
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMacrocyclic peptides as allosteric inhibitors of nicotinamide N-methyltransferase (NNMT)†
Matthijs J.
van Haren‡
 a,
Yurui
Zhang‡
a,
Vito
Thijssen
b,
Ned
Buijs
a,
Yongzhi
Gao
a,
Lukasz
Mateuszuk
c,
Filip A.
Fedak
c,
Agnieszka
Kij
c,
Roberto
Campagna
d,
Davide
Sartini
d,
Monica
Emanuelli
d,
Stefan
Chlopicki
ce,
Seino A. K.
Jongkees
*bf and
Nathaniel I.
Martin
*a
a,
Yurui
Zhang‡
a,
Vito
Thijssen
b,
Ned
Buijs
a,
Yongzhi
Gao
a,
Lukasz
Mateuszuk
c,
Filip A.
Fedak
c,
Agnieszka
Kij
c,
Roberto
Campagna
d,
Davide
Sartini
d,
Monica
Emanuelli
d,
Stefan
Chlopicki
ce,
Seino A. K.
Jongkees
*bf and
Nathaniel I.
Martin
*a
aBiological Chemistry Group, Institute of Biology Leiden, Leiden University, Sylviusweg 72, 2333 BE Leiden, The Netherlands. E-mail: n.i.martin@biology.leidenuniv.nl
bChemical Biology & Drug Discovery Group, Utrecht Institute for Pharmaceutical Sciences, Universiteitsweg 99, 3584 CG Utrecht, The Netherlands. E-mail: s.a.k.jongkees@vu.nl
cJagiellonian University, Jagiellonian Centre for Experimental Therapeutics (JCET), Bobrzynskiego 14, 30-348 Krakow, Poland
dDepartment of Clinical Sciences, Universitá Politecnica delle Marche, Via Ranieri 65, 60131 Ancona, Italy
eJagiellonian University Medical College, Chair of Pharmacology, Grzegorzecka 16, 31-531 Krakow, Poland
fDepartment of Chemistry and Pharmaceutical Sciences, Vrije Universiteit Amsterdam, De Boelelaan 1108, 1081 HZ Amsterdam, The Netherlands
First published on 19th August 2021
Abstract
Nicotinamide N-methyltransferase (NNMT) methylates nicotinamide to form 1-methylnicotinamide (MNA) using S-adenosyl-L-methionine (SAM) as the methyl donor. The complexity of the role of NNMT in healthy and disease states is slowly being elucidated and provides an indication that NNMT may be an interesting therapeutic target for a variety of diseases including cancer, diabetes, and obesity. Most inhibitors of NNMT described to date are structurally related to one or both of its substrates. In the search for structurally diverse NNMT inhibitors, an mRNA display screening technique was used to identify macrocyclic peptides which bind to NNMT. Several of the cyclic peptides identified in this manner show potent inhibition of NNMT with IC50 values as low as 229 nM. The peptides were also found to downregulate MNA production in cellular assays. Interestingly, substrate competition experiments reveal that these cyclic peptide inhibitors are noncompetitive with either SAM or NA indicating they may be the first allosteric inhibitors reported for NNMT.
Introduction
Nicotinamide N-methyltransferase (NNMT) is a cytosolic enzyme that catalyzes the methylation of nicotinamide (NA, vitamin B3) and a variety of other pyridines in the presence of S-adenosyl-L-methionine (SAM) to form 1-methyl nicotinamide (MNA) or the corresponding methylpyridinium ion.1,2 Recently, a number of reports have demonstrated that the role of NNMT is not limited to its involvement in xenobiotic metabolism, but rather reveal NNMT to be a master metabolic regulator in a variety of cancers.3–5 Additionally, NNMT overexpression is found to be a biomarker in an increasing number of cancers and is often linked to poor prognosis.6–10 Aside from its roles in cancer, NNMT is implicated in Parkinson's Disease (PD),11,12 vasoprotection,13,14 pulmonary arterial hypertension (PAH),15 diabetes16 and obesity.17,18 Interestingly, these involvements can be either protecting, as in PD, PAH, and endothelial function, or damaging as in cancer, diabetes, and obesity. To more fully understand the roles played by NNMT in healthy and disease states, specific NNMT inhibitors are needed. However, despite the increasing interest in NNMT, a limited number of NNMT inhibitors have been described to date and none have entered clinical trials.19–24Previous work in both our group and that of others has focused primarily on the design and optimization of bisubstrate inhibitors of NNMT that incorporate structural elements of both the NA and SAM substrates. In this report, we describe the use of an entirely different strategy for the development of NNMT inhibitors. Specifically, we applied a peptide-mRNA display technology known as the random nonstandard peptide integrated discovery (RaPID) system to screen a library of more than 1012 macrocyclic peptides binding to NNMT. This mRNA display selection technique has demonstrated promising results against a variety of protein targets and, together with similar screening methods, has led to the identification of numerous peptide macrocycles currently in clinical trials.25–27 In the present study, hits from this RaPID screen against NNMT were identified, synthesized by Fmoc solid phase peptide synthesis (SPPS), and tested for inhibition using a convenient LC-MS-based NNMT activity assay previously developed in our group.2 Potent NNMT inhibition was found for a number of the macrocyclic peptides identified. Interestingly, this inhibition was found not to be impacted by elevated concentrations of either NA or SAM substrates suggesting that these peptides function as allosteric inhibitors, the first to be reported for NNMT. In cellular assays, the cyclic peptides showed a reduction in the concentration of MNA indicating a target-specific effect.
Results and discussion
To identify macrocyclic peptide binders for NNMT, two parallel selections employing the RaPID system for reprogrammed mRNA display28,29 were performed using purified, N-terminal His-tagged NNMT as the target (Fig. 1). The two selections differed only in the stereochemistry of the initiating amino acid, L-tyrosine or D-tyrosine, as a way of increasing the conformational space of the library. Both initiating tyrosine amino acids also carried an N-terminal chloroacetyl moiety to give spontaneous macrocyclisation30 with a cysteine hard-coded after a stretch of 15 random amino acids. This head-to-sidechain thioether cyclized library showed exponential enrichment of target-binding sequences over the course of 6 rounds. Hits were identified by high-throughput sequencing of the output DNA from each round (see Fig. S1 in ESI†). Within these hits, no clear consensus sequences were visible but it was clear that hydrophobic and positively charged amino acids were overall enriched (see Fig. S2 in ESI†). Based on the results of the two selections performed, 17 unique peptides were selected for chemical synthesis and assessment as NNMT inhibitors (Table 1).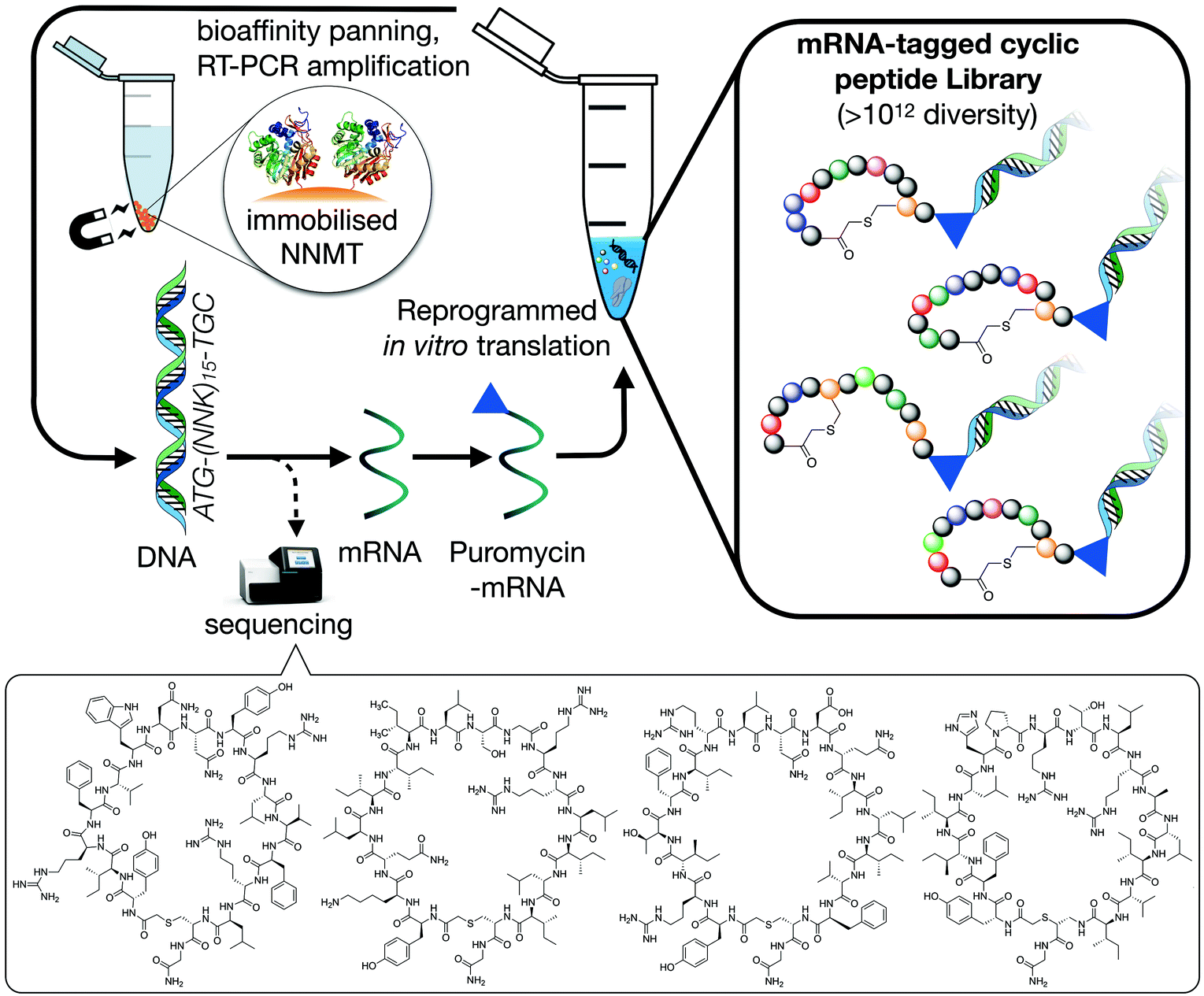 | ||
| Fig. 1 Schematic overview of the RaPID mRNA display system used to translate a random DNA library (>1012 library members, 17 residues), affording a large peptide library whose members were selected for binding affinity against NNMT. Selections initiated with either N-chloroacetyl-L-tyrosine or N-chloroacetyl-D-tyrosine were performed to introduce additional structural diversity in the library. The four cyclic peptides indicated at the bottom of the figure are the most potent NNMT inhibitors identified in the present study (see Table 1). | ||
| Peptide | Sequence | Abundancea (%) | IC50b (μM) |
|---|---|---|---|
| a Percentage of total sequences after the sixth round of enrichment. Peptides 1–8 originate from the selection initiated with L-tyrosine or and peptides 9–17 originate from the selection initiated with D-tyrosine. b Half-maximal inhibitory concentration of the compounds tested against human wild-type NNMT (full assay details provided in the ESI). Values reported in μM are based on triplicate data of at least 10 different concentrations. | |||
| 1 |

|
9.5 | 4.964 ± 0.509 |
| 2 |

|
1.4 | 1.584 ± 0.111 |
| 3 |

|
1.1 | 1.115 ± 0.066 |
| 4 |

|
1.1 | 0.772 ± 0.054 |
| 5 |

|
0.7 | 1.125 ± 0.076 |
| 6 |

|
0.6 | 0.674 ± 0.042 |
| 7 |

|
0.6 | 5.344 ± 0.432 |
| 8 |

|
0.5 | 2.238 ± 0.211 |
| 9 |

|
6.6 | 4.426 ± 0.428 |
| 10 |

|
6.241 ± 0.889 | |
| 11 |

|
5.7 | 1.131 ± 0.080 |
| 12 |

|
5.4 | 1.209 ± 0.113 |
| 13 |

|
2.6 | 0.241 ± 0.012 |
| 14 |

|
1.2 | 7.058 ± 0.530 |
| 15 |

|
1.1 | 0.437 ± 0.023 |
| 16 |

|
1.1 | 1.238 ± 0.100 |
| 17 |

|
0.9 | 0.229 ± 0.007 |
Using standard Fmoc-SPPS, the peptides were synthesized on rink amide resin as depicted in Scheme 1. The N-terminus of the linear peptide was subsequently treated with chloroacetyl chloride on the resin. After acidic cleavage and deprotection of the amino acid side chains, the peptides were cyclized in the presence of base and the macrocyclic peptides then purified using preparative HPLC. Notably, among the 17 peptides synthesized (Table 1) peptides 9 and 10 actually derive from the same sequence containing an additional Cys residue in the variable region of the sequence. For this reason, in peptides 9 and 10, one of the two Cys residues was replaced by Ala to allow for the controlled synthesis of a single macrocyclic species.
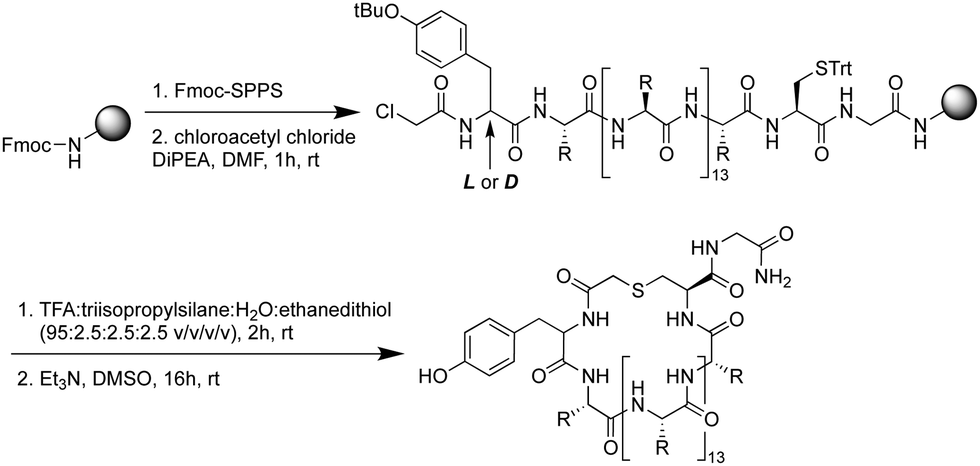 | ||
| Scheme 1 General synthesis route for the preparation of cyclic peptides identified from the mRNA display screen. The identified peptides identified all contain 18 amino acids (including a single additional C-terminal glycine for efficient synthesis). | ||
The cyclic peptides were subsequently tested for their inhibitory activity against NNMT using an LC-MS based method.2 The results given in Table 1 show that all 17 peptides identified and selected from the RaPID screenings demonstrate the capacity to inhibit NNMT. For 5 out of the 17 macrocyclic peptides potent inhibition (defined as an IC50 value below 1 μM) was observed (see ESI,† for full IC50 curves). Notably, no correlation could be found between the degree of enrichment in the RaPID selection and inhibitory activity. On the contrary, the most abundant peptides from the screen (peptides 1, 9 and 10) were found to be only moderate NNMT inhibitors with IC50 values around 5 μM. These findings suggested that the macrocyclic peptides may be interacting with NNMT at a site(s) not directly involved in the methylating activity of the enzyme.
To further investigate the mode of inhibition, macrocyclic peptide inhibitors with IC50 values below 2 μM were subsequently tested for competition with the substrate binding site(s). In this experiment, the concentration of either NA or SAM was increased 10-fold and the impact on the IC50 value of the macrocyclic peptide inhibitor determined. As can be seen in Fig. 2, none of the cyclic peptides saw a significant change in IC50 in the presence of elevated concentrations of either of the substrates. In comparison and as a control, two known bisubstrate inhibitors, compounds X21 and Y,20 previously reported to bind in the NNMT active site, were also included in the assay. In line with expectation, these bisubstrate NNMT inhibitors did show a marked increase in their IC50 values under higher concentrations of both substrates with a more pronounced competitive effect seen at increased SAM concentrations. These findings suggest that whereas the bisubstrate inhibitors bind in the NNMT active site, the cyclic peptides instead engage with NNMT at an allosteric binding site(s) and as such are not competitive with the NA and SAM substrates. To further assess the non-competitive mechanism of inhibition, the KM of SAM and Vmax of NNMT were determined in the presence of cyclic peptides 4 and 13 and bisubstrate inhibitor Y. The results of these investigations support the non-competitive mechanism of inhibition for cyclic peptides 4 and 13. Increasing concentrations of 4 or 13 had no significant effect on the KM value of SAM but did lead to a decrease in the Vmax of the enzyme. In contrast, the substrate competitive nature of compound Y was confirmed by the increased KM values observed with increasing inhibitor concentration but with no significant effect on Vmax (see Fig. S8 in the ESI†).
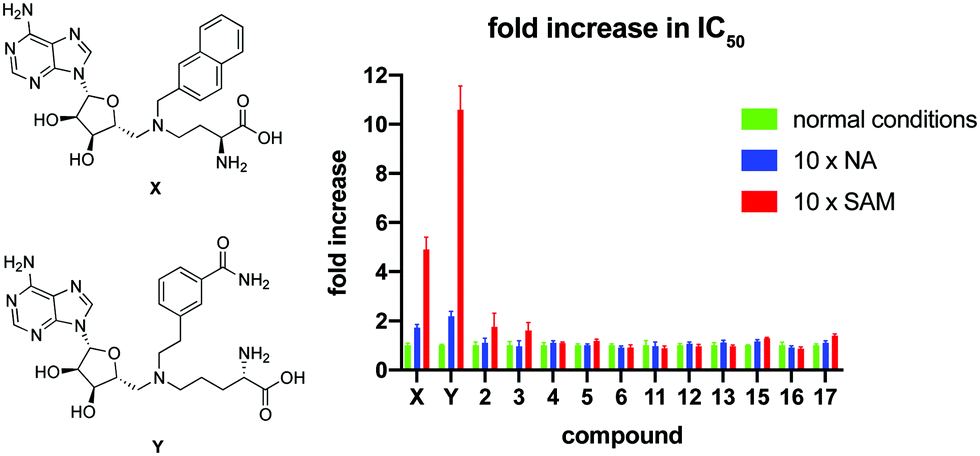 | ||
| Fig. 2 Results of the substrate competition experiment. The data is normalized per compound by setting the IC50 under normal assay conditions at 1. Data is based on duplicate data of at least 7 different concentrations. Structures of control compounds X and Y are presented on the left. Full IC50 curves are presented in Fig. S6 and S7 in the ESI.† | ||
Finally, the cyclic peptides were evaluated for their effect on MNA production in human aortic endothelial cells (HAEC) as well as in A549 lung carcinoma cells. Cyclic peptides 4 and 13 were selected as representative compounds with sequences initiating with either an L-tyrosine or a D-tyrosine residue, respectively. The recently published small molecule nicotinamide analogue 6-methylamino-nicotinamide (6-MANA), with a reported IC50 value of 588 ± 75 nM, was included as a reference compound.31 The compounds were incubated in the presence of 100 μM nicotinamide and 10 μM SAM for 1 h in A549 cells and 3 hours in HAEC cells. As illustrated in Fig. 3, the cyclic peptides produce a dose-dependent reduction of the concentration of MNA in both healthy cells and cancer cells. The observed effect is similar to that found for the small molecule reference inhibitor 6-MANA.
 | ||
| Fig. 3 Cellular activity of cyclic peptides 4 and 13 and small molecule reference compound 6-methylamino-nicotinamide (6-MANA) against A549 lung carcinoma cells (left) and human aortic endothelial cells (HAEC, right). Data is based on six replicates. The results indicate a significant reduction of MNA concentration compared to untreated cells. | ||
Conclusions
Using the RaPID mRNA display methodology, a set of macrocyclic peptides were identified with affinity for NNMT. While the hits identified from the RaPID selections did not reveal a clear consensus sequence, all peptides displayed a higher abundance of hydrophobic and positively charged amino acids. To investigate whether binding to NNMT resulted in the inhibition of its methylation activity, the most highly enriched cyclic peptides from both L-tyrosine and D-tyrosine initiating libraries were synthesized using Fmoc-SPPS and subsequently evaluated for their inhibitory activity against NNMT. From the screening hits, five macrocyclic peptides showed potent inhibition with IC50 values between 200 and 800 nM. Of note, while preparing our manuscript a publication appeared in the literature describing a similar strategy for generating peptidomimetic NNMT inhibitors.32 Interestingly, the most active peptides identified in that study share very little structural similarity to those identified in our investigations aside from the presence of a number of hydrophobic and positively charged residues. In addition, they appear to inhibit NNMT by binding to the enzyme active site as supported by structural studies. In contrast, none of the macrocyclic peptides identified in our study exhibit significant competition with the NNMT SAM or nicotinamide substrates, indicating that they may instead bind at an allosteric site on the enzyme. This is the first description of allosteric inhibitors of NNMT. Furthermore, in cell-based assays, administration of our macrocyclic peptides was found to result in a significant reduction in the production of MNA by endothelial HAEC cells and A549 lung carcinoma cells. To further elucidate the mode of binding and potential for further optimization of these macrocyclic peptide-based NNMT inhibitors, structural studies are currently being pursued, the results of which will be reported in due course.Experimental
Materials and methods
All reagents were purchased from Sigma Aldrich or Combi-blocks and used as received. HPLC-grade acetonitrile, peptide grade N,N-dimethylformamide (DMF) and dichloromethane (DCM) for peptide synthesis were purchased from Biosolve Chimie SARL and VWR, respectively. The ultrapure water was obtained from a Veolia Purelab flex3 water purification system. Standard Fmoc-protected amino acids and rink amide resin were purchased from P3 Biosystems.![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :5 (A/B) for 1 min, 95:5 to 5:95 (A/B) over 9 min, 5:95 to 2:98 (A/B) over 1 min, 2:98 (A/B) for 1 min, then reversion back to 95:5 (A/B) over 2 min, 95:5 (A/B) for 1 min. This system was connected to a Bruker micrOTOF-Q II mass spectrometer (ESI ionization) calibrated internally with sodium formate.
:5 (A/B) for 1 min, 95:5 to 5:95 (A/B) over 9 min, 5:95 to 2:98 (A/B) over 1 min, 2:98 (A/B) for 1 min, then reversion back to 95:5 (A/B) over 2 min, 95:5 (A/B) for 1 min. This system was connected to a Bruker micrOTOF-Q II mass spectrometer (ESI ionization) calibrated internally with sodium formate.
Reprogrammed mRNA display protocol
Acylation of N-chloroacetyl tyrosine (both L- and D-stereochemistry in separate reactions) onto tRNAfMetCAU was carried out using amino acids synthetically activated as cyanomethyl esters and in vitro transcribed tRNA and catalyst ‘enhanced flexizyme’ as previously reported,33,34 incubating for 2 hours on ice before purifying by ethanol precipitation and storing the dry pellet at −20 °C.Two parallel selections were carried out using His tag-immobilised NNMT based on a previously published method,35 one with D- and one with L-tyrosine initiation. Briefly, DNA encoding 15 randomised NNK codons followed by a section encoding a CGSGSGS linker was assembled by PCR, and subsequently transcribed in vitro using T7 RNA polymerase (NEB) at 1 mL scale with 25 pmol input DNA (starting diversity ∼1.5 × 1013) at 37 °C overnight. This was purified by PAGE, and the resulting library was ligated by T4 RNA ligase at room temperature for 30 min to a short oligonucleotide terminating in puromycin before purifying by ethanol precipitation with 0.25 mg mL−1 RNAse-free glycogen. Translation of 10 pmol of this puromycin-mRNA library in vitro using the PURExpress system (combining solution A from Δ (aa/tRNA) and solution B from Δ RF123 kits; NEB) was carried out in a 5 μL reaction at 37 °C for 30 min, with methionine omitted and with initiating acyl-tRNA added to 25 μM, gave a cyclic peptide library with covalent mRNA tag. Following translation, the reaction mix was allowed to stand at room temperature for 12 minutes, and then EDTA was added to 16.6 mM and the reaction mix was again incubated at 37 °C for 30 min. This was then reverse-transcribed by Protoscript II reverse transcriptase (NEB) at 42 °C for 1 hour, TBS-T and BSA added from concentrated stocks to 1× and 0.1% final concentrations (respectively), and a 0.5 μL sample diluted to 500 μL. Any background peptides and proteins that bind directly to Dynabeads His-tag isolation and pulldown resin (invitrogen) were removed by three sequential incubations with free resin (5, 2.5, and 2.5 μL) for 10 min at 4 °C. The supernatant following this last pre-clear step was added to immobilised NNMT saturating 0.4 μL of the same His-tag isolation and pulldown resin and incubated for 30 min at 4 °C with constant inversion. Nonspecifically-bound members of the library were removed by stringent washing on ice (3 × 20 μL TBS-T), and any surviving peptides were eluted by incubation in 50 μL RNAse free water at 95 °C for 5 min and then transferring the supernatant to a new tube while hot. The first aliquot of pre-clear beads was washed and eluted in the same way as the selection containing NNMT. All samples (1 μL each of input, positive, negative) were analysed by qPCR alongside a standard curve produced by reverse transcription of the input library, and recovery was calculated as the percentage of the input found in the positive or negative selection rounds after accounting for dilution factors. The remainder of the eluted specific binders were amplified by PCR to provide a new DNA template that served as the input for the subsequent round (round 1 carried out at double scale to increase initial diversity), and selection was continued until recovery indicated enrichment above the background negative selections (Fig. S1, ESI†).
The DNA output was used for sequencing of all rounds on the Illumina MiSeq platform using a 2 × 150 bp V2 reagent kit at the Utrecht UMC sequencing facility (USEQ). The resulting sequencing output files were analysed by Python script that searches for exact matches to the T7 promoter and puromycin ligation sequences, translates the coding sequence between these, and tallies at the peptide level the abundance of each unique hit.36
Fmoc-solid-phase peptide synthesis (SPPS)
:1 mixture of DMF/DCM for 5 min, drained, and then treated with 20 vol% piperidine (10 mL) in DMF for 65 seconds at 90 °C, drained and washed with DMF (3 × 5 mL). The resin was then treated with a solution of Fmoc-Xaa-OH (0.2 mol L−1, 2.5 mL, 5 eq.), DIC (1 mol L−1, 1 mL, 10 eq.) and Oxyma (1 mol L−1, 0.5 mL, 5 eq.) in DMF (4 mL) at 76 °C for 15 s before the temperature was increased to 90 °C for an additional 110 s before being drained. The resin was then treated again with the same amount of Fmoc-Xaa-OH, DIC and Oxyma in DMF (4 mL) at 76 °C for 15 s before the temperature was increased to 90 °C for an additional 110 s before being drained.
:2.5:2.5:2.5) under shaking for 2 hours at room temperature. The resin was filtered over cotton and washed with TFA (2 × 0.5 mL). The crude peptide was precipitated in a mixture of MTBE/Hexane (1:1). The peptide was pelleted by centrifugation (5 min at 4500 rpm), the pellet was washed twice with MTBE/Hexane (1:1) (50 mL), centrifiuged (5 min at 4500 rpm) and dried under a nitrogen flow.
Peptide 1. Rink Amide AM resin (146 mg, 100 μmol, 0.684 mmol g−1) was used for Fmoc solid phase peptide synthesis (Fmoc SPPS) according to general procedure A. After checking the crude peptide by LC-MS, a portion (25 μmol) of the peptide was capped with chloroacetyl chloride following general procedure B. The peptide was deprotected and cleaved from the resin according to general procedure C. Subsequently, the peptide was cyclized following general procedure D and purified by preparative HPLC (0–100%, buffer B) affording cyclic peptide 1 as a white solid (1.6 mg, 2.9%). HRMS (m/z): [M + 2H]2+ calculated for C103H173N29O21S2+, 1101.1468, found 1101.1462. LC-MS Rt 6.07 min (0 to 100% B over 12 min, 0.1% FA, λ = 214 nm).
Peptide 2. Rink Amide AM resin (146 mg, 100 μmol, 0.684 mmol g−1) was used for Fmoc solid phase peptide synthesis (Fmoc SPPS) according to general procedure A. After checking the crude peptide by LC-MS, a portion (25 μmol) of the peptide was capped with chloroacetyl chloride following general procedure B. The peptide was deprotected and cleaved from the resin according to general procedure C. Subsequently, the peptide was cyclized following general procedure D and purified by preparative HPLC (0–100%, buffer B) affording cyclic peptide 2 as a white solid (3.5 mg, 6.5%). HRMS (m/z): [M + 2H]2+ calculated for C101H171N31O22S2+,1092.1541, found 1092,1534. LC-MS Rt 5.57 min (0 to 100% B over 12 min, 0.1% FA, λ = 214 nm).
Peptide 3. Rink Amide AM resin (146 mg, 100 μmol, 0.684 mmol g−1) was used for Fmoc solid phase peptide synthesis (Fmoc SPPS) according to general procedure A. After checking the crude peptide by LC-MS, a portion (25 μmol) of the peptide was capped with chloroacetyl chloride following general procedure B. The peptide was deprotected and cleaved from the resin according to general procedure C. Subsequently, the peptide was cyclized following general procedure D and purified by preparative HPLC (0–100%, buffer B) affording cyclic peptide 3 as a white solid (4.2 mg, 7.8%). HRMS (m/z): [M + 2H]2+ calculated for C105H149N23O26S2+, 1090.0382, found 1090.0371. LC-MS Rt 7.01 min (0 to 100% B over 12 min, 0.1% FA, λ = 214 nm).
Peptide 4. Rink Amide AM resin (146 mg, 100 μmol, 0.684 mmol g−1) was used for Fmoc solid phase peptide synthesis (Fmoc SPPS) according to general procedure A. After checking the crude peptide by LC-MS, a portion (25 μmol) of the peptide was capped with chloroacetyl chloride following general procedure B. The peptide was deprotected and cleaved from the resin according to general procedure C. Subsequently, the peptide was cyclized following general procedure D and purified by preparative HPLC (0–100%, buffer B) affording cyclic peptide 4 as a white solid (3.6 mg, 6.7%). HRMS (m/z): [M + 2H]2+ calculated for C107H167N27O21S2+, 1069.1275, found 1069.1274. LC-MS Rt 7.01 min (0 to 100% B over 12 min, 0.1% FA, λ = 214 nm).
Peptide 5. Rink Amide AM resin (146 mg, 100 μmol, 0.684 mmol g−1) was used for Fmoc solid phase peptide synthesis (Fmoc SPPS) according to general procedure A. After checking the crude peptide by LC-MS, a portion (25 μmol) of the peptide was capped with chloroacetyl chloride following general procedure B. The peptide was deprotected and cleaved from the resin according to general procedure C. Subsequently, the peptide was cyclized following general procedure D and purified by preparative HPLC (0–100%, buffer B) affording cyclic peptide 5 as a white solid (5.1 mg, 9.1%). HRMS (m/z): [M + 2H]2+ calculated for C109H165N27O23S2+,1126.1146, found 1126.1140. LC-MS Rt 6.54 min (0 to 100% B over 12 min, 0.1% FA, λ = 214 nm).
Peptide 6. Rink Amide AM resin (146 mg, 100 μmol, 0.684 mmol g−1) was used for Fmoc solid phase peptide synthesis (Fmoc SPPS) according to general procedure A. After checking the crude peptide by LC-MS, a portion (25 μmol) of the peptide was capped with chloroacetyl chloride following general procedure B. The peptide was deprotected and cleaved from the resin according to general procedure C. Subsequently, the peptide was cyclized following general procedure D and purified by preparative HPLC (0–100%, buffer B) affording cyclic peptide 6 as a white solid (4.7 mg, 7.8%). HRMS (m/z): [M + 2H]2+ calculated for C117H167N31O24S2+,1211.1260, found 1211.1253. LC-MS Rt 6.35 min (0 to 100% B over 12 min, 0.1% FA, λ = 214 nm).
Peptide 7. Rink Amide AM resin (146 mg, 100 μmol, 0.684 mmol g−1) was used for Fmoc solid phase peptide synthesis (Fmoc SPPS) according to general procedure A. After checking the crude peptide by LC-MS, a portion (25 μmol) of the peptide was capped with chloroacetyl chloride following general procedure B. The peptide was deprotected and cleaved from the resin according to general procedure C. Subsequently, the peptide was cyclized following general procedure D and purified by preparative HPLC (0–100%, buffer B) affording cyclic peptide 7 as a white solid (6.5 mg, 12.2%). HRMS (m/z): [M + 2H]2+ calculated for C104H154N24O23S2+, 1069.5670, found 1069.5665. LC-MS Rt 5.90 min (0 to 100% B over 12 min, 0.1% FA, λ = 214 nm).
Peptide 8. Rink Amide AM resin (146 mg, 100 μmol, 0.684 mmol g−1) was used for Fmoc solid phase peptide synthesis (Fmoc SPPS) according to general procedure A. After checking the crude peptide by LC-MS, a portion (25 μmol) of the peptide was capped with chloroacetyl chloride following general procedure B. The peptide was deprotected and cleaved from the resin according to general procedure C. Subsequently, the peptide was cyclized following general procedure D and purified by preparative HPLC (0–100%, buffer B) affording cyclic peptide 8 as a white solid (5.7 mg, 10.4%). HRMS (m/z): [M + 2H]2+ calculated for C105H158N26O25S2+, 1107.5806, found 1107.5807. LC-MS Rt 5.91 min (0 to 100% B over 12 min, 0.1% FA, λ = 214 nm).
Peptide 9. Rink Amide AM resin (146 mg, 100 μmol, 0.684 mmol g−1) was used for Fmoc solid phase peptide synthesis (Fmoc SPPS) according to general procedure A. After checking the crude peptide by LC-MS, a portion (25 μmol) of the peptide was capped with chloroacetyl chloride following general procedure B. The peptide was deprotected and cleaved from the resin according to general procedure C. Subsequently, the peptide was cyclized following general procedure D and purified by preparative HPLC (0–100%, buffer B) affording cyclic peptide 9 as a white solid (4.9 mg, 9.1%). HRMS (m/z): [M + 2H]2+ calculated for C105H161N27O22S2+, 1092.1015, found 1092.1008. LC-MS Rt 6.07 min (0 to 100% B over 12 min, 0.1% FA, λ = 214 nm).
Peptide 10. Rink Amide AM resin (146 mg, 100 μmol, 0.684 mmol g−1) was used for Fmoc solid phase peptide synthesis (Fmoc SPPS) according to general procedure A. After checking the crude peptide by LC-MS, a portion (25 μmol) of the peptide was capped with chloroacetyl chloride following general procedure B. The peptide was deprotected and cleaved from the resin according to general procedure C. Subsequently, the peptide was cyclized following general procedure D and purified by preparative HPLC (0–100%, buffer B) affording cyclic peptide 10 as a white solid (5.4 mg, 10.2%). HRMS (m/z): [M + 2H]2+ calculated for C105H161N27O22S2+, 1092.1015, found 1092.1006. LC-MS Rt 6.18 min (0 to 100% B over 12 min, 0.1% FA, λ = 214 nm).
Peptide 11. Rink Amide AM resin (146 mg, 100 μmol, 0.684 mmol g−1) was used for Fmoc solid phase peptide synthesis (Fmoc SPPS) according to general procedure A. After checking the crude peptide by LC-MS, a portion (25 μmol) of the peptide was capped with chloroacetyl chloride following general procedure B. The peptide was deprotected and cleaved from the resin according to general procedure C. Subsequently, the peptide was cyclized following general procedure D and purified by preparative HPLC (0–100%, buffer B) affording cyclic peptide 11 as a white solid (6.1 mg, 10.8%). HRMS (m/z): [M + 2H]2+ calculated for C110H163N25O24S2+, 1125,1012, found 1125.1005. LC-MS Rt 6.67 min (0 to 100% B over 12 min, 0.1% FA, λ = 214 nm).
Peptide 12. Rink Amide AM resin (146 mg, 100 μmol, 0.684 mmol g−1) was used for Fmoc solid phase peptide synthesis (Fmoc SPPS) according to general procedure A. After checking the crude peptide by LC-MS, a portion (25 μmol) of the peptide was capped with chloroacetyl chloride following general procedure B. The peptide was deprotected and cleaved from the resin according to general procedure C. Subsequently, the peptide was cyclized following general procedure D and purified by preparative HPLC (0–100%, buffer B) affording cyclic peptide 12 as a white solid (4.4 mg, 8.5%). HRMS (m/z): [M + 2H]2+ calculated for C97H154N26O23S2+, 1041.5700, found 1041.5696. LC-MS Rt 6.59 min (0 to 100% B over 12 min, 0.1% FA, λ = 214 nm).
Peptide 13. Rink Amide AM resin (146 mg, 100 μmol, 0.684 mmol g−1) was used for Fmoc solid phase peptide synthesis (Fmoc SPPS) according to general procedure A. After checking the crude peptide by LC-MS, a portion (25 μmol) of the peptide was capped with chloroacetyl chloride following general procedure B. The peptide was deprotected and cleaved from the resin according to general procedure C. Subsequently, the peptide was cyclized following general procedure D and purified by preparative HPLC (0–100%, buffer B) affording cyclic peptide 13 as a white solid (5.2 mg, 10.0%). HRMS (m/z): [M + 2H]2+ calculated for C98H173N27O22S2+, 1056.1485, found 1056.1474. LC-MS Rt 6.21 min (0 to 100% B over 12 min, 0.1% FA, λ = 214 nm).
Peptide 14. Rink Amide AM resin (146 mg, 100 μmol, 0.684 mmol g−1) was used for Fmoc solid phase peptide synthesis (Fmoc SPPS) according to general procedure A. After checking the crude peptide by LC-MS, a portion (25 μmol) of the peptide was capped with chloroacetyl chloride following general procedure B. The peptide was deprotected and cleaved from the resin according to general procedure C. Subsequently, the peptide was cyclized following general procedure D and purified by preparative HPLC (0–100%, buffer B) affording cyclic peptide 14 as a white solid (4.7 mg, 8.7%). HRMS (m/z): [M + 2H]2+ calculated for C104H163N27O22S2+, 1087.1093, found 1087.1088. LC-MS Rt 5.64 min (0 to 100% B over 12 min, 0.1% FA, λ = 214 nm).
Peptide 15. Rink Amide AM resin (146 mg, 100 μmol, 0.684 mmol g−1) was used for Fmoc solid phase peptide synthesis (Fmoc SPPS) according to general procedure A. After checking the crude peptide by LC-MS, a portion (25 μmol) of the peptide was capped with chloroacetyl chloride following general procedure B. The peptide was deprotected and cleaved from the resin according to general procedure C. Subsequently, the peptide was cyclized following general procedure D and purified by preparative HPLC (0–100%, buffer B) affording cyclic peptide 15 as a white solid (5.9 mg, 10.6%). HRMS (m/z): [M + 2H]2+ calculated for C104H165N27O25S2+, 1112.1095, found 1112.1095. LC-MS Rt 6.87 min (0 to 100% B over 12 min, 0.1% FA, λ = 214 nm).
Peptide 16. Rink Amide AM resin (146 mg, 100 μmol, 0.684 mmol g−1) was used for Fmoc solid phase peptide synthesis (Fmoc SPPS) according to general procedure A. After checking the crude peptide by LC-MS, a portion (25 μmol) of the peptide was capped with chloroacetyl chloride following general procedure B. The peptide was deprotected and cleaved from the resin according to general procedure C. Subsequently, the peptide was cyclized following general procedure D and purified by preparative HPLC (0–100%, buffer B) affording cyclic peptide 16 as a white solid (4.7 mg, 9.0%). HRMS (m/z): [M + 2H]2+ calculated for C101H150N26O21S2+, 1047.5595, found 1047.5594. LC-MS Rt 6.10 min (0 to 100% B over 12 min, 0.1% FA, λ = 214 nm).
Peptide 17. Rink Amide AM resin (146 mg, 100 μmol, 0.684 mmol g−1) was used for Fmoc solid phase peptide synthesis (Fmoc SPPS) according to general procedure A. After checking the crude peptide by LC-MS, a portion (25 μmol) of the peptide was capped with chloroacetyl chloride following general procedure B. The peptide was deprotected and cleaved from the resin according to general procedure C. Subsequently, the peptide was cyclized following general procedure D and purified by preparative HPLC (0–100%, buffer B) affording cyclic peptide 17 as a white solid (5.6 mg, 10.9%). HRMS (m/z): [M + 2H]2+ calculated for C93H156N28O23S2+, 1032.5809, found 1032.5797. LC-MS Rt 6.01 min (0 to 100% B over 12 min, 0.1% FA, λ = 214 nm).
Inhibition studies
Expression and purification of full-length human wild-type NNMT protein (hNNMTwt) were performed as previously described.37 The purity of the enzyme was confirmed using sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) with Coomassie blue staining, and NNMT identity was confirmed using SDS-PAGE and Western blotting. Catalytic activity of the recombinant protein was evaluated with 1 unit of enzyme activity representing the formation of 1 nmol of MNA/h of incubation at 37 °C. The specific activity of the batch used in the inhibitory activity assays was 15064 units per mg of protein at a protein concentration of 8.4 mg mL−1. NNMT was used at a final concentration of 50 nM diluted in assay buffer (50 mM Tris buffer (pH 8.4) and 1 mM dithiothreitol). The compounds were dissolved in DMSO and diluted with water to concentrations ranging from 1 nM to 100 μM (DMSO was kept constant at 1.25% final concentration). The compounds were incubated with the enzyme for 10 min at room temperature before initiating the reaction with a mixture of NA and SAM at their KM values of 200 and 8.5 μM, respectively. The formation of MNA was measured after 30 min at room temperature. The reaction was quenched by addition of 30 μL of the sample to 70 μL of acetonitrile containing 50 nM deuteromethylated nicotinamide as internal standard. The enzymatic activity assays were performed using Multiple Reaction Monitoring (MRM) on a Shimadzu LC-20AD system with a Waters Acquity BEH Amide HILIC column (3.0 × 100 mm, 1.7 μm particle size, Waters, Milford) at 65 °C using water containing 300 mM formic acid and 550 mM NH4OH (pH 9.2) at 40% v/v and acetonitrile at 60% v/v isocratically at a flowrate of 0.6 mL min−1, with a runtime of 1.7 min. Calibration samples were prepared using 70 μL of internal standard d3-MNA at 50 nM in acetonitrile and 30 μL of an aqueous solution of reference standard MNA with concentrations ranging from 1 to 1024 nM. Ratios of the sums of the MNA and d3-MNA transitions were used to calculate concentrations of MNA. Concentrations of MNA were plotted against concentration of inhibitor and the results were subsequently normalized with the highest value in the concentration range defined as 100% inhibition. The percentage of inhibitory activity was plotted as a function of inhibitor concentration and fit using non-linear regression analysis of the sigmoidal dose–response curve generated using the normalized data and a variable slope in Graphpad Prism 8. IC50 curves are presented in Fig. S3–S5 in the ESI.†
Cell-based assays
Human aortic endothelial cell line (HAEC, ATCC, VA, USA) and human lung adenocarcinoma line (A549, ATCC, VA, USA) were cultured according to the provider's indications and seeded in 6-well or 24-well format. After 24 h-stabilization, when cells reached about 100% confluence, culture medium was removed and cells were pre-incubated for 1 h in normal Hank's buffer (HBSS). After the buffer change, cells were treated with NNMT peptide inhibitors or reference compound at concentrations of 1 and 10 μM. The compounds were incubated in the presence of nicotinamide (100 μM) and S-adenosyl-L-methionine 10 μM (Sigma Aldrich, MO, USA) for 3 hours in HAEC cells and for 1 hour in A549 cells. Effluent samples were collected after incubation and frozen (−80 °C) for further measurement of exogenous MNA. Cells were collected using scraper, centrifuged (2 × 500 G/5 min) and frozen for BCA protein assay.MNA measurement in buffer samples
The quantification of 1-methylnicotinamide (MNA) was performed using ultra-pressure liquid chromatography coupled to tandem mass spectrometry (UPLC-MS) according to previously described methodology with minor modifications.15 A UPLC-MS system comprised of an UPLC Ultimate 3000 (Dionex, Thermo Scientific, USA) connected to a TSQ Quantum Ultra mass spectrometer (Thermo Scientific, USA) equipped with a heated electrospray ionization interface (HESI-II Probe) was used. Chromatographic separation of analytes was carried out on an Aquasil C18 analytical column (4.6 mm × 150 mm, 5 μm; Thermo Scientific) under isocratic elution using acetonitrile with 0.1% of formic acid (A) and 5 mM ammonium formate in water (B) as mobile phases delivered at the flow rate of 0.8 ml min−1 (A:B, 80:20, v/v). 50 μl of effluent sample was used for the measurement of exogenous MNA. The internal standard (IS) containing MNA-d3 was added to each sample (5 μL) obtaining the final concentration of 500 ng mL−1. After sample mixing, the proteins were precipitated using 100 μL of acidified acetonitrile (0.1% of formic acid), and samples were mixed (10 min), cooled at 4 °C (15 min) and finally centrifuged (15 000 x g, 15 min, 4 °C). A clear supernatant was transferred to a chromatographic vial and directly injected (5 μL) into UPLC-MS system. The mass spectrometer was operated in the positive ionisation using selected reactions monitoring (SRM) mode monitoring the following ion transitions: m/z 137 → 94 for MNA and 140 → 97 for MNA-d3. The concentration of MNA was calculated based on the calibration curve plotted for the analyte as the relationship between the peak area ratios of analyte/IS to the nominal concentration of the analyte. The concentration of analytes was normalized to mg of proteins, which was assessed using PierceTM BCA Protein Assay Kit (Thermo Fisher, Waltham, MA, USA) and Synergy4 multiplate reader (BioTek, Winooski, VT, USA). MNA was obtained from Sigma-Aldrich (MO, USA). Deuterated standard MNA-d3 was synthesized by Dr Adamus (Technical University, Lodz, Poland). LC-MS–grade acetonitrile, ammonium formate and formic acid were purchased from Sigma-Aldrich. Ultrapure water was obtained from a Millipore system (Direct-Q 3UV).
Author contributions
NIM and SJ designed the research. MvH, YZ, DS, RC, ME, SC, SJ, and NIM designed the experiments. YZ, MvH, VT, NB, YG, LM, FF, AK and RC performed the experiments and analyzed the data. MvH, SJ, NIM wrote the manuscript. All authors read and approved the final manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank Utrecht Sequencing Facility for providing sequencing service and data. Utrecht Sequencing Facility is subsidized by the University Medical Center Utrecht, Hubrecht Institute, Utrecht University and The Netherlands X-omics Initiative (NWO project 184.034.019). Y. Zhang and Y. Gao are supported by the China Scholarship Council (CSC file numbers 201706210082 and 201506270162).Notes and references
- T. A. Alston and R. H. Abeles, Substrate specificity of nicotinamide methyltransferase isolated from porcine liver, Arch. Biochem. Biophys., 1988, 260, 601–608 CrossRef CAS.
- M. J. van Haren, J. Sastre Toraño, D. Sartini, M. Emanuelli, R. B. Parsons and N. I. Martin, A Rapid and Efficient Assay for the Characterization of Substrates and Inhibitors of Nicotinamide N-Methyltransferase, Biochemistry, 2016, 55, 5307–5315 CrossRef CAS PubMed.
- O. A. Ulanovskaya, A. M. Zuhl and B. F. Cravatt, NNMT promotes epigenetic remodeling in cancer by creating a metabolic methylation sink, Nat. Chem. Biol., 2013, 9, 300–306 CrossRef CAS.
- M. A. Eckert, F. Coscia, A. Chryplewicz, J. W. Chang, K. M. Hernandez, S. Pan, S. M. Tienda, D. A. Nahotko, G. Li, I. Blaženović, R. R. Lastra, M. Curtis, S. D. Yamada, R. Perets, S. M. McGregor, J. Andrade, O. Fiehn, R. E. Moellering, M. Mann and E. Lengyel, Proteomics reveals NNMT as a master metabolic regulator of cancer-associated fibroblasts, Nature, 2019, 569, 723–728 CrossRef CAS.
- A. Kanakkanthara, K. Kurmi, T. L. Ekstrom, X. Hou, E. R. Purfeerst, E. P. Heinzen, C. Correia, C. J. Huntoon, D. O’Brien, A. E. Wahner Hendrickson, S. C. Dowdy, H. Li, A. L. Oberg, T. Hitosugi, S. H. Kaufmann, S. J. Weroha and L. M. Karnitz, BRCA1 Deficiency Upregulates NNMT, Which Reprograms Metabolism and Sensitizes Ovarian Cancer Cells to Mitochondrial Metabolic Targeting Agents, Cancer Res., 2019, 79, 5920–5929 CrossRef CAS.
- Y. Xu, P. Liu, D.-H. Zheng, N. Wu, L. Zhu, C. Xing and J. Zhu, Expression profile and prognostic value of NNMT in patients with pancreatic cancer, Oncotarget, 2016, 7, 19975–19981 CrossRef.
- C. Chen, X. Wang, X. Huang, H. Yong, J. Shen, Q. Tang, J. Zhu, J. Ni and Z. Feng, Nicotinamide N-methyltransferase: a potential biomarker for worse prognosis in gastric carcinoma, Am. J. Cancer Res., 2016, 6, 649–663 CAS.
- Y. Wang, J. Zeng, W. Wu, S. Xie, H. Yu, G. Li, T. Zhu, F. Li, J. Lu, G. Y. Wang, X. Xie and J. Zhang, Nicotinamide N-methyltransferase enhances chemoresistance in breast cancer through SIRT1 protein stabilization, Breast Cancer Res., 2019, 21, 1–17 CrossRef.
- S. Holstein, S. Venz, H. Junker, R. Walther, M. B. Stope and U. Zimmermann, Nicotinamide N-methyltransferase and its precursor substrate methionine directly and indirectly control malignant metabolism during progression of renal cell carcinoma, Anticancer Res., 2019, 39, 5427–5436 CrossRef CAS.
- M. Mascitti, A. Santarelli, D. Sartini, C. Rubini, G. Colella, E. Salvolini, G. Ganzetti, A. Offidani and M. Emanuelli, Analysis of nicotinamide N-methyltransferase in oral malignant melanoma and potential prognostic significance, Melanoma Res., 2019, 29, 151–156 CrossRef CAS PubMed.
- R. B. Parsons, M.-L. Smith, A. C. Williams, R. H. Waring and D. B. Ramsden, Expression of Nicotinamide N-Methyltransferase (E.C. 2.1.1.1) in the Parkinsonian Brain, J. Neuropathol. Exp. Neurol., 2002, 61, 111–124 CrossRef CAS PubMed.
- R. B. Parsons, S. W. Smith, R. H. Waring, A. C. Williams and D. B. Ramsden, High expression of nicotinamide N-methyltransferase in patients with idiopathic Parkinson's disease, Neurosci. Lett., 2003, 342, 13–16 CrossRef CAS.
- L. Mateuszuk, A. Jasztal, E. Maslak, M. Gasior-Glogowska, M. Baranska, B. Sitek, R. Kostogrys, A. Zakrzewska, A. Kij, M. Walczak and S. Chlopicki, Antiatherosclerotic effects of 1-methylnicotinamide in apolipoprotein e/low-density lipoprotein receptor-deficient mice: A comparison with nicotinic acid, J. Pharmacol. Exp. Ther., 2016, 356, 514–524 CrossRef CAS PubMed.
- R. Campagna, Ł. Mateuszuk, K. Wojnar-Lason, P. Kaczara, A. Tworzydło, A. Kij, R. Bujok, J. Mlynarski, Y. Wang, D. Sartini, M. Emanuelli and S. Chlopicki, Nicotinamide N-methyltransferase in endothelium protects against oxidant stress-induced endothelial injury, Biochim. Biophys. Acta, Mol. Cell Res., 2021, 184, 119082 CrossRef.
- A. Fedorowicz, Ł. Mateuszuk, G. Kopec, T. Skórka, B. Kutryb-Zając, A. Zakrzewska, M. Walczak, A. Jakubowski, M. Łomnicka, E. Słomińska and S. Chlopicki, Activation of the nicotinamide N-methyltransferase (NNMT)-1-methylnicotinamide (MNA) pathway in pulmonary hypertension, Respir. Res., 2016, 17, 108 CrossRef PubMed.
- S. Brachs, J. Polack, M. Brachs, K. Jahn-Hofmann, R. Elvert, A. Pfenninger, F. Bärenz, D. Margerie, K. Mai, J. Spranger and A. Kannt, Genetic Nicotinamide N-Methyltransferase (NNMT) Deficiency in Male Mice Improves Insulin Sensitivity in Diet-Induced Obesity but Does Not Affect Glucose Tolerance, Diabetes, 2019, 68, 527–542 CrossRef CAS PubMed.
- D. Kraus, Q. Yang, D. Kong, A. S. Banks, L. Zhang, J. T. Rodgers, E. Pirinen, T. C. Pulinilkunnil, F. Gong, Y. Wang, Y. Cen, A. A. Sauve, J. M. Asara, O. D. Peroni, B. P. Monia, S. Bhanot, L. Alhonen, P. Puigserver and B. B. Kahn, Nicotinamide N-methyltransferase knockdown protects against diet-induced obesity, Nature, 2014, 508, 258–262 CrossRef CAS.
- R. Giuliante, C. Polidori, R. Rocchetti, D. Sartini, M. Emanuelli, I. Klöting, T. Bacchetti and G. Ferretti, Potential Involvement of Nicotinamide N -Methyltransferase in the Pathogenesis of Metabolic Syndrome, Metab. Syndr. Relat. Disord., 2015, 13, 165–170 CrossRef CAS.
- M. J. Van Haren, R. Taig, J. Kuppens, J. Sastre Toraño, E. E. Moret, R. B. Parsons, D. Sartini, M. Emanuelli and N. I. Martin, Inhibitors of nicotinamide: N -methyltransferase designed to mimic the methylation reaction transition state, Org. Biomol. Chem., 2017, 15, 6656–6667 RSC.
- N. Babault, A. Allali-Hassani, F. Li, J. Fan, A. Yue, K. Ju, F. Liu, M. Vedadi, J. Liu and J. Jin, Discovery of Bisubstrate Inhibitors of Nicotinamide N -Methyltransferase (NNMT), J. Med. Chem., 2018, 61, 1541–1551 CrossRef CAS.
- Y. Gao, M. J. van Haren, E. E. Moret, J. J. M. Rood, D. Sartini, A. Salvucci, M. Emanuelli, P. Craveur, N. Babault, J. Jin and N. I. Martin, Bisubstrate Inhibitors of Nicotinamide N -Methyltransferase (NNMT) with Enhanced Activity, J. Med. Chem., 2019, 62, 6597–6614 CrossRef CAS PubMed.
- R. L. Policarpo, L. Decultot, E. May, P. Kuzmič, S. Carlson, D. Huang, V. Chu, B. A. Wright, S. Dhakshinamoorthy, A. Kannt, S. Rani, S. Dittakavi, J. D. Panarese, R. Gaudet and M. D. Shair, High-Affinity Alkynyl Bisubstrate Inhibitors of Nicotinamide N -Methyltransferase (NNMT), J. Med. Chem., 2019, 62, 9837–9873 CrossRef CAS PubMed.
- D. Chen, L. Li, K. Diaz, I. D. Iyamu, R. Yadav, N. Noinaj and R. Huang, Novel Propargyl-Linked Bisubstrate Analogues as Tight-Binding Inhibitors for Nicotinamide N-Methyltransferase, J. Med. Chem., 2019, 62, 10783–10797 CrossRef CAS.
- A. Kannt, S. Rajagopal, M. S. Hallur, I. Swamy, R. Kristam, S. Dhakshinamoorthy, J. Czech, G. Zech, H. Schreuder and S. Ruf, Novel Inhibitors of Nicotinamide-N-Methyltransferase for the Treatment of Metabolic Disorders, Molecules, 2021, 26, 991 CrossRef CAS.
- A. Zorzi, K. Deyle and C. Heinis, Cyclic peptide therapeutics: past, present and future, Curr. Opin. Chem. Biol., 2017, 38, 24–29 CrossRef CAS PubMed.
- C. Morrison, Constrained peptides’ time to shine?, Nat. Rev. Drug Discovery, 2018, 17, 531–533 CrossRef CAS PubMed.
- A. A. Vinogradov, Y. Yin and H. Suga, Macrocyclic Peptides as Drug Candidates: Recent Progress and Remaining Challenges, J. Am. Chem. Soc., 2019, 141, 4167–4181 CrossRef CAS PubMed.
- T. Passioura and H. Suga, A RaPID way to discover nonstandard macrocyclic peptide modulators of drug targets, Chem. Commun., 2017, 53, 1931–1940 RSC.
- C. J. Hipolito and H. Suga, Ribosomal production and in vitro selection of natural product-like peptidomimetics: The FIT and RaPID systems, Curr. Opin. Chem. Biol., 2012, 16, 196–203 CrossRef CAS.
- Y. Goto, A. Ohta, Y. Sako, Y. Yamagishi, H. Murakami and H. Suga, Reprogramming the translation initiation for the synthesis of physiologically stable cyclic peptides, ACS Chem. Biol., 2008, 3, 120–129 CrossRef CAS.
- S. Ruf, M. S. Hallur, N. K. Anchan, I. N. Swamy, K. R. Murugesan, S. Sarkar, L. K. Narasimhulu, V. P. R. K. Putta, S. Shaik, D. V. Chandrasekar, V. S. Mane, S. V. Kadnur, J. Suresh, R. K. Bhamidipati, M. Singh, R. R. Burri, R. Kristam, H. Schreuder, J. Czech, C. Rudolph, A. Marker, T. Langer, R. Mullangi, T. Yura, R. Gosu, A. Kannt, S. Dhakshinamoorthy and S. Rajagopal, Novel nicotinamide analog as inhibitor of nicotinamide N-methyltransferase, Bioorg. Med. Chem. Lett., 2018, 28, 922–925 CrossRef CAS PubMed.
- K. Hayashi, S. Uehara, S. Yamamoto, D. R. Cary, J. Nishikawa, T. Ueda, H. Ozasa, K. Mihara, N. Yoshimura, T. Kawai, T. Ono, S. Yamamoto, M. Fumoto and H. Mikamiyama, Macrocyclic Peptides as a Novel Class of NNMT Inhibitors: A SAR Study Aimed at Inhibitory Activity in the Cell, ACS Med. Chem. Lett., 2021, 12, 1093–1101 CrossRef CAS.
- Y. Goto, T. Katoh and H. Suga, Flexizymes for genetic code reprogramming, Nat. Protoc., 2011, 6, 779–790 CrossRef CAS PubMed.
- H. Murakami, A. Ohta, H. Ashigai and H. Suga, A highly flexible tRNA acylation method for non-natural polypeptide synthesis, Nat. Methods, 2006, 3, 357–359 CrossRef CAS.
- Y. Hayashi, J. Morimoto and H. Suga, In vitro selection of anti-Akt2 thioether-macrocyclic peptides leading to isoform-selective inhibitors, ACS Chem. Biol., 2012, 7, 607–613 CrossRef CAS PubMed.
- S. A. K. Jongkees, S. Caner, C. Tysoe, G. D. Brayer, S. G. Withers and H. Suga, Rapid discovery of potent and selective glycosidase-inhibiting de novo peptides, Cell Chem. Biol., 2017, 24, 381–390 CrossRef CAS PubMed.
- Y. Peng, D. Sartini, V. Pozzi, D. Wilk, M. Emanuelli and V. C. Yee, Structural Basis of Substrate Recognition in Human Nicotinamide N -Methyltransferase, Biochemistry, 2011, 50, 7800–7808 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Details of the RaPID screens, structures and analytical data of cyclic peptides and IC50 curves. See DOI: 10.1039/d1cb00134e |
| ‡ Denotes equal contribution. |
| This journal is © The Royal Society of Chemistry 2021 |