology is found in the synthetic outcomes with respect to templating anion stoichiometry, mass spectrometry (fragmentation and ion mobility), NMR, and anion binding trends. The catenane is dense in anion-binding functionality and displayed improved anion recognition properties in competitive aqueous solvent mixtures when compared to the macrocyclic analogue. The geometric demands of the interlocked cavity results in an unusual halide selectivity preference for bromide. Further elaboration of imidazolium-dense structures may permit yet stronger and more selective anion coordination in competitive aqueous media.
C. J. S. thanks Johnson Matthey and the Engineering and Physical Sciences Research Council for a CASE Studentship.
Conflicts of interest
There are no conflicts to declare.Notes and references
- M. J. Langton, C. J. Serpell and P. D. Beer, Angew. Chem., Int. Ed., 2016, 55, 1974–1987 CrossRef CAS PubMed.
- M. Elias, A. Wellner, K. Goldin-Azulay, E. Chabriere, J. A. Vorholt, T. J. Erb and D. S. Tawfik, Nature, 2012, 491, 134–137 CrossRef CAS.
- K. M. Bąk, K. Porfyrakis, J. J. Davis and P. D. Beer, Mater. Chem. Front., 2020, 4, 1052–1073 RSC.
- J. Yoon, S. K. Kim, N. J. Singh and K. S. Kim, Chem. Soc. Rev., 2006, 35, 355–360 RSC.
- Z. Xu, S. K. Kim and J. Yoon, Chem. Soc. Rev., 2010, 39, 1457–1466 RSC.
- J. Cai and J. L. Sessler, Chem. Soc. Rev., 2014, 43, 6198–6213 RSC.
- E. Alcalde, N. Mesquida, L. Pérez-García, C. Alvarez-Rúa, S. García-Granda, E. E. García-Rodriguez, N. Mesquida and L. Pérez-García, Chem. Commun., 1999, 295–296 RSC.
- B. M. Rambo, H. Y. Gong, M. Oh and J. L. Sessler, Acc. Chem. Res., 2012, 45, 1390–1401 CrossRef CAS PubMed.
- C. J. Serpell, J. Cookson, A. L. Thompson and P. D. Beer, Chem. Sci., 2011, 2, 494–500 RSC.
- H. Zhou, Y. Zhao, G. Gao, S. Li, J. Lan and J. You, J. Am. Chem. Soc., 2013, 135, 14908–14911 CrossRef CAS PubMed.
- C. Kavitha, N. J. Singh, H. In-Chul, L. Jung Woo, S. K. Kwang, K. Chellappan, N. J. Singh, I.-C. Hwang, J. W. Lee and K. S. Kim, Angew. Chem., Int. Ed., 2005, 44, 2899–2903 CrossRef PubMed.
- Y. Chun, N. Jiten Singh, I. C. Hwang, J. Woo Lee, S. U. Yu and K. S. Kim, Nat. Commun., 2013, 4, 1–8 Search PubMed.
- C. J. Serpell, R. Chall, A. L. Thompson and P. D. Beer, Dalton Trans., 2011, 40, 12052–12055 RSC.
- G. T. Spence, C. J. Serpell, J. Sardinha, P. J. Costa, V. Félix and P. D. Beer, Chem. – Eur. J., 2011, 17, 12955–12966 CrossRef CAS PubMed.
- G. T. Spence, N. G. White and P. D. Beer, Org. Biomol. Chem., 2012, 10, 7282–7291 RSC.
- S. Dong, J. Yuan and F. Huang, Chem. Sci., 2014, 5, 247–252 RSC.
- N. Farahani, K. Zhu, C. A. O’Keefe, R. W. Schurko and S. J. Loeb, ChemPlusChem, 2016, 81, 836–841 CrossRef CAS PubMed.
- P. Langer, L. Yang, C. R. Pfeiffer, W. Lewis and N. R. Champness, Dalton Trans., 2019, 48, 58–64 RSC.
- W. B. Hu, W. J. Hu, X. L. Zhao, Y. A. Liu, J. S. Li, B. Jiang and K. Wen, Chem. Commun., 2015, 51, 13882–13885 RSC.
- A. Caballero, F. Zapata, N. G. White, P. J. Costa, V. Félix and P. D. Beer, Angew. Chem., Int. Ed., 2012, 51, 1876–1880 CrossRef CAS PubMed.
- S. S. Ramos, E. Alcalde, G. Doddi, P. Mencarelli, L. Perez-Garcia and L. Peréz-García, J. Org. Chem., 2002, 67, 8463–8468 CrossRef CAS PubMed.
- S. De, F. Joó, H. Horváth, A. Udvardy and C. E. Czégéni, J. Organomet. Chem., 2020, 918, 121308 CrossRef CAS.
- J. Li, P. Zhang, Y. Xu, Z. Su, Y. Qian, S. Li, T. Yu, P. J. Sadler and H. K. Liu, Dalton Trans., 2017, 46, 16205–16215 RSC.
- A. Fernández, D. Vázquez-García, A. García-Fernández, I. Marcos-Cives, C. Platas-Iglesias, S. Castro-García and M. Sánchez-Andújar, Inorg. Chem., 2018, 57, 7655–7664 CrossRef PubMed.
- J.-P. Sauvage and J. Weiss, J. Am. Chem. Soc., 1985, 107, 6108–6110 CrossRef CAS.
- C. A. Schalley, J. Hoernschemeyer, X. Lia, G. Silva and P. Weis, Int. J. Mass Spectrom., 2003, 228, 373–388 CrossRef CAS.
- M. Hutin, C. A. Schalley, G. Bernardinelli and J. R. Nitschke, Chem. – Eur. J., 2006, 12, 4069–4076 CrossRef CAS.
- W. Zhang, A. Abdulkarim, F. E. Golling, H. J. Räder and K. Müllen, Angew. Chem., Int. Ed., 2017, 56, 2645–2648 CrossRef CAS PubMed.
- A. Kruve, K. Caprice, R. Lavendomme, J. M. Wollschläger, S. Schoder, H. V. Schröder, J. R. Nitschke, F. B. L. Cougnon and C. A. Schalley, Angew. Chem., Int. Ed., 2019, 58, 11324–11328 CrossRef CAS PubMed.
- M. J. Hynes, J. Chem. Soc., Dalton Trans., 1993, 311–312 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Synthesis and characterisation, crystallography, and mass spectrometry. CCDC 2032606–2032608. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d0cc06299e |
| This journal is © The Royal Society of Chemistry 2021 |