
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCyclic peptide drugs approved in the last two decades (2001–2021)
Huiya
Zhang
and
Shiyu
Chen
 *
*
Biotech Drug Research Center, Shanghai Institute of Materia Medica, Chinese Academy of Sciences, Shanghai 201203, China. E-mail: chenshiyu@simm.ac.cn
First published on 5th November 2021
Abstract
In contrast to the major families of small molecules and antibodies, cyclic peptides, as a family of synthesizable macromolecules, have distinct biochemical and therapeutic properties for pharmaceutical applications. Cyclic peptide-based drugs have increasingly been developed in the past two decades, confirming the common perception that cyclic peptides have high binding affinities and low metabolic toxicity as antibodies, good stability and ease of manufacture as small molecules. Natural peptides were the major source of cyclic peptide drugs in the last century, and cyclic peptides derived from novel screening and cyclization strategies are the new source. In this review, we will discuss and summarize 18 cyclic peptides approved for clinical use in the past two decades to provide a better understanding of cyclic peptide development and to inspire new perspectives. The purpose of the present review is to promote efforts to resolve the challenges in the development of cyclic peptide drugs that are more effective.
Introduction
Since insulin was first used in the clinic almost 100 years ago,1 the spectrum of potency, specificity and safety of the peptide chain enhanced by cyclization has demonstrated the fundamental characteristics of cyclic peptides. Among the >60 FDA- and EMA-approved peptides,2 two-thirds are in the cyclic form and have an important role in the modern pharmaceutical industry.3 The constraint introduced by cyclization makes the peptide chain more conformationally stable, which improves the target protein binding affinity and reduces nonspecific binding due to fewer alternative conformations.4 Reduced conformational flexibility decreases the chance of the molecules fitting into the protease catalytic site and the proteomic resistance is improved.5 Cyclization also increases the efficacy of peptide chains by forming a larger interacting surface for intervening protein–protein interactions.6 Overall, peptide chain cyclization causes the cyclic peptides to be intrinsically different from linear peptides.7,8Cyclization improves not only the structural properties of peptide chains but also the pharmacokinetic properties for absorption and biological membrane permeability that is necessary for reaching protein targets.9 The constrained structure of peptides can cause them to have a lower energy barrier for adapting to the membrane environment and binding to transporting proteins, which increases both passive diffusion and active transport.10 Cyclic peptides can also be further improved by introducing noncanonical elements to improve both their pharmacokinetic and pharmacodynamic properties.11 The introduction of lipophilic side chains greatly improves their affinity to plasma carrier proteins, helps them to avoid renal clearance and improves their pharmacokinetic properties.12,13 Alternative strategies, including conjugation with albumin and immunoglobulin to extend the half-life, are also commonly adopted, and the frequency of administration can be reduced.14,15
However, cyclization makes the development of cyclic peptide drugs more challenging compared to the development of linear peptides.4,16 The majority of approved cyclic peptides are engineered from natural analogs by keeping the overall cyclic peptide structure to maintain affinity to the target protein.17,18 Canonical peptide chains are conventionally cyclized by head–tail amidation, disulfide bond formation between cysteines, and sidechain amine to C-terminal carboxyl group and sidechain carboxyl group to N-terminal amidations.19–21 More recently, peptide cyclization was advanced by the introduction of noncanonical elements that can induce cyclization of orthogonal reactive groups.22,23 The introduction of noncanonical elements in cyclic peptides expanded not only the chemical space of cyclic peptides but also the library diversity, which is very important for screening potent ligands.23 The combination of these developments in diverse chemical and biological technologies contributed to the rapid development of cyclic peptide drug discovery.4
In this review, we summarize the discovery and development of cyclic peptides that have been approved in the last two decades. Chemical structures, mechanisms of action and metabolic properties are analyzed to unveil the important factors for cyclic peptide drug development and utilization in clinical situations. We mainly focus on cyclic peptide therapeutics by excluding peptides with >20 amino acids that show properties similar to small proteins and peptidomimetics with <5 residues that show physical properties similar to small molecules. Taking advantage of many recently developed technologies for building cyclic peptide libraries with a diversity of billions by translating genetic variations, cyclic peptides with a length in this range can be routinely screened and rapidly synthesized for hit discovery and affinity maturation to obtain cyclic peptides suitable for drug development. The opportunities and perspectives for cyclic peptide drug development are also discussed.
Targeting intracellular proteins
Developing cell permeable cyclic peptides that can reach intracellular targets is a challenging task.10 Unlike small molecules that enter cells primarily through passive diffusion, peptide-based molecules greater than 1000 Da have distinct physical properties, mechanisms, and cell permeability ability, and currently available theories for improving the ADMET (absorption, distribution, metabolism, excretion, and toxicity) properties of small molecules do not apply to the development of cyclic peptides.24 Available theories for designing cell permeable peptides, such as passive diffusion, endocytosis and carrier-mediated transport with (CPPs),25,26 are limited in many aspects. It is well known that CPPs bind to several extracellular receptors and can be actively imported.27,28 It is necessary to incorporate specific elements in the sequence of the peptide chain, and these chemical alterations can impact the conformation of peptide drugs and further reduce the target binding affinity. Improving the hydrophilic and lipophilic characteristics of peptides with modification strategies such as N-methylation, side chain lipidation,29,30 and introduction of D-amino acid substituents31 can all increase the general ability of peptides to pass through lipid bilayer-based cell membranes. However, the fundamental difficulty associated with developing cell-permeable peptides is the lack of knowledge on the mechanisms associated with cell penetration.10 Five properties, including hydrogen-bonding potential, conformation, charge, size, and hydrophobicity, were observed to influence cell permeability.24 The extra stability of cyclization introduced into cyclic peptides makes them ideal candidates for entering cells through endocytosis, which requires a resistance to the variety of enzymes present in lysosomes.32Romidepsin (1, Table 1 and Fig. 1a) and voclosporin (2, Table 1 and Fig. 1a) are the only two cyclic peptides approved in the past two decades that target intracellular protein targets.33,34 Romidepsin is a natural bicyclic peptide obtained from the bacterium Chromobacterium violaceum in 1994 and approved by the FDA in 2009 for cutaneous T-cell lymphoma (CTCL).35,36 Romidepsin is a rather small depsipeptide that contains a head–tail lactone cyclization and a pair of disulfide bridges.37 Although romidepsin can be chemically synthesized, the primary source of romidepsin was manufactured by fermentation. Romidepsin is a prodrug in which the disulfide is reduced to two thiols in the intracellular matrix.38 Once reduced, thiols can chelate zinc in the zinc-dependent active site of histone deacetylase (HDAC) enzymes, thereby inhibiting histone deacetylases and inducing cell apoptosis.39 The half-life of intravenously administered romidepsin is approximately 3.5 hours.40 In rats, the primary route of elimination of romidepsin and its metabolites is the bile with subsequent excretion in feces, and approximately 20% hepatic clearance is metabolized.41 Because it does not rely on enzymatic degradation or hepatic or renal excretion, the drug is safe to use in patients with any degree of hepatic or renal impairment.42 Recently, Istodax was withdrawn as a treatment for peripheral T-cell lymphoma, while it remains on the market for treatment of patients with cutaneous T-cell lymphoma (CTCL) who have received at least one prior systemic therapy.43
| No. | Trade name | Generic name | Target | Indication | Approval | PDB | Cycle # | Ring size (AA) | Cyclization type | Hydrophobica | Polar | Basic | Acidic | N-Me | Route of admin. | Company | Sales 2020 |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| a Based on the physical properties of the sidechains of the canonical amino acids, Ala, Gly, Ile, Leu, Met, Phe, Val, Pro and cystine are categorized as hydrophobic; Asn, Gln, Ser, Thr, Trp and Tyr are categorized as polar; Arg, His and Lys are categorized as basic; and Asp and Glu are categorized as acidic. The noncanonical amino acids are categorized similarly as the canonical analogs. | |||||||||||||||||
| 1 | Istodax | Romidepsin | Histone deacetylases | Anticancer | 2009 | 3RQD | 2 | 3–2 | Lactone and disulfide | 4 | 0 | 0 | 0 | 0 | IV fusion | Celgene Inc. | N. A. |
| 2 | Lupkynis | Voclosporin | Calcineurin | Lupus nephritis | 2021 | 3ODI | 1 | 11 | Head–tail | 11 | 0 | 0 | 0 | 7 | Oral | Aurinia Inc. | N. A. |
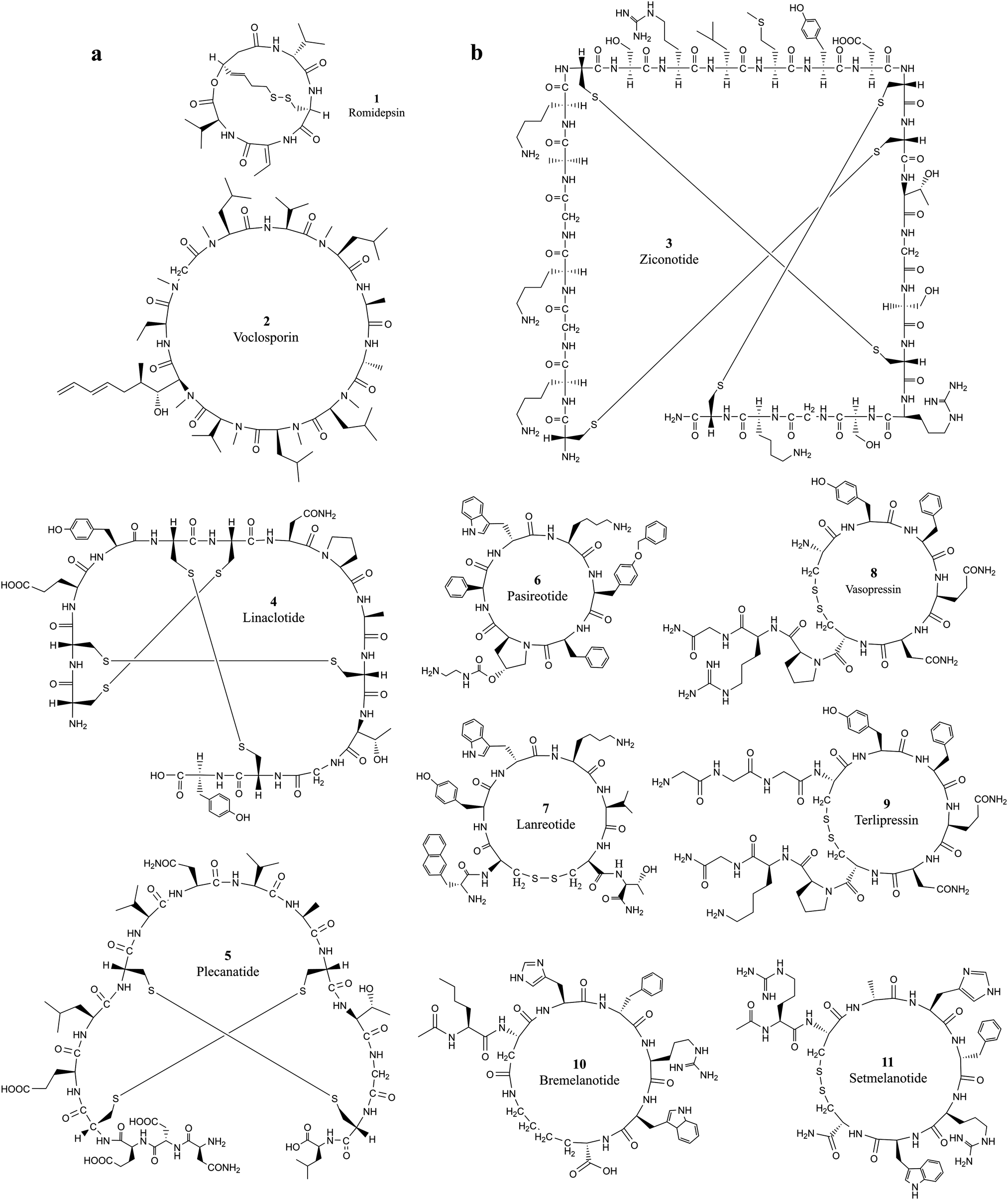 | ||
| Fig. 1 The chemical structures of cyclic peptides from Tables 1 and 2 that target (a) intracellular proteins and (b) extracellular proteins. | ||
Voclosporin was recently approved by the FDA for the treatment of lupus nephritis (LN) on January 22, 2021.44 Voclosporin was developed from ciclosporin with enhanced potency and metabolic stability.45 The rationale of the optimization is to replace the butenyl group of the first residue 4-[(E)-2-butenyl]-4,N-dimethyl-l-threonine (Bmt1) in cyclosporin A (CsA) with 4-[(2E,4EZ)-2,4-pentadienyl]-4,N-dimethyl-l-threonine (E-MePmt1) to optimize its fit against the hydrophobic Cn surface to increase the binding affinity.46,47 Voclosporin is 11 residues long containing a single D-amino acid with head–tail cyclization. Some of the amino acid substrates become N-methylated by S-adenosyl methionine.48 Voclosporin plays an important role in inhibiting T-cell proliferation and preventing the release of proinflammatory cytokines by blocking the activity of the calcium-regulated serine-threonine phosphatase calcineurin.49
Targeting outer membrane proteins
Half of the cyclic peptides approved in the last twenty years target extracellular protein targets, which primarily interact with G protein-coupled receptors (GPCRs). So far most GPCR-targeting therapeutics are small molecules;50 in contrast, the endogenous ligands for many GPCRs are peptides (comprising 50 or fewer amino acids),51 which suggests that this class of molecules could be therapeutically useful for being modified to develop peptide-based therapeutics. However, the short half-life of most naturally occurring linear and temporary peptide ligands severely hinders their development as drugs. The goal of developing peptide-based therapeutics from endogenous ligands is to preserve and exploit the native properties of high affinity, selectivity and potency while at the same time improving the unfavorable pharmacokinetic properties including the short half-life, rapid degradation, high levels of clearance, and even the oral bioavailability by overcoming gastrolienal degradation and poor membrane permeability.52,53 Although peptide therapy with a short half-life is desirable in some cases, extending the half-life of peptide therapies is one of the primary goals of drug development.54 Cyclization, a common strategy applied to rigidify the conformation of the peptide chain, can benefit the development of peptide-based therapeutics by increasing the plasma half-life and reducing enzymatic metabolism and renal elimination. In the past two decades, nine cyclic peptides that target membrane proteins were approved for clinic use (Table 2 and Fig. 1b). The ring size and amino acid composition were also summarized for comparison with the cyclic peptide therapeutics targeting intracellular proteins, and a trend in the population of more hydrophobic amino acids in the cell-penetrating peptides can be observed.| No. | Trade name | Generic name | Target | Indication | Approval | PDB | Cycle # | Ring size (AA) | Cyclization type | Hydrophobic | Polar | Basic | Acidic | Route of admin. | Company | Sales 2020 |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 3 | Prialt | Ziconotide | Calcium channel | Severe and chronic pain | 2004 | 3 | 15–13–10 | Disulfide | 10 | 5 | 6 | 1 | Intrathecal | TerSera LLC | N. A. | |
| 4 | Linzess | Linaclotide | Guanylate cyclase | Irritable bowel syndrome/chronic constipation | 2012 | 3 | 6–9–9 | Disulfide | 6 | 4 | 0 | 1 | Oral | AbbVie & IronWood Inc., Forest Inc. | $ 931 M | |
| 5 | Trulance | Plecanatide | Guanylate cyclase | Chronic idiopathic constipation/irritable bowel syndrome | 2017 | 2 | 9–9 | Disulfide | 8 | 3 | 0 | 3 | Oral | Salix Pharm. | N. A. | |
| 6 | Signifor | Pasireotide | Somatostatin receptor | Cushing's disease | 2012 | 1 | 6 | Head–tail | 3 | 1 | 2 | 0 | SC, IM | Recordati Inc. | $ 67 M | |
| 7 | Somatuline | Lanreotide | Somatostatin receptor | Acromegaly and symptoms caused by neuroendocrine tumors | 2007 | 1SOC | 1 | 6 | Disulfide | 3 | 3 | 1 | 0 | IM, SC | Biomeasure Inc. | $ 1145 M |
| 8 | Vasostrict | Vasopressin | Vasopressin receptor | Anti-diuretic hormone deficiency | 2014 | 1JK4 | 1 | 6 | Disulfide | 4 | 3 | 1 | 0 | IV, IM, SC | Par sterile products LLC | $ 785 M |
| 9 | Teripressin/glypressin | Terlipressin | Vasopressin receptor | Management of low blood pressure | 2009 | 1 | 6 | Disulfide | 7 | 3 | 1 | 0 | IV | New Medicon Ltd. Ferring Ltd. | N. A. | |
| 10 | Vyleesi | Bremelanotide | Melanocortin receptors | Hypoactive sexual desire disorder | 2019 | 1 | 6 | Asp–Lys | 3 | 1 | 2 | 0 | SC | Palatin Technologies Inc. | $ 4.7 M | |
| 11 | Imcivree | Setmelanotide | Melanocortin 4 receptor | Obesity | 2020 | 7AUE | 1 | 7 | Disulfide | 3 | 2 | 3 | 0 | SC | Rhythm Inc. | N. A. |
Ziconotide (3, Table 2 and Fig. 1b) is a synthetic version of ω-conotoxin MVIIA (ω-MVIIA) isolated from the venom of the snail Conus magus. ω-MVIIA contains 25 residues, and 6 of the amino acids are cysteines that are linked in three pairs of disulfide bonds.55 The NMR structure revealed that ω-MVIIA has a stable antiparallel β-sheet structure in solution.56,57 As a potent and selective blocker of N-type calcium channels, ziconotide was approved for the treatment of severe chronic pain in 2004.58 Evidence suggests that ziconotide delivers its antinociceptive efficacy by reducing the release of pronociceptive neurotransmitters in the dorsal horn of the spinal cord, thereby inhibiting pain signal transmission.59 Ziconotide has only limited ability to cross the blood–brain barrier. To achieve optimal analgesic efficacy with reduced potential for serious side-effects, ziconotide must be administered intrathecally.59,60
Linaclotide (4, Table 2 and Fig. 1b), an orally administered 14-amino acid synthetic peptide, is a first-in-class, high-affinity guanylate cyclase C agonist (GCCA) developed in the 1990s.61 Linaclotide is homologous to the paracrine peptide hormones guanylin and uroguanylin62 and was modified to confer a higher binding affinity as a guanylate cyclase C (GC-C) agonist along the longitudinal axis of the gastrointestinal tract regardless of changes in pH between 5 and 8.63,64 The active conformation of linaclotide is stabilized by three intramolecular disulfide bridges and developed for the treatment of irritable bowel syndrome with constipation (IBS-C).65 Linaclotide is minimally absorbed by the intestine and occurs through the local activation of intestinal GC-C receptors on the intestinal epithelium. Linaclotide was degraded rapidly in mouse small intestinal fluid during in vitro assays with a calculated first-order half-life of 3 minutes.66
Plecanatide (5, Table 2 and Fig. 1b) is a 16-amino-acid peptide nearly structurally identical to human uroguanylin apart from the substitution of Asp3 with Glu3.63 Plecanatide is constrained by two pairs of disulfide bridges and adopts active conformations for binding guanylate cyclase-C (GC-C) receptors at pH 5.0, the pH value of the duodenum and proximal jejunum.67 Activation of GC-C receptors in the GI tract stimulates intracellular production of cyclic guanosine monophosphate (cGMP), which then activates cystic fibrosis transmembrane conductance regulators (CFTRs) and cGMP-dependent protein kinase G-II.68 The activation of both CFTRs and cGMP-dependent protein kinase G-II produces an efflux of chloride ions from enterocytes lining the GI tract, resulting in an efflux of water into the intestinal lumen.63,64,69,70 As with most orally ingested peptides, plecanatide is degraded by intestinal enzymes, and a very little amount of the active drug enters systemic circulation.71
Pasireotide (6, Table 2 and Fig. 1b) is the second-generation analog of somatostatin, a naturally occurring inhibitory hormone that interacts with G protein-coupled somatostatin receptors and blocks the release of several other hormones, including growth hormone, thyroid-stimulating hormone (TSH), insulin and glucagon.72 Pasireotide has a 40-fold increased affinity to somatostatin receptor 5 compared to other somatostatin analogs.73 Pasireotide is a hexapeptide with head-to-tail cyclization. Pasireotide binds with a higher affinity to somatostatin receptor 1 (SSTR1, 30-fold), SSTR3 (5-fold), and SSTR5 (39-fold) and with the same affinity to SSTR2 when compared with octreotide, a higher affinity to SSTR1 (19-fold), SSTR3 (9-fold), and SSTR5 (106-fold), and the same affinity to SST2R (2-fold) when compared with lanreotide.18,19 Pasireotide was approved for the treatment of Cushing's disease by the EMA74 and FDA in 2012.75 Pasireotide LAR was approved by the FDA for the treatment of acromegaly in December 2014.76
Lanreotide (7, Table 2 and Fig. 1b) is also an octapeptide analog of somatostatin, and it was cyclized by a pair of disulfides formed between cysteine 2 and cysteine 7. Several strategies were developed for extending the half-life of lanreotide.77 Slow-release formulation, lanreotide sustained release (lanreotide SR), has an extended half-life of 4.5 days.78 Lanreotide embedded in the microspheres of biodegradable polymers can be administered at a dose of 30 or 60 mg every 7–14 days.79 Lanreotide was approved for acromegaly treatment in Europe in the 1990s. The FDA approved lanreotide for the treatment of acromegaly in 2007, metastatic gastroenteropancreatic neuroendocrine tumors (GEP-NETs) in 2014 and for the treatment of carcinoid syndrome in 2017.80
Vasopressin (8, Table 2 and Fig. 1b) is an antidiuretic hormone developed from a natural nonapeptide with nine amino acids cyclized by a disulfide bond between Cys4 and Cys9. Vasopressin was discovered in 1928,81 and its sequence was elucidated in 1951.82–84 In humans, vasopressin is encoded by the mRNA for preproneurophysin II. Vasopressin is nonselective with affinity for activating vasopressin V1a, V2, and V1b, and oxytocin receptors. Vasopressin mediates vasoconstriction by activating the V1 receptor, consequently promoting antidiuresis activity and exerting procoagulant activity on vascular smooth muscle.85 Vasopressin was branded as Pitressin and marketed for clinical use almost 100 years ago. Par Sterile Products received new drug applications (NDAs) from the FDA for branding Vasostrict in hypotensive adult patients with vasodilatory shock.86 Vasopressin shows great advantages over other medications in the treatment of septic shock by increasing blood pressure.87
Terlipressin (9, Table 2 and Fig. 1b) is a synthetic long-acting vasopressin analog with a half-life of 6 hours in contrast to 10 min for vasopressin.88 Terlipressin acts on vasopressin receptors V1a, V1b and V2, and has an improved safety profile and fewer side effects than vasopressin.89,90 Terlipressin is metabolized by endopeptidases to form lysine vasopressin by removing the triglycine residues at the N-terminal end of the cyclic peptide. Terlipressin has similar effects, but this drug has been used in far fewer patients.87
Bremelanotide (10, Table 2 and Fig. 1b) is a heptapeptide cyclized between the sidechains of Asp2 and Lys7 to form the lactam analog of α-melanocyte-stimulating hormone (α-MSH).91 Bremelanotide was originally developed as a potential sunless tanning agent and accidentally discovered to cause sexual arousal. Bremelanotide is a non-selective agonist of melanocortin receptors, primarily of the MC3 and MC4 receptors.92 Activation of G-protein coupled MC receptors will cause increased intracellular production of cAMP, calcium mobilization and receptor internalization, and subsequently activates dopamine terminals in the medial preoptic area (MPOA).93,94 Dopamine (DA) released in the MPOA mediates a variety of physiological effects, including the activation of sexual arousal. Bremelanotide has a half-life of 2.7 hours and is administered by subcutaneous injection before anticipated sexual activity for the treatment of generalized hypoactive sexual desire disorder (HSDD) in premenopausal women.95
Setmelanotide (11, Table 2 and Fig. 1b) is a disulfide cyclized octapeptide that preferentially agonizes melanocortin 4 receptor (MC4R) with an EC50 of 0.27 nM.96 Terlipressin and setmelanotide are the only two approved cyclic peptides that were developed from the natural linear hormone by introducing a cyclization bridge to lock the cyclic conformation of the partial sequence bound to the target. Setmelanotide is a nonselective melanocortin receptor initially developed from α-MSH by Ipsen.96 Setmelanotide improves energy homeostasis by producing satiety signaling and regulating behavioral and metabolic processes.97–99 Setmelanotide can effectively decrease weight in obese individuals with MC4R deficiency.100,101 Setmelanotide can also reduce hunger and body weight in individuals with obesity due to pro-opiomelanocortin (POMC) deficiency or leptin receptor deficiency.102,103
Antimicrobial cyclic peptides
The high potency and low toxicity of cyclic peptides make them ideal for developing antimicrobials for targeting microbial proteins that have no or low similarity to human proteins.104 In the past two decades, four antibacterial and three antifungal cyclic peptides were approved for clinical use (Table 3 and Fig. 2). All of these cyclic peptides were developed from natural metabolic products that were evolved and produced by organisms for eliminating competitors.105 The application of cyclic peptides in developing antimicrobials demonstrates the strength of their stable structure in binding to relatively small targets, which can only be achieved by large proteins in traditional beliefs.106 The action of cyclic peptides depends on a combination of secondary structure, charge, and hydrophobicity and amphipathicity properties, distinct from small molecules’ binding mode.107 In this section, the structures and action mechanisms of selected antimicrobials will be described and discussed for unveiling the importance of cyclic peptides.| No. | Trade name | Generic name | Target | Indication | Approval | PDB | Route of admin. | Cmaxa (nM) | HT (h) | Company | Sales 2020 |
|---|---|---|---|---|---|---|---|---|---|---|---|
| a Pharmacokinetic data was cited from FDA approval summaries. | |||||||||||
| 12 | Cubicin | Daptomycin | Membrane pore formation | Antibiotic | 2005 | 1T5N | IV | 11![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 3400 3400 |
7.7 | Merck Sharp & Dohme Corp. | $ 152 M |
| 13 | Vibativ | Telavancin | Cell wall synthesis | Antibiotic | 2009 | IV fusion | 57400 |
8 | Cumberland Inc. | N. A. | |
| 14 | Dalvance | Dalbavancin | Cell wall synthesis | Antibiotic | 2014 | 3RUL | IV fusion | 158000 |
346 | Allergan Ltd. | N. A. |
| 15 | Orbactiv | Oritavancin | Cell wall synthesis | Antibiotic | 2014 | IV fusion | 77000 |
245 | Melinta Inc. | N. A. | |
| 16 | Cancidas | Caspofungin | 1,3-Beta-glucan synthase | Antifungal | 2001 | 2N2Q | IV | 8450 | 27 | MERCK & CO., Inc. | $ 430 M |
| 17 | Mycamine | Micafungin | 1,3-Beta-glucan synthase | Antifungal | 2005 | IV | 7815 | 13.4 | Astellas Inc. | $ 349 M | |
| 18 | Eraxis | Anidulafungin | 1,3-Beta-glucan synthase | Antifungal | 2006 | IV fusion | 9500 | 50.3 | Pfizer | N. A. | |
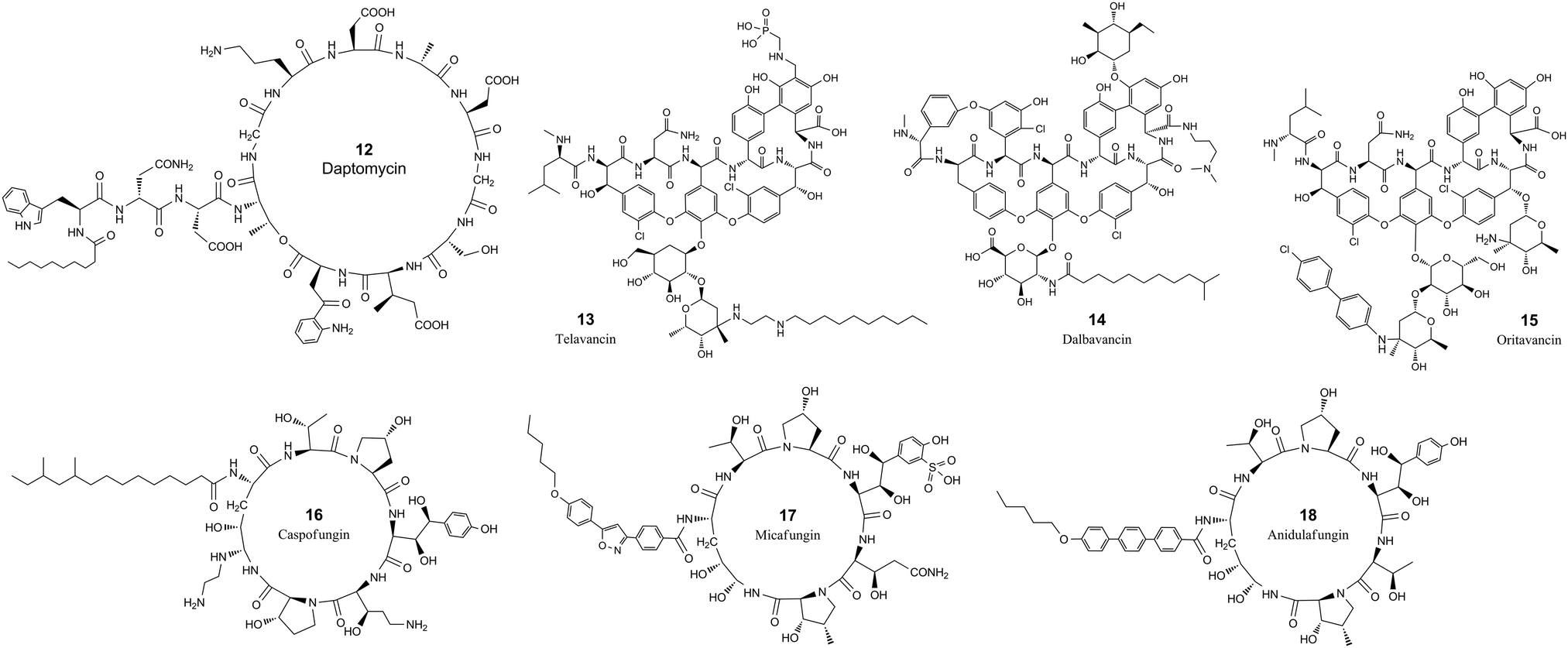 | ||
| Fig. 2 The chemical structures of cyclic peptide antimicrobial agents from Table 3. | ||
Daptomycin (12, Table 3 and Fig. 2) is a cyclic lipopeptide antibiotic isolated from Streptomyces roseosporus.108,109 Daptomycin is 13 residues in length with two noncanonical amino acids, 10 of which are arranged in a cyclic format by forming a lactone structure between the sidechain of Thr4 and the C-terminus. Daptomycin acts by inserting its decanoic acid into the cell wall of Gram-positive bacteria in a phosphatidylglycerol-dependent fashion.110 Aggregated daptomycin creates holes in the membrane of targeted bacteria and disrupts the curvature of the membrane, resulting in ion leakage and a loss of membrane potential. Consequently, the cell activities of protein, DNA, and RNA synthesis are inhibited and apoptotic cell death is induced.111 The half-life of daptomycin in adult patients is around 8 hours and the majority (78%) remains unchanged and eliminated by excretion into the urine.112
Telavancin (13, Table 3 and Fig. 2) is a semisynthetic derivative of vancomycin obtained by alkylating the vancosamine nitrogen with a decylaminoethyl side chain and a hydrophilic phosphonomethylaminomethyl group on the cyclic peptide residue for achieving improved potency and faster action.113–117 Telavancin inhibits Gram-positive bacterial cell wall synthesis by binding to the D-Ala-D-Ala terminus of peptidoglycan in the growing cell wall and interfering with the polymerization and cross-linking of peptidoglycan. The MIC90 of telavancin against S. aureus and coagulase-negative staphylococci strains is ≤1 μg ml−1, regardless of methicillin resistance.118 Telavancin was also reported to bind to the cell wall precursor lipid II and disrupts the membrane barrier function.119 Telavancin can also inhibit liver enzymes cytochrome P450 3A4 and 3A5, and should not be prescribed for patients with pre-existing moderate to severe renal impairment.120 Telavancin has poor oral bioavailability and is administered by intravenous infusion with a half-life of approximately 6.5 h.121
Dalbavancin (14, Table 3 and Fig. 2) is a novel second-generation lipoglycopeptide antibiotic medication that is more potent than vancomycin and also targets cell wall biosynthesis.122 The vancomycin family of natural antimicrobial glycopeptides are highly modified heptapeptides that have five invariant amino acids, while amino acids 1 and 3 are highly differentiated.123 The constancy of the peptide is responsible for binding to the D-alanyl-D-alanine motif, and the sidechain functional groups are derivatized to improve the properties of the peptide.122 Dalbavancin possesses a potent and rapid anti-bactericidal activity in vitro against a broad spectrum of both resistant and susceptible Gram-positive bacteria, including Staphylococcus aureus, MRSA, enterococci, and streptococci.124,125 Precursor glycopeptides of dalbavancin is manufactured by fermentation of a selected Nonomuraea strain, followed by carboxyl amidation and saponification for increased potency against staphylococci, particularly CoNS.126 The lipophilic side chain of dalbavancin has the common function of anchoring in the bacterial membrane, which is attributed to the increased affinity of dalbavancin for its target. The final dalbavancin is a compound mixture composed of five subtypes, with approximately 90% as B0,127 which has a longer half-life of approximately 1 week compared with that of vancomycin which is only 4 to 6 hours.128,129 In May 2014, dalbavancin was approved by the FDA for treating Gram-positive skin infections.130
Oritavancin (15, Table 3 and Fig. 2) is a synthetic analog of vancomycin derived from chloroeremomycin, and it differs by the addition of a lipophilic N-4-(4-chlorophenyl)benzyl side chain.131 The heptapeptide core shares the same structure as vancomycin, and the glycosylation is the same as that of chloroeremomycin. This structural alteration imparts improved activity against both vancomycin-susceptible Enterococcus (VSE) and vancomycin-resistant Enterococcus (VRE).132,133 The lipophilic side chain in oritavancin anchors the compound to the cell membrane through hydrophobic interactions and at the same time stabilizes the oritavancin dimer.133–135 Oritavancin shares a similar mechanism of action to bind the D-Ala-D-Ala stem termini in Gram-positive bacteria for inhibiting transglycosylation during peptidoglycan synthesis.132,136 Oritavancin is stable in vivo and is excreted as unchanged.137 Oritavancin has a very long half-life of 393 hours, which saves dosing frequency in the treatment of acute bacterial skin and skin structure infections (ABSSSIs).138
Caspofungin, micafungin and anidulafungin (16, 17 & 18, Table 3 and Fig. 2) all belong to the cyclic hexa-lipopeptide family. They share the same cyclic peptide core structure by targeting the enzyme 1,3-β-glucan synthase for synthesizing glucan in the cell wall.139,140 The cyclization was formed by amidation between the sidechain of the first N-terminal residue of lysine and the C-terminus of the peptide chain.141 All three antifungal drugs have clinical efficacy against invasive candidiasis and other forms of systemic mycoses.142 Caspofungin is a semisynthetic analog of pneumocandin B0, a natural lipophilic cyclic peptide that was isolated from the fungus Glarea lozoyensis in 1985 and approved for treating yeast and fungal infections under certain conditions in 2001.143,144 Caspofungin has a half-life of about 9 days and the majority undergoes ring-opening metabolic hydrolysis and N-acetylation.145 Only around 1.4% is excreted unchanged in urine.146 Micafungin is a semisynthetic analog of the natural cyclic hexapeptide FR901379, by enzymatic deacylation of the N-terminal palmitoyl group and chemical reacylation to the optimal N-acyl isoxazole analog.147 Micafungin has a poor oral bioavailability and is only intended for parenteral use with a half-life of around 15 hours.148 The majority of micafungin is metabolized by enzymatic modification of sidechains and excreted to the feces.149 Anidulafungin is also a semisynthetic echinocandin-class cyclic hexapeptide. Anidulafungin is semisynthesized from echinocandin B by diacylation to remove the linoleoyl side chain, followed by three more steps, including reacylation with the terphenylacyl chain.150,151 Anidulafungin can inhibit invasive candidiasis via noncompetitive inhibition of beta-(1,3)-glucan-synthase and has good clinical efficacy.152 Anidulafungin has a low oral bioavailability of 2–7% and is administered parenterally.153 The half-life of anidulafungin is 27 hours, and the majority is metabolized to a linear peptide by ring-opening hydrolysis. 10% of the administered anidulafungin remains unchanged and 30% of the radioactivity of administered anidulafungin is excreted in the feces and 1% in the urine.154
A lesson from the withdrawn cyclic peptide drug
In the past two decades, Omontys (peginesatide) was the only cyclic peptide drug withdrawn. Peginesatide contains two identical cyclic peptides of 21 amino acids attached to a 40 kDa polyethylene glycol (PEG) chain. In the bone marrow, peginesatide binds to the erythropoietin receptor (EpoR) for stimulating the production of red blood cells from erythroid progenitor cells (erythropoiesis).155,156 The FDA approved peginesatide for the treatment of anemia in adults on dialysis for chronic kidney disease in March 2012. Peginesatide was withdrawn from the US market in February 2013 due to serious Adverse Drug Reaction (sADR) of a high rate of anaphylactic reactions observed in 28 out of 20000 patients during the first dose IV administration.157 It is unclear what causes peginesatide sADR, but it is unlikely that it has a connection with the cyclic peptide motifs itself.158
Perspectives and conclusions
Cyclic peptides have received increasing attention as a unique category of substances with many advantageous characteristics as drugs in comparison to small molecules and macromolecular antibodies. Natural peptides have been the major source of approved cyclic peptides in the last century due to the large number of cyclic peptides present in nature. The generation of specific biological functions by natural evolution also contributed to the drug development of cyclic peptides. Cyclic peptide hormones, such as oxytocin and vasopressin, are potent and selective agonizers and were used in clinics for a long time. Antagonizing cyclic peptides, such as the large number of toxins used by diverse species against competitors and prays, are widely present in nature and are a rich source for drug development.In the past two decades, the primary trend in peptide drug discovery has been the shift of naturally isolated cyclic peptides toward cyclic peptide analogs optimized for improving potency, stability and pharmacokinetic properties. By incorporating aromatic naphthalene and D-tryptophan into lanreotide, its nanotubes with supramolecular packing were stabilized, which contributed to its extended half-life and ability to suppress hormone levels and activities. From a technical perspective, cyclic peptide drug development benefited from rapid lead discovery with adjustable and expandable optimization by chemical synthesis, which was the milestone in peptide drug development. The mutation of the lipophilic side chain of oritavancin prolonged its half-life and enhanced coverage against several vancomycin-resistant bacteria. Fermentation of cyclic peptides also benefited from modern molecular biology technologies, and the primary sequences of enzymes could be rapidly mutated and optimized for achieving a higher production yield. Increased expression of echinocandin B deacylase in the production host significantly increased the transcription and bioconversion efficiency of anidulafungin. The convenience to incorporate nonnatural elements helped to improve not only the potency, but also the stability and pharmacokinetic/pharmacodynamic properties. Noncanonical amino acids were introduced to improve proteomic resistance. Amino acids with hydrophobic long side chains were incorporated for extending plasma retention time and introducing extra biological activities of cyclic peptides. The population of Fmoc based solid-phase peptide synthesis technologies also greatly reduced the cost of production and promoted cyclic peptide research in individual groups.
The combination of modern recombinant technologies and peptide chemistry has solved the major problems of cyclic peptide production, mutation and optimization of the primary sequences. However, the space of optimizing natural cyclic peptides for achieving cyclic peptide drugs is limited due to the expanded desire to develop cyclic peptide ligands targeting diverse proteins. Recent development of cyclic peptide discovery was characterized by the generation of combinatory libraries that can cover a much larger chemical space and the development of novel screening methodologies for identifying binders and inhibitors.4,159,160 Split-and-mix cyclic peptide libraries could be synthesized with millions of diversities and screened against various targets.161–167 However, the difficulties associated with sequence identification of cyclic peptides by partial Edman degradation or mass spectrometry limited the applications of one bead-one peptide library. Directed evolutions with genetically displayed cyclic peptide recombinant libraries are increasingly playing more important roles in next-generation cyclic peptide drug discovery. Display systems such as mRNA display,168–171 DNA display172 and phage display22,173,174 offer the advantages of large library diversity and rapid screening, and have resulted in the discovery of a good portion of cyclic peptides in clinic development. Compared with the typical diversity of only a few thousands for conventional natural and synthetic cyclic peptide libraries, the diversity of cyclic peptide libraries built by genetic recombinant technologies is several orders higher. The introduction of various cyclization strategies, noncanonical amino acids and even functional moieties have further increased the functionality of recombinant libraries, such as chemical stability, metabolic stability and uniform conformation. The development of selection strategies also enabled the discovery of cyclic peptides that have special properties such as cell membrane permeation ability and oral bioavailability.
A second trend in cyclic peptide drug discovery is the development of cyclic peptide conjugates to achieve a selective delivery of different effector molecules to target tissue. Besides the drug itself, which induces the desired biological functions, and the linker, which controls drug release, cyclic peptides have been investigated intensively to facilitate sufficient target selectivity for engineering cyclic peptide drug conjugates (PDCs). Cyclic peptides, possessing the characteristics of both antibodies and small molecules, have the capacity to penetrate tissue freely and deeply, and selectively and effectively interact with receptors that are specific for certain cell types. Furthermore, PDCs are more advanced for low immunogenicity, large-scale synthesis using solid-phase synthesis, low production cost, and relatively good pharmacokinetics. When conjugated with chemotherapeutic or cytotoxic agents, PDCs have effects on tumor growth inhibition and elimination. Cyclic peptides are also conjugated with positron-emitting radioisotopes and gamma-emitting radioisotopes for developing positron emission tomography (PET) and single-photon emission computed tomography (SPECT) imaging agents.
In the coming decades, the position of cyclic peptides in the pharmaceutical industry is expected to outperform small molecules and antibodies in certain applications due to their structural complexity, large surface binding area and relatively small size. Orally available cell penetrating peptides will be one of the most important focuses of cyclic peptide studies, and the reduced production cost of cyclic peptides will contribute to the growth of cyclic peptide development.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by Shanghai Pujiang Program, Science and Technology Commission of Shanghai Municipality (#20PJ1415900 to S. C.).References
- M. Bliss, Rewriting Medical History: Charles Best and the Banting and Best myth, J. Hist. Med. Allied Sci., 1993, 48(3), 253–274 CrossRef CAS PubMed.
- S. S. Usmani, et al., THPdb: Database of FDA-approved peptide and protein therapeutics, PLoS One, 2017, 12(7), e0181748 CrossRef.
- A. Zorzi, K. Deyle and C. Heinis, Cyclic peptide therapeutics: past, present and future, Curr. Opin. Chem. Biol., 2017, 38, 24–29 CrossRef CAS PubMed.
- C. Sohrabi, A. Foster and A. Tavassoli, Methods for generating and screening libraries of genetically encoded cyclic peptides in drug discovery, Nat. Rev. Chem., 2020, 4(2), 90–101 CrossRef CAS.
- D. J. Craik, Chemistry. Seamless proteins tie up their loose ends, Science, 2006, 311(5767), 1563–1564 CrossRef.
- A. Angelini, et al., Bicyclic peptide inhibitor reveals large contact interface with a protease target, ACS Chem. Biol., 2012, 7(5), 817–821 CrossRef CAS.
- M. Empting, An Introduction to Cyclic Peptides, in Cyclic Peptides: From Bioorganic Synthesis to Applications, ed. J. Koehnke, J. Naismith and W. A. van der Donk, 2018, The Royal Society of Chemistry Search PubMed.
- S. D. Appavoo, et al., Conformational Control of Macrocycles by Remote Structural Modification, Chem. Rev., 2019, 119(17), 9724–9752 CrossRef CAS.
- A. T. Bockus, C. M. McEwen and R. S. Lokey, Form and function in cyclic peptide natural products: a pharmacokinetic perspective, Curr. ToMed. Chem., 2013, 13(7), 821–836 CAS.
- P. G. Dougherty, A. Sahni and D. Pei, Understanding Cell Penetration of Cyclic Peptides, Chem. Rev., 2019, 119(17), 10241–10287 CrossRef CAS PubMed.
- A. A. Vinogradov, Y. Yin and H. Suga, Macrocyclic Peptides as Drug Candidates: Recent Progress and Remaining Challenges, J. Am. Chem. Soc., 2019, 141(10), 4167–4181 CrossRef PubMed.
- R. E. Kontermann, Half-life extended biotherapeutics, Expert Opin. Biol. Ther., 2016, 16(7), 903–915 CrossRef CAS.
- S. C. Penchala, et al., A biomimetic approach for enhancing the in vivo half-life of peptides, Nat. Chem. Biol., 2015, 11(10), 793–798 CrossRef CAS.
- K. Chen, J. Xie and X. Chen, RGD-human serum albumin conjugates as efficient tumor targeting probes, Mol. Imaging, 2009, 8(2), 65–73 CrossRef CAS.
- M. Yu, et al., Battle of GLP-1 delivery technologies, Adv. Drug Delivery Rev., 2018, 130, 113–130 CrossRef CAS PubMed.
- D. Gang, D. W. Kim and H. S. Park, Cyclic Peptides: Promising Scaffolds for Biopharmaceuticals, Genes, 2018, 9, 11 CrossRef.
- M. A. Abdalla and L. J. McGaw, Natural Cyclic Peptides as an Attractive Modality for Therapeutics: A Mini Review, Molecules, 2018, 23, 8 CrossRef.
- E. Muratspahić, et al., Harnessing cyclotides to design and develop novel peptide GPCR ligands, RSC Chem. Biol., 2020, 1(4), 177–191 RSC.
- C. J. White and A. K. Yudin, Contemporary strategies for peptide macrocyclization, Nat. Chem., 2011, 3(7), 509–524 CrossRef CAS.
- H. Y. Chow, et al., Ligation Technologies for the Synthesis of Cyclic Peptides, Chem. Rev., 2019, 119(17), 9971–10001 CrossRef CAS.
- J. N. Lambert, J. P. Mitchell and K. D. Roberts, The synthesis of cyclic peptides, J. Chem. Soc., Perkin Trans. 1, 2001,(5), 471–484 RSC.
- C. Heinis, et al., Phage-encoded combinatorial chemical libraries based on bicyclic peptides, Nat. Chem. Biol., 2009, 5(7), 502–507 CrossRef CAS PubMed.
- S. Chen, et al., Peptide ligands stabilized by small molecules, Angew. Chem., Int. Ed., 2014, 53(6), 1602–1606 CrossRef CAS PubMed.
- N. J. Yang and M. J. Hinner, Getting across the cell membrane: an overview for small molecules, peptides, and proteins, Methods Mol. Biol., 2015, 1266, 29–53 CrossRef CAS.
- C. Bechara and S. Sagan, Cell-penetrating peptides: 20 years later, where do we stand?, FEBS Lett., 2013, 587(12), 1693–1702 CrossRef CAS.
- M. Kristensen, D. Birch and H. Morck, Nielsen, Applications and Challenges for Use of Cell-Penetrating Peptides as Delivery Vectors for Peptide and Protein Cargos., Int. J. Mol. Sci., 2016, 17(2), 185 CrossRef.
- I. Ruseska and A. Zimmer, Internalization mechanisms of cell-penetrating peptides, Beilstein J. Nanotechnol., 2020, 11, 101–123 CrossRef CAS.
- S. G. Patel, et al., Cell-penetrating peptide sequence and modification dependent uptake and subcellular distribution of green florescent protein in different cell lines, Sci. Rep., 2019, 9(1), 6298 CrossRef.
- A. C. Rand, et al., Optimizing PK properties of cyclic peptides: the effect of side chain substitutions on permeability and clearance(), MedChemComm, 2012, 3(10), 1282–1289 RSC.
- T. Rezai, et al., Conformational flexibility, internal hydrogen bonding, and passive membrane permeability: successful in silico prediction of the relative permeabilities of cyclic peptides, J. Am. Chem. Soc., 2006, 128(43), 14073–14080 CrossRef CAS.
- E. Biron, et al., Improving oral bioavailability of peptides by multiple N-methylation: somatostatin analogues, Angew. Chem., Int. Ed., 2008, 47(14), 2595–2599 CrossRef CAS PubMed.
- Z. Qian, et al., Early endosomal escape of a cyclic cell-penetrating peptide allows effective cytosolic cargo delivery, Biochemistry, 2014, 53(24), 4034–4046 CrossRef CAS PubMed.
- Approval Date(s) and History, Letters, Labels, Reviews for ISTODAX. Available from: https://www.accessdata.fda.gov/scripts/cder/daf/index.cfm?event=overview.process&ApplNo=022393.
- Approval Date(s) and History, Letters, Labels, Reviews for LUPKYNIS. Available from: https://www.accessdata.fda.gov/scripts/cder/daf/index.cfm?event=overview.process&ApplNo=213716.
- Drug Approval Package for Istodax. Available from: https://www.accessdata.fda.gov/drugsatfda_docs/nda/2009/022393s000TOC.cfm.
- H. Ueda, et al., FR901228, a novel antitumor bicyclic depsipeptide produced by Chromobacterium violaceum No. 968. I. Taxonomy, fermentation, isolation, physico-chemical and biological properties, and antitumor activity, J. Antibiot., 1994, 47(3), 301–310 CrossRef CAS.
- K. W. Li, et al., Total synthesis of the antitumor depsipeptide FR-901,228, J. Am. Chem. Soc., 1996, 118(30), 7237–7238 CrossRef CAS.
- B. C. Valdez, et al., Romidepsin targets multiple survival signaling pathways in malignant T cells, Blood Cancer J., 2015, 5, e357 CrossRef CAS.
- H. Nakajima, et al., FR901228, a potent antitumor antibiotic, is a novel histone deacetylase inhibitor, Exp. Cell Res., 1998, 241(1), 126–133 CrossRef CAS PubMed.
- S. Woo, et al., Population pharmacokinetics of romidepsin in patients with cutaneous T-cell lymphoma and relapsed peripheral T-cell lymphoma, Clin. Cancer Res., 2009, 15(4), 1496–1503 CrossRef CAS.
- Product monograph of istodax. Available from: https://www.bms.com/assets/bms/ca/documents/productmonograph/ISTODAX_EN_PM.pdf.
- S. J. Harrison, et al., A focus on the preclinical development and clinical status of the histone deacetylase inhibitor, romidepsin (depsipeptide, Istodax((R))), Epigenomics, 2012, 4(5), 571–589 CrossRef CAS PubMed.
- Bristol Myers Squibb Statement on Istodax® (romidepsin) Relapsed/Refractory Peripheral T-cell Lymphoma U.S. Indicatio. Available from: https://news.bms.com/news/corporate-financial/2021/Bristol-Myers-Squibb-Statement-on-Istodax-romidepsin-Relapsed-Refractory-Peripheral-T-cell-Lymphoma-U.S.-Indication/default.aspx.
- Drug Approval Package for lupkynis. Available from: https://www.accessdata.fda.gov/drugsatfda_docs/nda/2021/213716Orig1s000TOC.cfm.
- Voclosporin, in Meyler's Side Effects of Drugs, ed. J. K. Aronson, 2016, Sixteenth Edition, Elsevier, Oxford. pp. 500–501 Search PubMed.
- E. Anglade, L. J. Aspeslet and S. L. Weiss, A new agent for the treatment of noninfectious uveitis: rationale and design of three LUMINATE (Lux Uveitis Multicenter Investigation of a New Approach to Treatment) trials of steroid-sparing voclosporin, Clin. Ophthalmol., 2008, 2(4), 693–702 CrossRef CAS.
- A. Kuglstatter, et al., Structural basis for the cyclophilin A binding affinity and immunosuppressive potency of E-ISA247 (voclosporin), Acta Crystallogr., Sect. D: Biol. Crystallogr., 2011, 67(Pt 2), 119–123 CrossRef CAS.
- A. Lawen, Biosynthesis of cyclosporins and other natural peptidyl prolyl cis/trans isomerase inhibitors, Biochim. Biophys. Acta, 2015, 1850(10), 2111–2120 CrossRef CAS.
- C. Schultz, Voclosporin as a treatment for noninfectious uveitis, Ophthalmol. Eye Dis., 2013, 5, 5–10 CAS.
- A. S. Hauser, et al., Trends in GPCR drug discovery: new agents, targets and indications, Nat. Rev. Drug Discovery, 2017, 16(12), 829–842 CrossRef CAS.
- W. K. Chan, et al., GLASS: a comprehensive database for experimentally validated GPCR-ligand associations, Bioinformatics, 2015, 31(18), 3035–3042 CrossRef CAS PubMed.
- C. W. Gruber, M. Muttenthaler and M. Freissmuth, Ligand-based peptide design and combinatorial peptide libraries to target G protein-coupled receptors, Curr. Pharm. Des., 2010, 16(28), 3071–3088 CrossRef CAS PubMed.
- E. Muratspahic, M. Freissmuth and C. W. Gruber, Nature-Derived Peptides: A Growing Niche for GPCR Ligand Discovery, Trends Pharmacol. Sci., 2019, 40(5), 309–326 CrossRef CAS PubMed.
- A. Zorzi, et al., Acylated heptapeptide binds albumin with high affinity and application as tag furnishes long-acting peptides, Nat. Commun., 2017, 8, 16092 CrossRef CAS.
- D. Chung, et al., Determination of disulfide bridge pattern in omega-conopeptides, Int. J. Pept. Protein Res., 1995, 46(3-4), 320–325 CrossRef CAS.
- V. J. Basus, et al., Solution structure of omega-conotoxin MVIIA using 2D NMR spectroscopy, FEBS Lett., 1995, 370(3), 163–169 CrossRef CAS.
- T. Kohno, et al., Three-dimensional structure in solution of the calcium channel blocker omega-conotoxin MVIIA, Biochemistry, 1995, 34(32), 10256–10265 CrossRef CAS PubMed.
- Approval Date(s) and History, Letters, Labels, Reviews for PRIALT. Available from: https://www.accessdata.fda.gov/scripts/cder/daf/index.cfm?event=overview.process&ApplNo=021060.
- J. G. McGivern, Ziconotide: a review of its pharmacology and use in the treatment of pain, Neuropsychiatr. Dis. Treat., 2007, 3(1), 69–85 CrossRef CAS.
- Drug Approval Package for Prialt. Available from: https://www.accessdata.fda.gov/drugsatfda_docs/nda/2004/21-060_Prialt.cfm.
- M. Vazquez Roque and M. Camilleri, Linaclotide, a synthetic guanylate cyclase C agonist, for the treatment of functional gastrointestinal disorders associated with constipation, Exp. Rev. Gastroenterol. Hepatol., 2011, 5(3), 301–310 CrossRef PubMed.
- A. P. Bryant, et al., Linaclotide is a potent and selective guanylate cyclase C agonist that elicits pharmacological effects locally in the gastrointestinal tract, Life Sci., 2010, 86(19-20), 760–765 CrossRef CAS.
- K. Shailubhai, et al., Plecanatide, an oral guanylate cyclase C agonist acting locally in the gastrointestinal tract, is safe and well-tolerated in single doses, Dig. Dis. Sci., 2013, 58(9), 2580–2586 CrossRef CAS PubMed.
- K. Shailubhai, et al., Plecanatide and dolcanatide, novel guanylate cyclase-C agonists, ameliorate gastrointestinal inflammation in experimental models of murine colitis, World J. Gastrointest. Pharmacol. Ther., 2015, 6(4), 213–222 CrossRef PubMed.
- Y. Yang, et al., Linaclotide in irritable bowel syndrome with constipation: A Phase 3 randomized trial in China and other regions, J. Gastroenterol. Hepatol., 2018, 33(5), 980–989 CrossRef CAS.
- R. H. Thomas and K. Allmond, Linaclotide (Linzess) for Irritable Bowel syndrome With Constipation and For Chronic Idiopathic Constipation, Proc. Trans., 2013, 38(3), 154–160 Search PubMed.
- A. Brancale, et al., Therapeutically targeting guanylate cyclase-C: computational modeling of plecanatide, a uroguanylin analog, Pharmacol. Res. Perspect., 2017, 5(2), e00295 CrossRef.
- M. K. Ahsan, et al., Linaclotide activates guanylate cyclase-C/cGMP/protein kinase-II-dependent trafficking of CFTR in the intestine, Physiol. Rep., 2017, 5(11), e13299 CrossRef PubMed.
- R. W. Busby and S. Ortiz, Clarification of linaclotide pharmacology presented in a recent clinical study of plecanatide, Dig. Dis. Sci., 2014, 59(5), 1066–1067 CrossRef.
- A. Jarmuz, et al., Emerging treatments in Neurogastroenterology: Perspectives of guanylyl cyclase C agonists use in functional gastrointestinal disorders and inflammatory bowel diseases, Neurogastroenterol. Motil., 2015, 27(8), 1057–1068 CrossRef CAS.
- Z. T. Al-Salama and Y. Y. Syed, Plecanatide: First Global Approval, Drugs, 2017, 77(5), 593–598 CrossRef CAS.
- T. J. O'Toole and S. Sharma, Physiology, Somatostatin, StatPearls, 2021, Treasure Island (FL) Search PubMed.
- R. M. Paragliola and R. Salvatori, Novel Somatostatin Receptor Ligands Therapies for Acromegaly, Front. Endocrinol., 2018, 9, 78 CrossRef.
- European Medicines Agency recommends approval of new medicine for Cushing's disease. Available from: https://www.ema.europa.eu/en/documents/press-release/european-medicines-agency-recommends-approval-new-medicine-cushings-disease_en.pdf.
- Drug Approval Package for Signifor. Available from: https://www.accessdata.fda.gov/drugsatfda_docs/nda/2012/200677Orig1s000TOC.cfm.
- Drug Approval Package for Signifor LAR. Available from: https://www.accessdata.fda.gov/drugsatfda_docs/nda/2014/203255Orig1s000TOC.cfm.
- J. M. Kuhn, et al., Pharmacokinetic study and effects on growth hormone secretion in healthy volunteers of the new somatostatin analogue BIM 23014, Eur. J. Clin. Pharmacol., 1993, 45(1), 73–77 CrossRef CAS PubMed.
- I. Morange, et al., Slow release lanreotide treatment in acromegalic patients previously normalized by octreotide, J. Clin. Endocrinol. Metab., 1994, 79(1), 145–151 CAS.
- J. M. Kuhn, et al., Pharmacokinetic and pharmacodynamic properties of a long-acting formulation of the new somatostatin analogue, lanreotide, in normal healthy volunteers, Br. J. Clin. Pharmacol., 1994, 38(3), 213–219 CrossRef CAS.
- Drug Approval Package for lanreotide. Available from: https://www.accessdata.fda.gov/drugsatfda_docs/nda/2007/022074s000TOC.cfm.
- Editorials, Separation of the active principles of the posterior lobe of the pituitary gland, J. Am. Med. Assoc., 1928, 90(8), 618–619 CrossRef.
- R. A. Turner, J. G. Pierce and V. V. Du, The purification and the amino acid content of vasopressin preparations., J. Biol. Chem., 1951, 191(1), 21–28 CrossRef CAS.
- R. Acher and J. Chauvet, La structure de la vasopressine de boeuf, Biochem. Biophys. Acta, 1954, 14, 421–429 CAS.
- V.d. Vigneaud, H. C. Lawler and E. A. Popenoe, Enzymatic cleavage of glycinamide from vasopressin and a proposed structure for this pressor-antidiuretic hormone of the posterior pituitary, J. Am. Chem. Soc., 1953, 75(19), 4880–4881 CrossRef.
- J. Demiselle, et al., Vasopressin and its analogues in shock states: a review, Ann. Intensive Care, 2020, 10(1), 9 CrossRef.
- Drug Approval Package for Vasostrict. Available from: https://www.accessdata.fda.gov/drugsatfda_docs/nda/2014.204485Orig1s000TOC.cfm.
- A. Delmas, et al., Clinical review: vasopressin and terlipressin in septic shock patients, Crit. Care, 2005, 9(2), 212–222 CrossRef PubMed.
- J. W. Czaczkes, C. R. Kleeman and M. Koenig, Physiologic Studies of Antidiuretic Hormone by Its Direct Measurement in Human Plasma, J. Clin. Invest., 1964, 43, 1625–1640 CrossRef CAS.
- J. Bosch, D. Lebrec and S. A. Jenkins, Development of analogues: successes and failures, Scand. J. Gastroenterol., Suppl., 1998, 226, 3–13 CrossRef CAS PubMed.
- K. Wisniewski, et al., New, potent, selective, and short-acting peptidic V1a receptor agonists, J. Med. Chem., 2011, 54(13), 4388–4398 CrossRef CAS.
- Z. R. Belkin, J. M. Krapf and A. T. Goldstein, Drugs in early clinical development for the treatment of female sexual dysfunction, Expert Opin. Invest. Drugs, 2015, 24(2), 159–167 CrossRef CAS.
- S. H. King, et al., Melanocortin receptors, melanotropic peptides and penile erection, Curr. Top. Med. Chem., 2007, 7(11), 1098–1106 CrossRef PubMed.
- S. Sharma, et al., Current Mechanistic and Pharmacodynamic Understanding of Melanocortin-4 Receptor Activation, Molecules, 2019, 24, 10 Search PubMed.
- J. Pfaus, F. Giuliano and H. Gelez, Bremelanotide: an overview of preclinical CNS effects on female sexual function, J. Sex Med., 2007, 4(suppl 4), 269–279 CrossRef CAS PubMed.
- Highlights Of Prescribing Information For Vyleesi. Available from: https://www.accessdata.fda.gov/drugsatfda_docs/label/2019/210557s000lbl.pdf.
- K. Y. Chen, et al., RM-493, a melanocortin-4 receptor (MC4R) agonist, increases resting energy expenditure in obese individuals, J. Clin. Endocrinol. Metab., 2015, 100(4), 1639–1645 CrossRef CAS.
- R. D. Cone, Studies on the physiological functions of the melanocortin system, Endocr. Rev., 2006, 27(7), 736–749 CrossRef CAS PubMed.
- A. S. Garfield, et al., A neural basis for melanocortin-4 receptor-regulated appetite, Nat. Neurosci., 2015, 18(6), 863–871 CrossRef CAS PubMed.
- G. W. Kim, et al., Regulation of appetite to treat obesity, Expert Rev. Clin. Pharmacol., 2011, 4(2), 243–259 CrossRef CAS.
- E. Pilitsi, et al., Pharmacotherapy of obesity: available medications and drugs under investigation, Metabolism, 2019, 92, 170–192 CrossRef CAS PubMed.
- T. H. Collet, et al., Evaluation of a melanocortin-4 receptor (MC4R) agonist (Setmelanotide) in MC4R deficiency, Mol. Metab., 2017, 6(10), 1321–1329 CrossRef CAS.
- P. Kuhnen, et al., Proopiomelanocortin Deficiency Treated with a Melanocortin-4 Receptor Agonist, N. Engl. J. Med., 2016, 375(3), 240–246 CrossRef.
- K. Clement, et al., MC4R agonism promotes durable weight loss in patients with leptin receptor deficiency, Nat. Med., 2018, 24(5), 551–555 CrossRef CAS PubMed.
- Microbiology, Chapter 14 | Antimicrobial Drugs. Available from: https://openstax.org/books/microbiology/pages/14-introduction.
- M. M. Mullis, et al., Diversity, Ecology, and Prevalence of Antimicrobials in Nature, Front. Microbiol., 2019, 10, 2518 CrossRef PubMed.
- J. R. Knox and R. F. Pratt, Different modes of vancomycin and D-alanyl-D-alanine peptidase binding to cell wall peptide and a possible role for the vancomycin resistance protein, Antimicrob. Agents Chemother., 1990, 34(7), 1342–1347 CrossRef CAS PubMed.
- A. Falanga, et al., Cyclic Peptides as Novel Therapeutic Microbicides: Engineering of Human Defensin Mimetics, Molecules, 2017, 22, 7 CrossRef PubMed.
- V. Miao, et al., Daptomycin biosynthesis in Streptomyces roseosporus: cloning and analysis of the gene cluster and revision of peptide stereochemistry, Microbiology, 2005, 151(Pt 5), 1507–1523 CrossRef CAS PubMed.
- J. N. Steenbergen, et al., Daptomycin: a lipopeptide antibiotic for the treatment of serious Gram-positive infections, J. Antimicrob. Chemother., 2005, 55(3), 283–288 CrossRef CAS.
- J. Pogliano, N. Pogliano and J. A. Silverman, Daptomycin-mediated reorganization of membrane architecture causes mislocalization of essential cell division proteins, J. Bacteriol., 2012, 194(17), 4494–4504 CrossRef CAS PubMed.
- A. C. Papanicolaou, et al., Evidence for right-hemisphere involvement in recovery from aphasia, Arch. Neurol., 1988, 45(9), 1025–1029 CrossRef CAS PubMed.
- B. H. Dvorchik, et al., Daptomycin pharmacokinetics and safety following administration of escalating doses once daily to healthy subjects, Antimicrob. Agents Chemother., 2003, 47(4), 1318–1323 CrossRef CAS PubMed.
- K. Sterling, et al., Specific mitochondrial binding sites for triiodothyronine, Trans. Assoc. Am. Physicians, 1979, 92, 334–345 CAS.
- S. S. Hegde, et al., Pharmacodynamics of telavancin (TD-6424), a novel bactericidal agent, against Gram-positive bacteria, Antimicrob. Agents Chemother., 2004, 48(8), 3043–3050 CrossRef CAS PubMed.
- Food and Drug Administration, FDA labelling information, FDA website, 2009. Available from: http://www.accessdata.fda.gov/drugsatfda_docs/label/2009/022110s000lbl.pdf.
- S. Laohavaleeson, J. L. Kuti and D. P. Nicolau, Telavancin: a novel lipoglycopeptide for serious Gram-positive infections, Expert Opin. Invest. Drugs, 2007, 16(3), 347–357 CrossRef CAS PubMed.
- P. Plotkin, et al., Telavancin (vibativ), a new option for the treatment of Gram-positive infections, Proc. Trans., 2011, 36(3), 127–138 Search PubMed.
- R. E. Mendes, et al., Update on the telavancin activity tested against European staphylococcal clinical isolates (2009–2010), Diagn. Microbiol. Infect. Dis., 2011, 71(1), 93–97 CrossRef CAS PubMed.
- C. S. Lunde, et al., Telavancin disrupts the functional integrity of the bacterial membrane through targeted interaction with the cell wall precursor lipid II, Antimicrob. Agents Chemother., 2009, 53(8), 3375–3383 CrossRef CAS PubMed.
- L. J. Scott, Telavancin: a review of its use in patients with nosocomial pneumonia, Drugs, 2013, 73(16), 1829–1839 CrossRef CAS PubMed.
- V. Al Jalali and M. Zeitlinger, Clinical Pharmacokinetics and Pharmacodynamics of Telavancin Compared with the Other Glycopeptides, Clin. Pharmacokinet., 2018, 57(7), 797–816 CrossRef CAS PubMed.
- A. Y. Chen, M. J. Zervos and J. A. Vazquez, Dalbavancin: a novel antimicrobial, Int. J. Clin. Pract., 2007, 61(5), 853–863 CrossRef CAS PubMed.
- A. Malabarba and R. Ciabatti, Glycopeptide derivatives, Curr. Med. Chem., 2001, 8(14), 1759–1773 CrossRef CAS PubMed.
- B. S. Zuckerman, H. Amaro and W. Beardslee, Mental health of adolescent mothers: the implications of depression and drug use, J. Dev. Behav. Pediatr., 1987, 8(2), 111–116 CrossRef CAS.
- R. E. Mendes, et al., Activity of oritavancin tested against uncommonly isolated Gram-positive pathogens responsible for documented infections in hospitals worldwide, J. Antimicrob. Chemother., 2014, 69(6), 1579–1581 CrossRef CAS PubMed.
- A. Malabarba and B. P. Goldstein, Origin, structure, and activity in vitro and in vivo of dalbavancin, J. Antimicrob. Chemother., 2005, 55(suppl_2), ii15–ii20 CrossRef CAS PubMed.
- M. Stogniew, Dalbavancin compositions for treatment of bacterial infections, US, 2004 Search PubMed.
- A. Leighton, et al., Tolerability, pharmacokinetics, and serum bactericidal activity of intravenous dalbavancin in healthy volunteers, Antimicrob. Agents Chemother., 2004, 48(3), 940–945 CrossRef CAS PubMed.
- E. Seltzer, et al., Once-weekly dalbavancin versus standard-of-care antimicrobial regimens for treatment of skin and soft-tissue infections, Clin. Infect. Dis., 2003, 37(10), 1298–1303 CrossRef CAS PubMed.
- Drug Approval Package for Dalvance. Available from: https://www.accessdata.fda.gov/drugsatfda_docs/nda/2014/021883Orig1s000TOC.cfm.
- G. G. Zhanel, F. Schweizer and J. A. Karlowsky, Oritavancin: mechanism of action, Clin. Infect. Dis., 2012, 54(suppl 3), S214–S219 CrossRef CAS PubMed.
- G. J. Patti, et al., Vancomycin and oritavancin have different modes of action in Enterococcus faecium, J. Mol. Biol., 2009, 392(5), 1178–1191 CrossRef CAS PubMed.
- A. Belley, et al., Ultrastructural effects of oritavancin on methicillin-resistant Staphylococcus aureus and vancomycin-resistant Enterococcus, Antimicrob. Agents Chemother., 2009, 53(2), 800–804 CrossRef CAS PubMed.
- N. E. Allen and T. I. Nicas, Mechanism of action of oritavancin and related glycopeptide antibiotics, FEMS Microbiol. Rev., 2003, 26(5), 511–532 CrossRef CAS PubMed.
- A. Belley, et al., Oritavancin kills stationary-phase and biofilm Staphylococcus aureus cells in vitro, Antimicrob. Agents Chemother., 2009, 53(3), 918–925 CrossRef CAS PubMed.
- S. J. Kim, et al., Oritavancin exhibits dual mode of action to inhibit cell-wall biosynthesis in Staphylococcus aureus, J. Mol. Biol., 2008, 377(1), 281–293 CrossRef CAS PubMed.
- D. J. Cada and D. E. Baker, Oritavancin diphosphate, Hosp. Pharm., 2014, 49(11), 1049–1060 CrossRef CAS.
- L. D. Saravolatz and G. E. Stein, Oritavancin: A Long-Half-Life Lipoglycopeptide, Clin. Infect. Dis., 2015, 61(4), 627–632 CrossRef CAS PubMed.
- M. B. Kurtz, et al., Morphological effects of lipopeptides against Aspergillus fumigatus correlate with activities against (1,3)-beta-D-glucan synthase, Antimicrob. Agents Chemother., 1994, 38(7), 1480–1489 CrossRef CAS PubMed.
- N. Robbins, G. D. Wright and L. E. Cowen, Antifungal Drugs: The Current Armamentarium and Development of New Agents, Microbiol. Spectr., 2016, 4, 5 Search PubMed.
- W. Huttel, Echinocandins: structural diversity, biosynthesis, and development of antimycotics, Appl. Microbiol. Biotechnol., 2021, 105(1), 55–66 CrossRef PubMed.
- D. W. Denning, Echinocandins: a new class of antifungal, J. Antimicrob. Chemother., 2002, 49(6), 889–891 CrossRef CAS PubMed.
- Drug Approval Package for Cancidas. Available from: https://www.accessdata.fda.gov/drugsatfda_docs/nda/2001/21227_Cancidas.cfm.
- J. M. Balkovec, et al., Discovery and development of first in class antifungal caspofungin (CANCIDAS(R))–a case study, Nat. Prod. Rep., 2014, 31(1), 15–34 RSC.
- G. Kofla and M. Ruhnke, Pharmacology and metabolism of anidulafungin, caspofungin and micafungin in the treatment of invasive candidosis: review of the literature, Eur. J. Med. Res., 2011, 16(4), 159–166 CrossRef CAS PubMed.
- D. W. Denning, Echinocandin antifungal drugs, Lancet, 2003, 362(9390), 1142–1151 CrossRef CAS.
- S. Hashimoto, Micafungin: a sulfated echinocandin, J. Antibiot., 2009, 62(1), 27–35 CrossRef CAS PubMed.
- R. E. Wasmann, et al., Clinical Pharmacokinetics and Pharmacodynamics of Micafungin, Clin. Pharmacokinet., 2018, 57(3), 267–286 CrossRef CAS PubMed.
- J. Houst, J. Spizek and V. Havlicek, Antifungal Drugs, Metabolites, 2020, 10, 3 CrossRef PubMed.
- D. W. Denning, Echinocandins and pneumocandins-a new antifungal class with a novel mode of action, J. Antimicrob. Chemother., 1997, 40(5), 611–614 CrossRef CAS PubMed.
- L. Shao, et al., Efficient bioconversion of echinocandin B to its nucleus by overexpression of deacylase genes in different host strains, Appl. Environ. Microbiol., 2013, 79(4), 1126–1133 CrossRef CAS PubMed.
- S. L. Davis and J. A. Vazquez, Anidulafungin: an evidence-based review of its use in invasive fungal infections, Core Evid., 2008, 2(4), 241–249 Search PubMed.
- Summary Of Product Characteristics For Ecalta. Available from: https://www.ema.europa.eu/en/documents/product-information/ecalta-epar-product-information_en.pdf.
- T. J. Walsh, et al., Treatment of aspergillosis: clinical practice guidelines of the Infectious Diseases Society of America, Clin. Infect. Dis., 2008, 46(3), 327–360 CrossRef CAS PubMed.
- J. M. Green, et al., Peginesatide and erythropoietin stimulate similar erythropoietin receptor-mediated signal transduction and gene induction events, Exp. Hematol., 2012, 40(7), 575–587 CrossRef CAS PubMed.
- R. Verma, et al., The Erythropoiesis Stimulating Agent Omontys/Peginesatide up-Modulates Erythroid Progenitor Formation, and Cell Surface Erythropoietin Receptor Levels, Blood, 2012, 120(21), 2089 CrossRef.
- C. L. Bennett, et al., Anaphylaxis and hypotension after administration of peginesatide, N. Engl. J. Med., 2014, 370(21), 2055–2056 CrossRef PubMed.
- T. Hermanson, C. L. Bennett and I. C. Macdougall, Peginesatide for the treatment of anemia due to chronic kidney disease - an unfulfilled promise, Expert Opin. Drug Saf., 2016, 15(10), 1421–1426 CrossRef CAS PubMed.
- K. Shinbara, et al., Methodologies for Backbone Macrocyclic Peptide Synthesis Compatible With Screening Technologies, Front. Chem., 2020, 8, 447 CrossRef CAS PubMed.
- J. Valentine and A. Tavassoli, Genetically Encoded Cyclic Peptide Libraries: From Hit to Lead and Beyond, Methods Enzymol., 2018, 610, 117–134 CAS.
- T. Liu, et al., Synthesis and screening of a cyclic peptide library: discovery of small-molecule ligands against human prolactin receptor, Bioorg. Med. Chem., 2009, 17(3), 1026–1033 CrossRef CAS PubMed.
- W. M. Hewitt, et al., Cell-permeable cyclic peptides from synthetic libraries inspired by natural products, J. Am. Chem. Soc., 2015, 137(2), 715–721 CrossRef CAS PubMed.
- W. Tegge, W. Bautsch and R. Frank, Synthesis of cyclic peptides and peptide libraries on a new disulfide linker, J. Pept. Sci., 2007, 13(10), 693–699 CrossRef CAS PubMed.
- V. Adebomi, et al., CyClick Chemistry for the Synthesis of Cyclic Peptides, Angew. Chem., Int. Ed., 2019, 58(52), 19073–19080 CrossRef CAS PubMed.
- R. Wills, V. Adebomi and M. Raj, Site-Selective Peptide Macrocyclization, ChemBioChem, 2021, 22(1), 52–62 CrossRef CAS PubMed.
- B. Li, et al., Construction of Natural-Product-Like Cyclophane-Braced Peptide Macrocycles via sp(3) C–H Arylation, J. Am. Chem. Soc., 2019, 141(23), 9401–9407 CrossRef PubMed.
- X. Hu, et al., Construction of Peptide Macrocycles via Radical-Mediated Intramolecular C–H Alkylations, Org. Lett., 2021, 23(3), 716–721 CrossRef CAS PubMed.
- Z. Ma and M. C. T. Hartman, In Vitro Selection of Unnatural Cyclic Peptide Libraries via mRNA Display, in Ribosome Display and Related Technologies: Methods and Protocols, ed. J. A. Douthwaite and R. H. Jackson, 2012, Springer New York, New York, NY, pp. 367–390 Search PubMed.
- T. Passioura and H. Suga, A RaPID way to discover nonstandard macrocyclic peptide modulators of drug targets, Chem. Commun., 2017, 53(12), 1931–1940 RSC.
- T. Passioura, et al., Display Selection of Exotic Macrocyclic Peptides Expressed under a Radically Reprogrammed 23 Amino Acid Genetic Code, J. Am. Chem. Soc., 2018, 140(37), 11551–11555 CrossRef CAS PubMed.
- R. Takatsuji, et al., Ribosomal Synthesis of Backbone-Cyclic Peptides Compatible with In Vitro Display, J. Am. Chem. Soc., 2019, 141(6), 2279–2287 CrossRef CAS PubMed.
- D. Neri and R. A. Lerner, DNA-Encoded Chemical Libraries: A Selection System Based on Endowing Organic Compounds with Amplifiable Information, Annu. Rev. Biochem., 2018, 87, 479–502 CrossRef CAS PubMed.
- S. Chen, et al., Identification of highly selective covalent inhibitors by phage display, Nat. Biotechnol., 2021, 39(4), 490–498 CrossRef CAS PubMed.
- A. Glas, et al., Constrained peptides with target-adapted cross-links as inhibitors of a pathogenic protein–protein interaction, Angew. Chem., Int. Ed., 2014, 53(9), 2489–2493 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2022 |