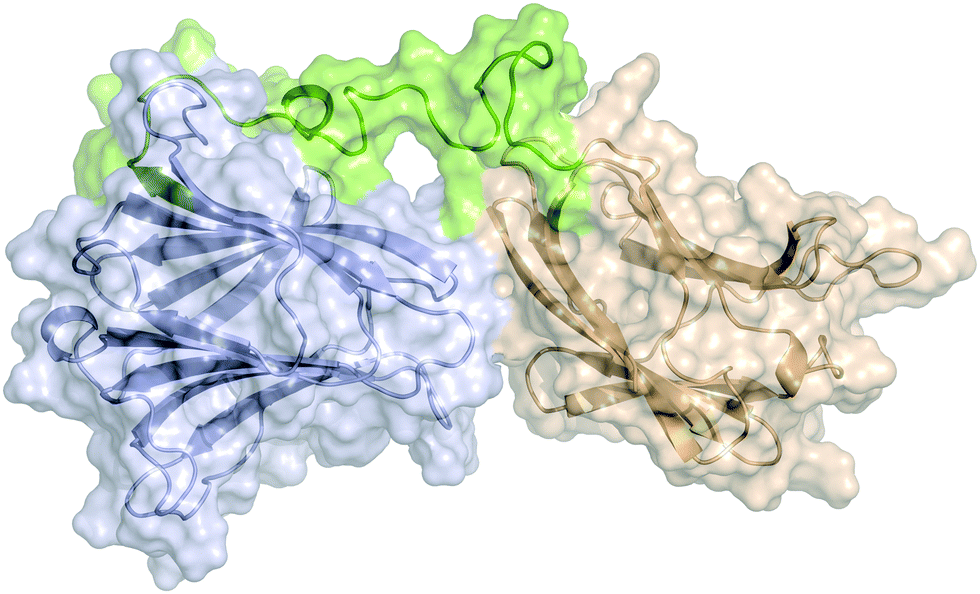
The two domains of human galectin-8 bind sialyl- and fucose-containing oligosaccharides in an independent manner. A 3D view by using NMR†
Received
11th March 2021
, Accepted 10th April 2021
First published on 12th April 2021
Abstract
The interaction of human galectin-8 and its two separate N-terminal and C-terminal carbohydrate recognition domains (CRD) to their natural ligands has been analysed using a synergistic combination of experimental NMR and ITC methods, and molecular dynamics simulations. Both domains bind the minimal epitopes N-acetyllactosamine (1) and Galβ1–3GalNAc (2) in a similar manner. However, the N-terminal and C-terminal domains show exquisite and opposing specificity to bind either Neu5Ac- or Fuc-containing ligands, respectively. Moreover, the addition of the high-affinity ligands specific for one of the CRDs does not make any effect on the binding at the alternative one. Thus, the two CRDs behave independently and may simultaneously target different molecular entities to promote clustering through the generation of supramolecular assemblies.
Introduction
Galectins are an extensive family of lectins all characterized by their affinity to bind β-galactosides.1,2 Galectins display a multitude of biological functions in the extracellular and intercellular compartments, mostly mediated by their multimeric or oligomerization nature.3,4 From the structural perspective, galectins are classified in three separate groups according to their structure and domain organization: prototype galectins (-1, -2, -5, -7, -10, -11 -13, -14, and -15) which exist as homodimers, chimera-type Gal-3 composed of a single C-terminal domain bound to a short non-lectin N-terminal linker, and tandem-repeat galectins (-4, -6, -8, -9, -12) with two non-identical carbohydrate recognition domains (CRD) linked together by a short peptide linker.5 Due to the bivalent nature of tandem-repeat galectins and the oligomerization tendency of prototype and chimera-types, galectins are able to cross-link glycosylated receptors on the cell surface and form supramolecular structures.6 Given their involvement in a variety of pathologies, several galectins have been identified as potential targets for drug discovery projects.7 Among galectins, galectin-8 (Gal-8) is expressed across a wide number of organs in the human body (including lung, kidney, spleen, liver and heart).8 It has been suggested that its critical role for mediating bacterial infection and autophagy is related, at the molecular level, with its bivalent nature.9 Previous functional and structural analysis have shown that the N- and C-terminal domains of Gal-8 display different carbohydrate specificity.10,11 Indeed, the N-terminal CRD (Gal-8N) preferentially binds sulfated and sialylated saccharides, whereas the C-terminal domain (Gal-8C), mostly recognizes polylactosamine, histo-blood group antigen-like structures and binds to the autophagic receptor NDP52 in the opposite side of the carbohydrate-binding site.9,12–14 Noteworthy, glycan arrays showed that the heterodimer, full-length Gal-8 (Gal-8FL), binds a broader scope of structures than their isolated CRDs.15 Altogether these results strongly suggest the importance of analysing the whole protein structure when studying the biological functions of Gal-8.16 Despite this, its structural analysis has been limited to mutant forms in which the peptide linker between both domains was mostly eliminated and replaced with a histidine-methionine spacer.9,17 Herein, we report a comprehensive structural analysis of the binding of Gal-8FL to its natural ligands using a synergistic combination of NMR, ITC experiments and molecular dynamics simulations. Our results show that both CRDs of Gal-8FL bind minimal epitopes such as N-acetyllactosamine (LacNAc, 1) or Galβ1-3GalNAc (2) in a similar manner. In contrast, they show exquisite and opposing specificity to bind either Neu5Ac- or Fuc-containing ligands, such as 3 and 5–6, respectively. Moreover, the addition of ligands which are specific for one of the CRDs did not provide any effect at the alternative CRD. Thus, these results experimentally demonstrate, for the first time, that both Gal-8FL CRDs behave independently in terms of binding affinity and specificity and, therefore, they may target different entities simultaneously to promote clustering through the formation of large supramolecular entities.
Results and discussion
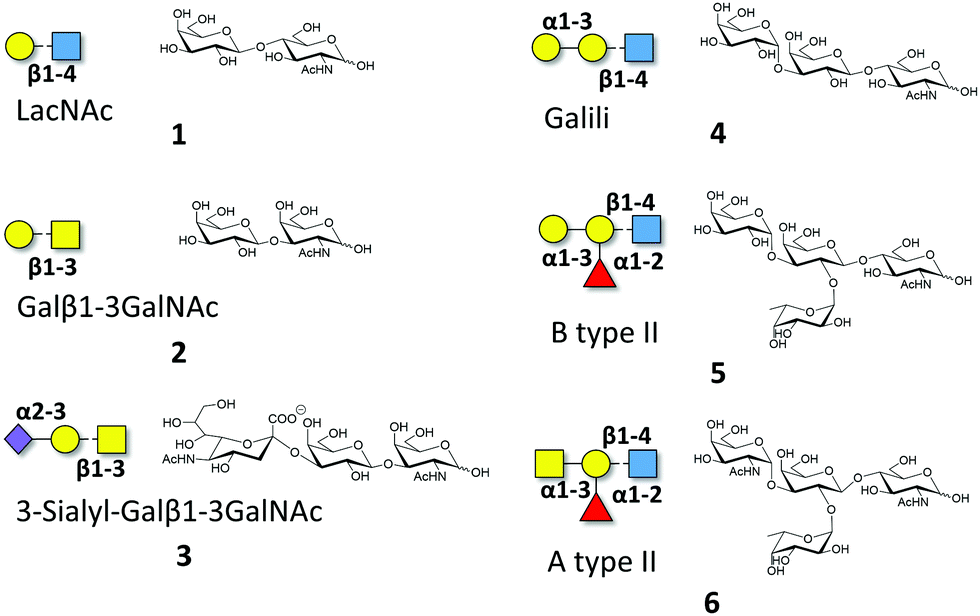
Macroscopic methods, such as glycan arrays, fluorescence anisotropy and frontal affinity chromatography have been previously employed to deduce the different binding specificities of the two separate Gal-8 CRDs, highlighting the potential of Gal-8FL to trigger clustering effects in glycan arrays.11,15 However, a detailed structural and chemical analysis of the corresponding binding processes involving complex ligands for Gal-8FL remains absent. Thus, the interaction between the full-length protein and a repertoire of β-galactose-containing saccharides was systematically analyzed to provide a solid framework to understand the recognition process. Gal-8FL and its two isolated CRDs were employed as lectins and a variety of chemical entities (1–6) displaying diverse epitopes and binding affinities were used as ligands (Fig. 1). In particular, blood group antigens 5 and 6 were selected as preferred ligands of Gal-8C, and their minimal epitopes 4 and 1 were analyzed for comparison. In contrast, ligand 2 and its sialylated counterpart 3 were picked out as higher selective Gal-8N ligands, due to the preference of the N-terminal CRD for Galβ1–3GalNAc vs. LacNAc.15
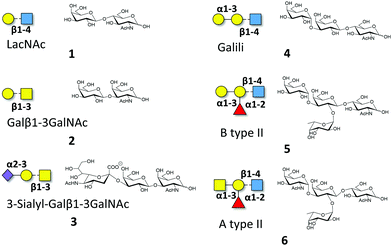 |
| | Fig. 1 Schematic representation of the oligosaccharides whose interaction with Gal-8FL is described herein. | |
Isothermal titration calorimetry
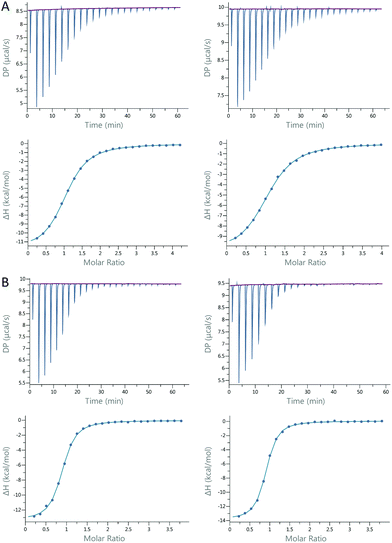
ITC experiments were carried out to determine the binding constants and thermodynamic profiles of the interaction between Gal-8 and ligands 1–6 (Table 1).18 All experiments were performed for Gal-8FL and the single N- and C-CRDs. The affinities are in the low micromolar range, with LacNAc (1) and Galβ1–3GalNAc (2) being the exceptions. The affinity of LacNAc (1) for both CRDs was very similar and remained almost unaltered in the full-length form (ΔG = −4.22 ± 0.01 kcal mol−1). Galβ1–3GalNAc 2 showed similar affinities for Gal-8N, Gal-8C and Gal8-FL (ΔG = −4.96 ± 0.06 kcal mol−1). In contrast, the Galili trisaccharide (4), bearing an extra galactose α1–3-linked to type II LacNAc, binds significantly stronger (ca. 1 kcal mol−1) to Gal-8C and Gal-8FL, with the affinity towards Gal-8N remaining somewhat identical to that of LacNAc (ΔΔG = −0.2 kcal mol−1). Interestingly, the introduction of the sialyl moiety present in 3 resulted in a much increased affinity (ca. 3 kcal mol−1) for Gal-8N in comparison with its unsialylated analog 2, whereas it prevented the binding to Gal-8C. Contrarily, histo-blood group antigen B type II (5), with an additional α1–2Fuc linked to Galili trisaccharide, displayed a striking rise in affinity (>1 kcal mol−1) towards Gal-8C. Similar results were obtained with HBGA A type II (6). Interestingly, no interaction was shown for either of the blood group antigens to Gal-8N. Of note, 3, 5 and 6, which are highly specific for Gal-8N or Gal-8C, bind to Gal-8FL with similar affinities to those measured for its corresponding individual domains, thus reflecting the specific binding to just one single CRD. In fact, almost identical thermodynamic signatures were obtained for the binding between the 3-sialyl-Galβ1-3GalNAc 3 or the BGA A type II 6 and glycan-pre-loaded Gal-8FL species using 6 or 3, respectively (Fig. 2 and Table 1). These results confirm the high specificity of both N- and C-CRDs of Gal-8FL and suggest that the two binding sites in the full-length protein do not show any cooperativity.
Table 1
K
D and thermodynamics parameters for the interactions of ligands 1–6 with Gal-8FL, Gal-8 N and Gal-8C, as determined by ITC. Average values and standard deviations are indicated. The thermodynamic parameters for the binding of ligands 3 and 6 to Gal-8FL glycan pre-loaded species (runs in which one CRD of Gal-8FL was saturated with its high-affinity ligand, 3 and 6 for the N- and C-CRD, respectively, and the interaction of the opposite CRD with 6 and 3, respectively, was measured) are indicated in parenthesis. The low binding affinity of Gal-8C, Gal-8N and Gal-8FL towards LacNAc, Galβ1–3GalNAc and Galili prevented the accurate analysis of the data. Thus, N = 1 or N = 2 were employed as fixed values. ΔH and −TΔS contributions were not estimated for these ligands24
|
|
LacNAc (1) |
Galβ1–3GalNAc(2) |
Neu5Acα2–3Galb1–3GalNAc (3) |
Galili (4) |
B type II (5) |
A type II (6) |
| Gal8-FL |
K
D (μM) |
804 ± 2 |
229 ± 24 |
4.3 ± 0.4 (3.0 ± 0.2) |
165 ± 4/68 ± 2 |
36.7 ± 0.6 |
12 ± 1 (17 ± 1) |
| ΔG (kcal mol−1) |
−4.22 ± 0.01 |
−4.96 ± 0.06 |
−7.33 ± 0.05 (−7.52 ± 0.03) |
5.16 ± 0.4/−5.68 ± 0.01 |
−6.05 ± 0.01 |
−6.71 ± 0.05 (−6.50 ± 0.04) |
| ΔH (kcal mol−1) |
— |
— |
−13.5 ± 0.1 (−14.1 ± 0.1) |
— |
−10.9 ± 0.2 |
−11.9 ± 0.2 (−10.7 ± 0.4) |
| −TΔS (kcal mol−1) |
— |
— |
6.2 ± 0.2 (6.6 ± 0.2) |
— |
4.8 ± 0.2 |
5.1 ± 0.1 (4.20 ± 0.01) |
|
n
|
2 |
2 |
0.92 ± 0.06 (0.77 ± 0.08) |
1/2 |
1.36 ± 0.02 |
1.09 ± 0.05 (1.07 ± 0.02) |
|
|
| Gal-8N |
K
D (μM) |
621 ± 43 |
186 ± 5 |
2.2 ± 0.1 |
444 ± 34 |
— |
— |
| ΔG (kcal mol−1) |
−4.38 ± 0.04 |
−5.08 ± 0.01 |
−7.72 ± 0.04 |
−4.57 ± 0.04 |
— |
— |
| ΔH (kcal mol−1) |
— |
— |
−13 ± 1 |
— |
— |
— |
| −TΔS (kcal mol−1) |
— |
— |
5 ± 1 |
— |
— |
— |
|
n
|
1 |
1 |
0.90 ± 0.09 |
1 |
— |
— |
|
|
| Gal-8C |
K
D (μM) |
844 ± 135 |
348 ± 216 |
— |
115.5 ± 0.5 |
18.4 ± 0.1 |
13.45 ± 0.05 |
| ΔG (kcal mol−1) |
−4.02 ± 0.09 |
−4.9 ± 0.3 |
— |
−5.37 ± 0.01 |
−6.46 ± 0.01 |
−6.65 ± 0.01 |
| ΔH (kcal mol−1) |
— |
— |
— |
— |
−10.7 ± 0.1 |
−11.2 ± 0.1 |
| −TΔS (kcal mol−1) |
— |
— |
— |
— |
4.3 ± 0.1 |
4.6 ± 0.1 |
|
n
|
1 |
1 |
— |
1 |
1.26 ± 0.03 |
1.13 ± 0.01 |
 |
| | Fig. 2 (A) Energetic profile of the binding of A type II (6) to the C-CRD of Gal-8FL (left) compared to the binding of A type II (6) to the C-CRD in the presence of 3-sialyl-Galβ1–3GalNAc (3) in the N-CRD of Gal-8FL (right). (B) Energetic profile of the binding of 3-sialyl-Galβ1–3GalNAc (3) to the N-CRD of Gal-8FL (left) compared to the binding of 3-sialyl-Galβ1–3GalNAc (3) to the N-CRD in the presence of A type II (6) in the C-CRD of Gal-8FL. | |
NMR and molecular recognition
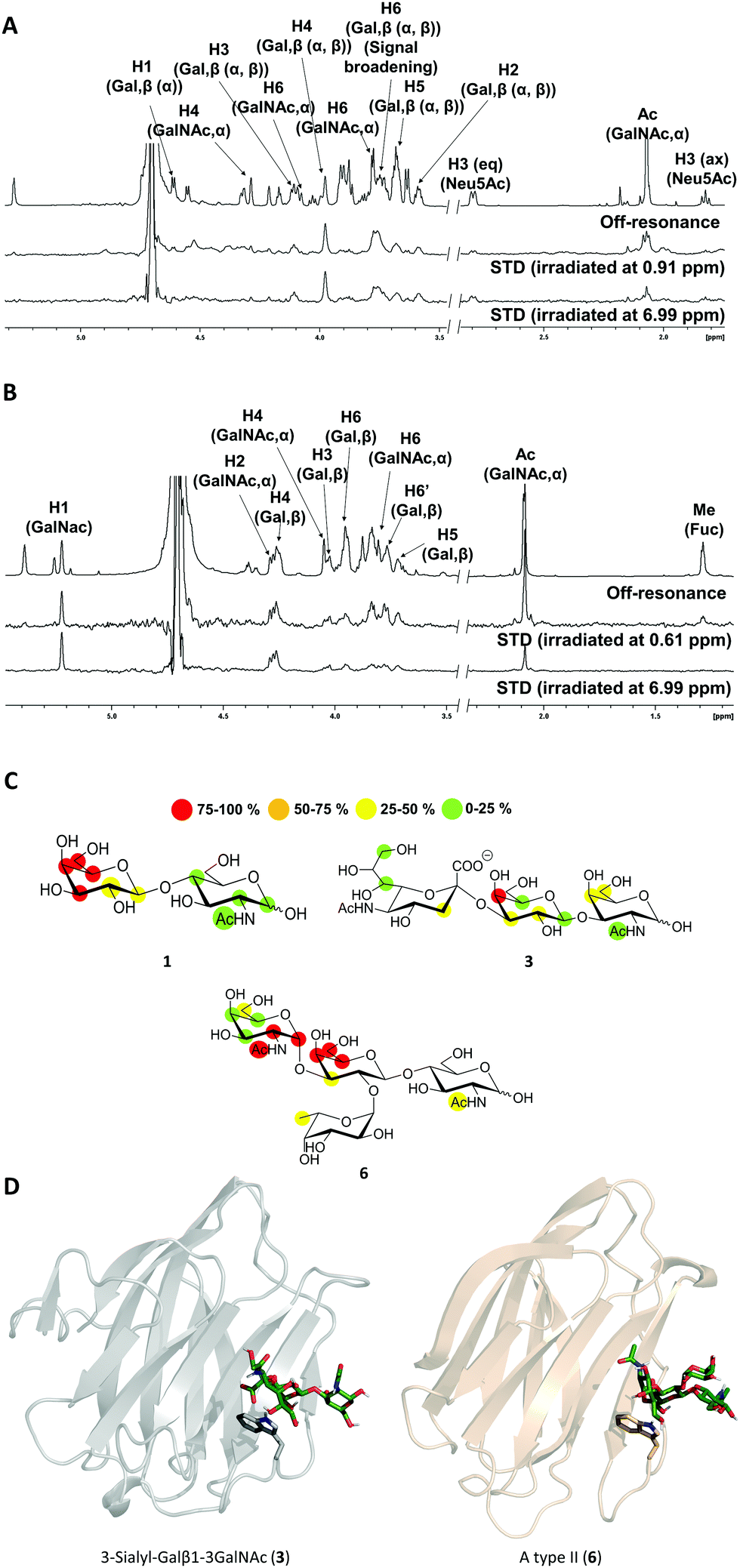
The high flexibility and instability to protease activity of the full-length protein has prevented crystallization studies of Gal-8FL so far.19 In fact, only the structural details of truncated Gal-8FL forms, lacking the peptide linker between both domains, have been reported.9,17 Although these mutants resulted active and bind to β-galactosides, the interplay between the binding to both subdomains was not examined. Thus, in order to investigate the binding features of Gal8-FL and to provide insights into the possible cooperativity between the two domains, the molecular recognition event was analyzed by using NMR experiments assisted by MD simulations.20 Particularly, the interaction of Gal-8FL with its high-affinity ligands 3 and 6, together with LacNAc 1, the canonical ligand for galectins, was analyzed by ligand-based and protein-based NMR experiments. For 1, the simple analysis of broadening effects of the sugar signals in the 1H-NMR spectrum in the presence of the protein, coupled with 1H-STD-NMR experiments, was sufficient to determine the ligand binding epitope.21 In fact, substantial signal broadening was observed for H3, H4 and H5 of the β-galactose ring. In a similar fashion, signals for H3, H4 and H5 showed up in the 1H-STD-NMR spectrum, although in this case higher temperatures were required to increase the on–off exchange and in turn, to enhance the intensity of STD signals (Fig. 3).22
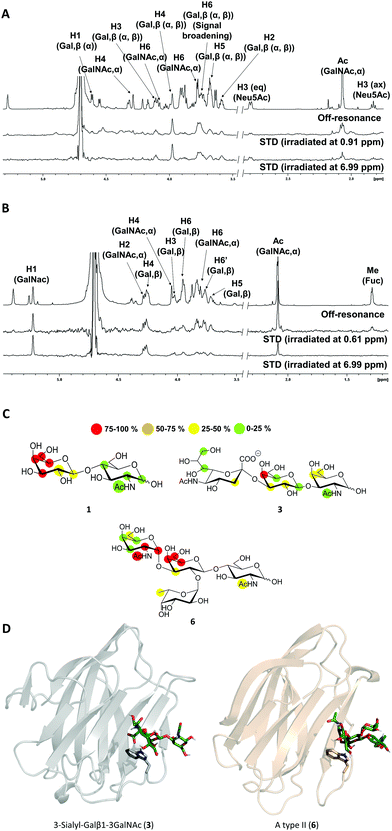 |
| | Fig. 3 (A and B) 1H-STD-NMR spectra of ligands 3 and 6, respectively. First spectrum: reference spectrum with annotation for those signals showing STD intensity; second spectrum: STD spectrum with irradiation of the protein on the aliphatic region; third spectrum: STD spectrum with irradiation of the protein on the aromatic region. (C) Epitope mapping for ligands 1, 3 and 6 determined by 1H-STD NMR experiments. (D) 3D models for Gal-8N/3 and Gal-8C/6 complexes. | |
These results were in complete agreement with the canonical binding mode shared for all galectins towards Lac/LacNAc moieties, and more specifically with the X-ray crystallographic data described for the complexes between Gal-8N and Gal-8FL with lactose (pdb entries 3AP412 and 3VKL,17 respectively), where the β-galactose unit, on the top of a Trp residue, establishes the main interactions with the lectin.23 Similar results were obtained for 3, where both signal broadening (H4 and H6) and STD signals (H2, H3, H4 and H5) were detected for the central β-galactose (Fig. 3). Additionally, STD intensities were observed for H4 and H6 of the β-GalNAc moiety as well as for H3s of αNeu5Ac, showing their close proximity to the protein surface. Indeed, weak STD signals were detected for H7–H9 of Neu5Ac suggesting that only the exo–syn conformation of Neu5Acα2–3Galβ linkage was populated in the bound state. As described for 1 and 3, the histo-blood group antigen 6 showed STD signals for the central β-Gal (H4, H5 and H6) and for αGalNAc (H1 and H2). In contrast, no STD signals were detected for the Fuc moiety (Fig. 3). Thus, the results obtained so far allow describing the binding of these saccharides to Gal8-FL as a simple bimolecular process, where β-galactosides 1, 3 and 6 interact with the CRDs of the protein following a lactose-type binding mode.
Thus, in light of our NMR experimental data, initial 3D models of the interaction of 3 and 6 with the individual Gal-8 CRDs were built. The ligands were docked in the binding site of the protein by pair-fitting to lactose (crystal structures: PDB ID 3AP4 for Gal-8N and PDB IDs 3OJB and 3VKL for Gal-8C) and subjected to molecular dynamics (MD) simulations (Fig. 3D).
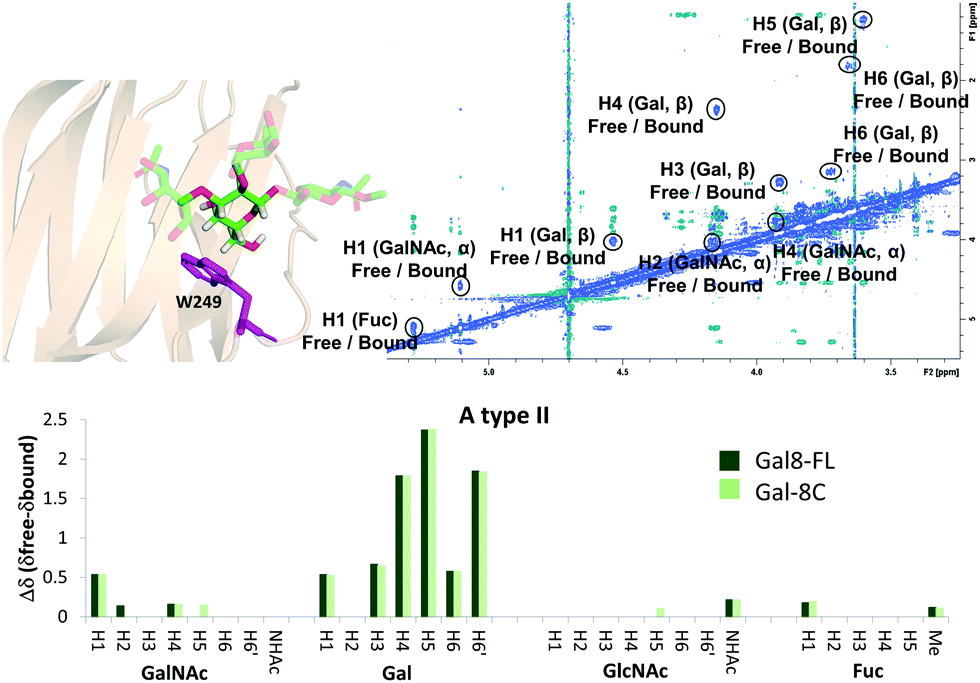
The binding mode described above was also supported by 2D-trROESY experiments.25 Fittingly, for the complexes formed between Gal-8FL and ligands 3 and 6, the chemical exchange process between the free and bound sugars was under the slow-exchange regime in the chemical shift time scale. Thus, 2D-trROESY experiments resulted ideal to reveal the chemical shifts of the protons of bound 3 and 6, allowing to guess their chemical environment in the bound state. For ligand 3, a striking up-field shift for H4 of the β-galactose in the bound-state was detected. This fact is in full agreement with the binding mode described above, where the β-galactose residue lies on the top of W86. Moreover, H1 of GalNAc and H3s of Neu5Ac also showed important chemical shift perturbations, strongly supporting their proximity to the lectin in the bound state. Interestingly, H3ax and H3eq of Neu5Ac showed opposite chemical shift perturbation, nicely demonstrating their precise and disparate chemical environment in the bound state. For ligand 6 (A type II), H4, H5 and H6 of the central β-galactose were the most up-field shifted in the presence of Gal-8FL, while smaller changes were detected for other protons of the molecule (Fig. 4). Interestingly, tr-ROESY experiments of ligands 3 and 6, employing either Gal-8C or Gal-8N isolated domains, respectively, offered almost identical results (see ESI†), suggesting that the binding kinetics of these high-affinity ligands remained domain-independent in Gal-8FL.
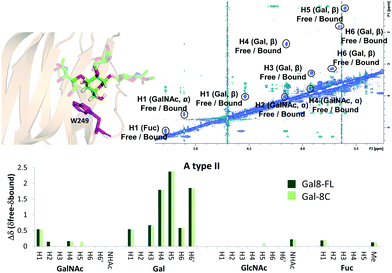 |
| | Fig. 4 Above: 2D-NMR ROESY spectrum of Gal-8FL![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :A type II (1:10). A molecular model of the interaction, highlighting W249 and the β-Gal residue was included. Below: comparison between the chemical shifts obtained for the interaction of ligand 6 with Gal-8FL and Gal-8C. :A type II (1:10). A molecular model of the interaction, highlighting W249 and the β-Gal residue was included. Below: comparison between the chemical shifts obtained for the interaction of ligand 6 with Gal-8FL and Gal-8C. | |
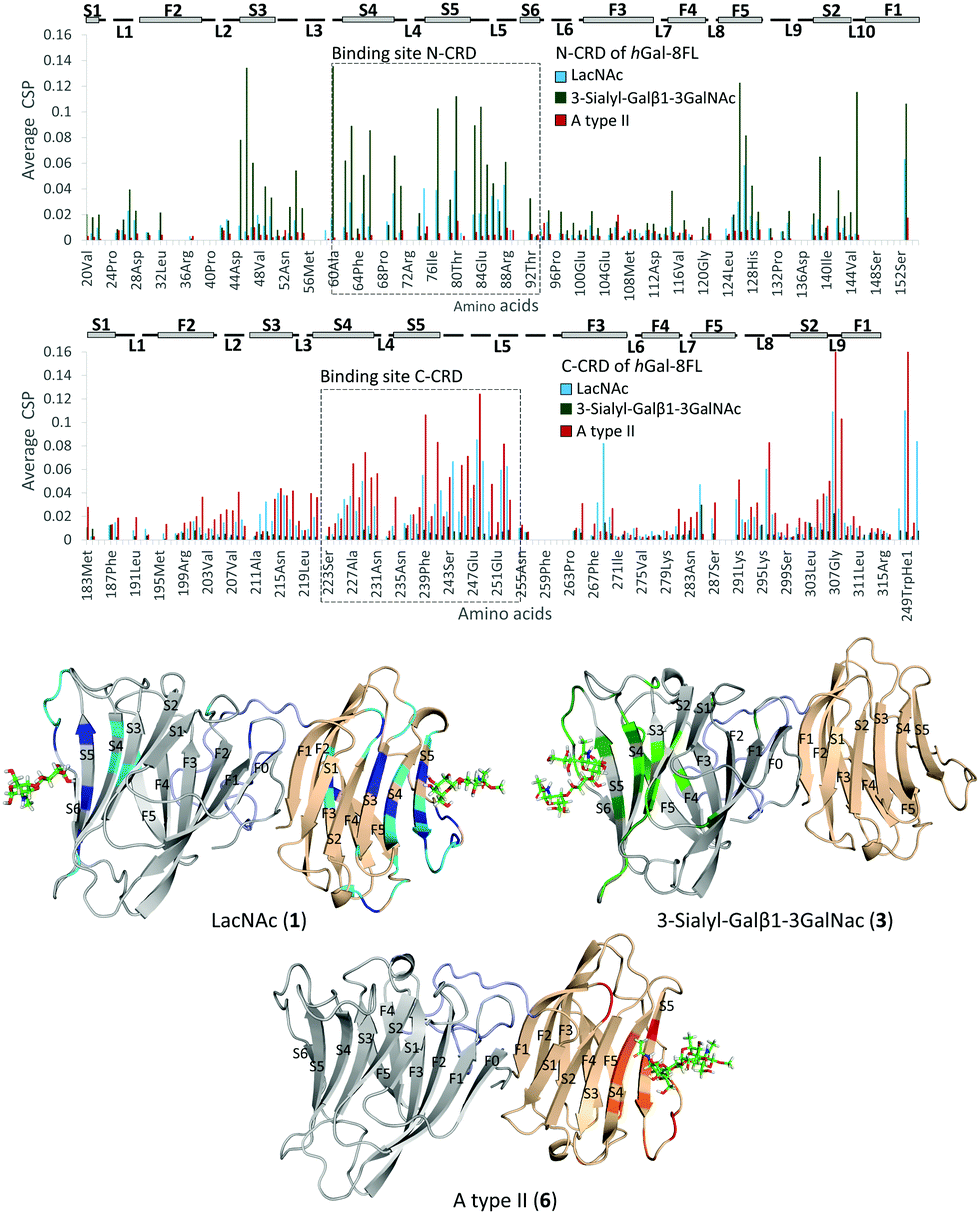
Additional NMR experiments were performed from the point of view of the protein to obtain complementary structural information of the complexes.26 Although Gal-8FL (30kDa) is a medium-size protein, signal overlapping and broadening was remarkably low at 308 K and 15N-HSQC experiments allowed monitoring the chemical-shift perturbation (CSP) induced upon ligand binding (Fig. 5).27 Indeed, the 15N-HSQC spectra of Gal-8FL and their CRDs, Gal-8N and Gal-8C, were almost completely superimposable (see ESI,† for additional details), thus showing the similarities in the chemical environment and dynamics of both CRDs in the isolated and full-length isoforms. Upon systematic addition of 1 (from 0.25 to 60 equivalents), a progressive CSP of the 1H–15N cross-peaks of the NH amides of Gal-8FL was observed, a common trend for ligands in the fast exchange regime. These results show that LacNAc 1 binds to Gal-8FL in the high μM–mM affinity range, in agreement with the KD (804 μM) determined by ITC experiments. Analysis of the CSP showed that the most perturbed residues are those located in protein regions comprising both the N- and C-CRD binding sites, thus supporting the fact that LacNAc 1 binds indiscriminately to both CRDs of Gal-8FL. Among the N-CRD residues, the largest changes were detected for residues in strands S4, S5 and loop L5. Further CSP were observed for amino acids away from the binding site, such as those on F5, L2 and the loop connecting with the linker. Similarly, the most perturbed residues of the C-CRD comprised those belonging to the binding site, more specifically in strands S3–S5 and loops L5 and L9. A completely different behavior was observed for ligands 3 and 6.
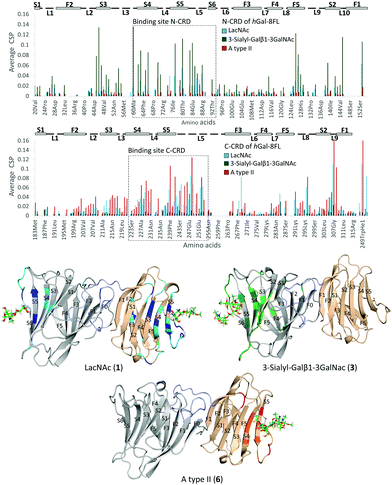 |
| | Fig. 5 Top, CSP analysis for 1 (blue, 60 equivalents), 3 (green, 10 equivalents) and 6 (red, 8 equivalents). Below, 3D models for the interaction of Gal-8FL with 1, top-left; 3, top-right; 6, below. The most perturbed residues are highlighted in dark (perturbated >2σ) and light (perturbated >σ) colors. | |
In both cases, 10 equivalents of ligand or less were sufficient to saturate the protein and broadening of signals was clearly noticeable at intermediate ligand amounts (see ESI,† Fig. S10 and S11). This behavior clearly pointed to binding events in the intermediate-slow exchange regime in the chemical shift timescale, in agreement with the higher binding affinity determined by ITC experiments. Interestingly, the CSP observed for 3 was definitely limited to N-CRD residues. This observation clearly supports the exclusive binding of 3 to the N-CRD, without altering other protein regions. In particular, amino acids in strands S4–S6 and L5 loop, certainly involved in the recognition of the β-galactose residue, showed clear CSP. Additionally, residues in strands S3–S2 and loops L3 and L10 were significantly perturbed, accounting for the accommodation of the sialyl moiety. Indeed, the interaction of Gal8 N-term with the glycans of the receptor binding domain of SARS CoV2 has been shown to involve the sialyl epitope.28 These results, together with the ligand-based NMR experiments29 described above, highlight the direct participation of the Neu5Ac moiety in lectin binding. Noteworthy, binding specificity completely switched in the case of the histo-blood group antigen A type II 6. In this case, the observed CSP were constrained to the C-CRD. As described for 1, these CSP mainly involved amino acids in strands S4–S5 and L5 loop. Further changes were also detected for amino acids on L9, thus reflecting the presence of the GalNAcα epitope in 6 and its direct involvement in the recognition process.
MD Simulations of Gal-8FL in complex with ligands 1, 3 and 6
Atomistic models for full-length Gal-8 (Gal-8FL) in the apo and bound states with ligands 1, 3, and 6 were constructed from the X-ray structures of N- and C-terminal CRDs and an extended peptide linker connecting both domains (see ESI†). Molecular dynamics (MD) simulations were performed on these models to yield an accumulated simulation time of 100 μs (see ESI†). In all cases, unstructured folding of the flexible linker peptide was observed. The most representative structure of these simulations obtained after clustering analysis is depicted in Fig. 6. This structure is highly conserved in the apo and bound states. A systematic analysis of all the interactions between the peptide linker and each CRD in apo Gal-8FL revealed the regions in close contact with the linker (Fig. S12, ESI†). The N-terminal CRD showed a larger number of highly dynamic interactions with the peptide linker. However, and applying the methodology previously reported by our group to analyse allosteric networks,30 it was observed that the correlated motion of binding residues quickly dissipated nearby each binding sites without affecting the interdomain peptide (Fig. S14, ESI†). In agreement with the measured CSP, the residues most frequently involved in the calculated allosteric pathways (i.e. the residues mostly affected by ligand binding) are concentrated in the β-strands at the canonical binding sites of N- and C-CRDs of Gal-8FL. Thus, the results support the independent behaviour of both domains in Gal-8FL.
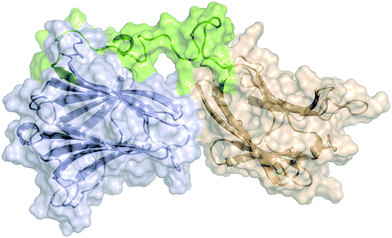 |
| | Fig. 6 Model structure for Gal-8FL representing the most populated cluster (67%) derived from MD simulations over an accumulated time of 100 μs. N-terminal and C-terminal CRDs are shown in blue and light brown, respectively. The interdomain peptide linker is shown in green. Ligands have been omitted for clarity. | |
Simulations (consisting on many independent replicas) were long enough to witness ligand unbinding events, which in turn can be related to binding affinity (i.e. ligands with higher affinity would remain bound longer throughout the simulation). Thus, the relative binding affinities for 1, 3 and 6, both in absence and presence of other ligands simultaneously bound to the adjacent domain, were qualitatively estimated from the fraction of the total simulation time in which each ligand is bound in its canonical binding pose to the corresponding CRD (Table 2). Such calculated ratios were compared to the experimental dissociation constants (KD) measured by ITC. 3-Sialyl-Galβ1–3GalNAc 3 was experimentally determined to have the smallest dissociation constant and the highest specificity towards the N-terminal domain. Accordingly, this ligand stays the longest bound to this domain. As a negative control, 3 escaped almost immediately when the binding sites for each ligand were exchanged, attesting the observed regioselectivity. Regarding the C-term CRD, 6 presents a higher experimental affinity than LacNAc, which is reproduced by its consistently larger residence time in the MD simulations. LacNAc is known to bind both CRDs although with lower affinities, showing a slightly higher preference towards the N-terminal domain. The calculated residence times of this ligand bound to the N- and C-terminal domains, also agree with the experimentally observed trend.
Table 2 MDs Residence Times. The residence time is defined as the fraction of simulation time in which the ligand is bound in its canonical binding pose before leaving the active site accumulated through 20 μs (except for the negative control system, Gal-8 N-term:6-C-term:3, for which the accumulated time is 5 μs)
| Ligand |
MDs Residence time |
| N-terminal domain |
C-terminal domain |
N-term |
C-term |
| LacNAc (1) |
LacNAc (1) |
40% |
13% |
|
Apo
|
A type II (6) |
— |
45% |
| 3-Sialyl-Galβ1-3GalNAc (3) |
apo
|
66% |
— |
| 3-Sialyl Galβ1-3GalNAc (3) |
A type II (6) |
77% |
25% |
| Negative control: |
| A type II (6) |
3-Sialyl-Galβ1-3GalNAc (3) |
32% |
1% |
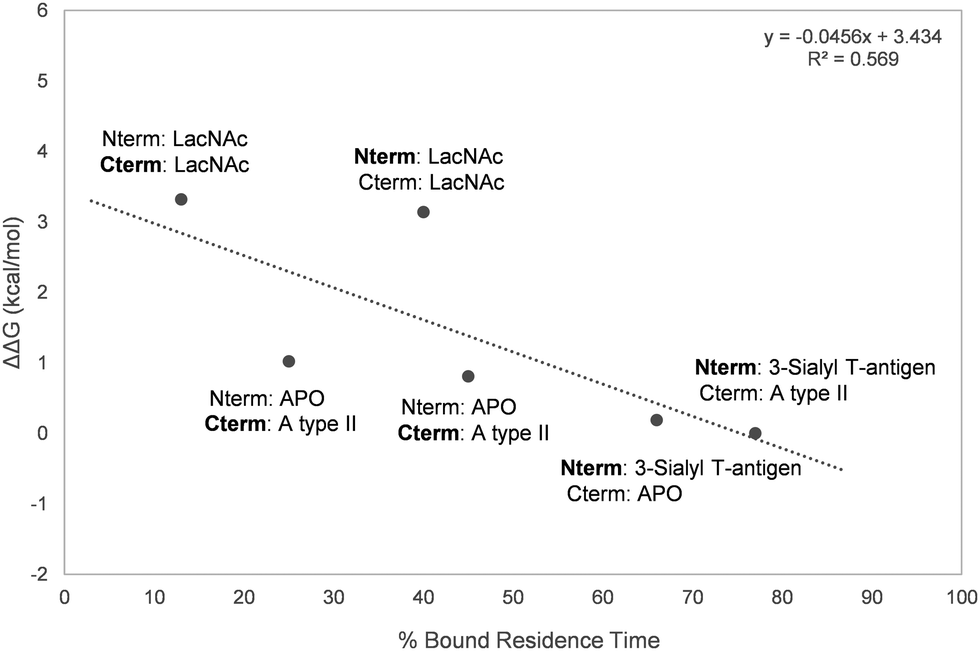
Overall, a moderate correlation between experimental relative ΔG of binding and the calculated residence times was found (Fig. 7), proving the ability of MD simulations to satisfactorily estimate the relative binding affinities of the studied ligands to Gal-8.
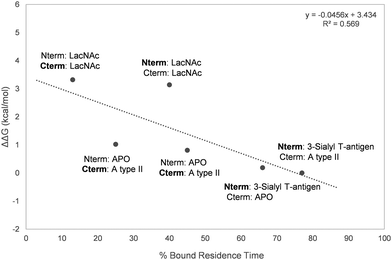 |
| | Fig. 7 Representation of the percentage of bound residence time versus the experimentally measured ΔΔG. The domain for which the residence time of the bound ligand is calculated is marked in bold. | |
Conclusions
The two, N-terminal and C-terminal, domains of the full-length human galectin-8, show an excellent and divergent specificity to bind different saccharide antigens. Our results demonstrate that the N-terminal domain nicely binds sialyl containing saccharides, such as the 3-sialyl-Galβ1–3GalNAc and does not recognize the histo-blood group A or B saccharides. In contrast, the C-terminal domain displays a complete opposite trend, as demonstrated by detailed NMR investigations, both from the ligand and lectin perspective, combined with ITC measurements and complemented with MD simulations. The independence of both CRDs in the full-length protein is fully sustained by our results, especially when saturation at one binding site (N-term or C-term) does not produce any effect at the alternative one (C-term or N-term). Thus, the two domains interact with their ligands in an autonomous manner and may bind to diverse molecular entities in a synchronised mode to foster clustering and generating supramolecular assemblies.
Experimental
Ligands
Compounds 1 (LacNAc), 2 (Galβ1–3GalNAc) and 3 (3-sialyl-Galβ1–3GalNAc) were purchased from Carbosynth (references OA08244, OA01686 and, OS09250 respectively). Compounds 4 (Galili), 5 (histo blood B type II antigen) 6 (A type II) were purchased from Elycityl (references GLY074-2, GLY038-2 and GLY035-2).
Expression of unlabelled Gal8-FL, Gal-8N and Gal-8C
Each gene encoding either full-length isoform 1 (1–317 amino acids) or the carbohydrate recognition domains (N-terminal CRD, 1–155 amino acids and C-terminal CRD, 184–317) of human Gal-8 were inserted into a pET21b expression vector. Transformation of BL21 (D3) E. coli competent cells with the expression vector was performed by heat shock method (42 °C for 90 s, 5 minutes in ice). After being incubated overnight on agar plates in the presence of ampicillin at 37 °C, one single colony harbouring the expression construct was inoculated into 200 mL Luria Broth (LB) medium containing 100 μg mL−1 ampicillin and it was cultured overnight at 37 °C with shaking. A certain volume of the pre-culture was added to 2 L of new LB medium with ampicillin In order to get an OD600 of 0.1. Cells were grown at 37 °C until OD600 was between 0.6–1.2. Then, protein expression was induced with 1 mM Isopropyl β-D-1-thio-galactopyranoside (IPTG) and cell culture grow for another 3 h at 37 °C. Afterwards, cells were harvested by centrifugation at 5500 rpm for 20 min. The obtained pellets were purified following the protocols explained below.
Expression of 15N labelled Gal-8FL, Gal-8N and Gal-8C
Each gene encoding the carbohydrate recognition domains (N-terminal CRD, 1-155 amino acids and C-terminal CRD, 184–317) of human Gal-8 were inserted into a pET21b expression vector. Transformation of BL21 (D3) E. coli competent cells with the expression vector was performed by heat shock method (42 °C for 90 s, 5 minutes in ice). After being incubated overnight on agar plates in the presence of ampicillin at 37 °C, one colony harbouring the expression construct was inoculated into 3 mL Luria Broth (LB) me-dim containing 100 μg mL−1 ampicillin and then, cultured with shaking for 6 h at 37 °C. The culture was centrifuged for 5 minutes at 4400 rpm and the subsequent pellet resuspended in 1 mL of M9 medium, containing ampicillin. It was then transferred into a flask with 200 mL of the same ampicillin-containing M9 medium and incubated overnight at 37 °C with shaking. In order to get an OD600 of 0.1 a certain volume of the culture was added to 2 L of new M9 labelled (15N-NH4Cl as nitrogen source) medium with ampicillin. Cells were grown at 37 °C until OD600 was between 0.6–1.2, induced with 1 mM Isopropyl β-D-1-thio-galactopyranoside (IPTG) and then grown for another 3 h at 37 °C. Finally, cells were harvested by centrifugation at 5500 rpm for 20 min. The obtained pellets were purified following the protocols explained below in the Purification of Gal-8FL and Gal-8N and Purification of Gal-8C sections.
Expression of 13C, 15N labelled Gal-8N
13C, 15N labelled Gal-8N was expressed and purified following the protocol described in the Expression of 15N labelled Gal-8FL, Gal-8N and Gal-8C section and Purification of Gal-8FL and Gal-8N section, respectively, with the only variation being the addition of 13C-glucose as carbon source as well as 15N-NH4Cl as nitrogen source.
Purification of Gal-8FL and Gal-8N
The pellet obtained in the expression process was resuspended in 10 mL of lysis buffer/g of pellet with the lysis buffer having 2 mM EDTA, 4 mM β-mercaptoethanol and 0.1% NaN3 in phosphate-buffered saline (50 mM sodium phosphate, 150 mM NaCl, pH 7.4) and was then left in an ice bath for 30 minutes with shaking. The cell suspension was lysed by sonication in ice (60% amplitude, 12 × 20 s, with 59 seconds intervals between each burst). The crude extract was clarified by ultracentrifugation at 35000 rpm for 30 min at 4 °C. The soluble fraction was loaded onto 5 mL α-lactose-agarose resin (Sigma–Aldrich) previously equilibrated with lysis buffer. The column loaded was washed with 120 mL of equilibration buffer and then the lectin was eluted with 7 mL of elution buffer having 150 mM α-lactose and 1 mM DTT in phosphate-buffered saline (50 mM sodium phosphate, 150 mM NaCl, pH 7.4). Full-length hGal-8 and N-terminal CRD purity was checked by 4–12% SDS–PAGE and by LC-MS. To eliminate the lactose from the protein sample, a series of dialysis and washes with centrifuge filters (Sartorius Vivaspin 6 5000 MWCO) using fresh buffer (PBS 1×, 1 mM DTT and NaN3 0.1% pH 7.4) were performed. The absence of lactose was checked by NMR. The addition of the reducing agent in the buffer of hGal-8 is justified by the presence of cysteine residues exposed to the solvent that could cause the formation of non-specific dimers or aggregates through inter-molecular disulphide bonds.
Purification of Gal-8C
The pellet obtained in the expression process was resuspended in 10 mL of lysis buffer/g of pellet with the lysis buffer having 1 mM PMSF in phosphate-buffered saline (50 mM sodium phosphate, 150 mM NaCl, pH 7.4) and was then left in an ice bath for 30 minutes with shaking. The cell suspension was lysed by sonication in ice (60% amplitude, 12 × 20 s, with 59 seconds intervals between each burst). Protein was purified through a HisTrap Ni-NTA column (GE Healthcare) followed by size exclusion chromatography on a Superdex 75 HiLoad 26/600 column (GE Healthcare).
NMR experiments
General information. 500 μL were used as total volume for all NMR samples, using a precision NMR tube with 5 mm outer diameter (New Era Enterprises, Vineland, USA). All pH measurements of the buffers were performed with pH-meter Crison Basic 20 (Crison Instruments SA, Barcelona, Spain) and adjusted with the required amount of NaOH and HCl or NaOD and DCl.
Backbone assignment of Gal-8FL
Backbone assignment of Gal-8FL was establish by minimum chemical shift procedure, in which each resonance of the full-length protein was linked to the least shifted signals of Gal-8N and Gal-8C isolated domains. To this end, 15N Gal-8C backbone assignment deposited in the Biological Magnetic Resonance Data Bank (BMRB entry 25453)31 was employed, whereas the Gal-8N backbone assignment was kindly provided by Dr S. T. D. Hsu. The unambiguous identification of 73% of the NH signals of Gal-8N was accomplished by the acquisition and assignment of standard 3D NMR experiments. 3D HNCACB, HN(CO)CACB, HNCA, HN(CO)CA, HNCO and HN(CA)CO experiments were acquired using Bruker AVANCE 2 800 MHz spectrometer equipped with cryoprobe and using a 560 μM sample of Gal-8N in the presence of 150 mM lactose. The spectra were processed with Bruker Topspin 3.5 and analysed via CARANMR 1.9.1.
Saturation transfer difference (STD) NMR
STD experiments were acquired using Bruker AVANCE 2 800 MHz spectrometer equipped with cryoprobe. All samples were prepared in deuterated phosphate-buffered saline (50 mM sodium phosphate, 150 mM NaCl, pH 7.4) with 1 mM of dithiothreitol-d10 (DTT-d10) and 0.1% NaN3. All experiments were acquired at 308 K. In the case of high-affinity ligands 1:30 ratios of ligand:Gal-8 were used with the concentration of unlabelled protein set at 30 and 40 μM for ligands 3 and 6, respectively. Higher equivalents of ligand (ratio 1:100) were used to amplify the STD effect for ligands in fast-exchange regime. An in-house 1D-STD sequence using 90° PC9 pulses for protein saturation and 100 ms T1rho protein filter was employed for STD experiments. Acquisition was done with 4608 scans, 2 s of total saturation time and 3 s of relaxation delay. The on- and off-resonance spectra were registered in an interleaved mode with the same number of scans. On-resonance saturation was set between 0.61 and 0.91 ppm for the aliphatic region and in 6.99 ppm for the aromatic, while the off-resonance saturation was set at −30 ppm. The STD NMR spectra were obtained by subtracting the on-resonance spectrum from the off-resonance spectrum. The analysis of the spectra was carried out using the proton signal with the strongest STD effect as reference (100% of STD effect). On this basis the relative STD intensities for the others protons of the molecule were calculated.
2D-ROESY NMR experiments
ROESY experiments were acquired using Bruker AVANCE 2 800 MHz spectrometer equipped with cryoprobe. All samples were prepared in deuterated phosphate-buffered saline (50 mM sodium phosphate, 150 mM NaCl, pH 7.4) with 1 mM of dithiothreitol-d10 (DTT-d10) and 0.1% NaN3. A protein concentration of 100 μM was used with a protein:ligand ratio of 1:10 for ligand 3 and 1:8 for ligand 6. All experiments were acquired at 298 K. An in-house ROESY sequence using a 100 ms mixing time was used.
Chemical shift perturbation (CSP) analysis
15N-HSQC experiments were acquired using Bruker AVANCE 2 800 MHz spectrometer equipped with cryoprobe. Samples containing 100 μM 15N labelled Gal-8 in 90% phosphate-buffered saline (50 mM sodium phosphate, 150 mM NaCl, pH 7.4)/10% D2O with 1 mM of dithiothreitol (DTT) and 0.1% NaN3 were prepared. All experiments were acquired at 308 K. The ligands were titrated in the protein samples (0.25–0.5–1–3–6–9–12–15–20–25–30–40–60 equivalents for ligand 1, 0.5–1–1.5–2.5–5–10 for ligand 3 and 0.25–0.5–0.75–1–1.5–2–5–8 for ligand 6) and 15N-HSQC experiments were acquired at each intermediate point. An in-house TROSY sequence was used with 256 (T1) × 1536 (T2) complex data points in 15N and 1H dimensions, respectively. The CSP data were analysed using CcpNmr Analysis software. The CSP of the protein-backbone NH groups were calculated using the formula: Δδ (ppm) = [(ΔδH2 + (0.14·ΔδN)2)/2]1/2 and the results were graphically plotted.
Isothermal titration calorimetry (ITC)
Isothermal titration calorimetry experiments were performed using MicroCal PEAQ-ITC calorimeter. All experiments were acquired at 298 K with every run being performed at least in duplicate. Gal-8 and ligand samples (1–6) were prepared in phosphate-buffered saline (50 mM sodium phosphate, 150 mM NaCl, pH 7.4) with 1 mM TCEP as reducing agent. The concentration of the protein solution was set at around 100 μM and that of the sugar stock between 3–12 mM. During the automated experiment, small amounts of the sugar solution (1.20–2.40 μL) were titrated into a cell containing the protein solution and the dispersed heat was detected. The association constants and the thermodynamic parameters were obtained from data fitting to a single-site (Gal-8, Gal-8C and Gal-8FL with selective ligands) or 1:2 (Gal-8FL with non-selective ligands) binding model. In the case of low-affinity ligands (LacNAc, Galβ1–3GalNAc and Galili) n values were fixed to n = 1 or n = 2 (for the interaction Gal-8FL/LacNAc and Gal-8FL/Galβ1–3GalNAc) for data fitting. Final data were shown in Table 1 as average ± standard deviation of two independent runs. When the standard deviation calculated from two independent measurements was lower than the precision given by the instrument, the latter value was used to determine the accuracy of the final displayed data. Examples of titration profiles for each complex with data fitting to single-site binding model are reported in the ESI.†
Model building
Full-length human galectin-8 was constructed from the crystallographic coordinates of the N- (PDB code: 4BMB; residues 1–150) and C-terminal (PDB code: 3OJB; residues 184–317) CRDs. The unresolved peptide linker (residues 151–183) was built in an extended conformation using the tleap module of Amber 18. The three fragments were manually assembled using PyMol and the resulting full-length structure was refined through MD simulations (see below).
Molecular dynamics
Molecular dynamics (MD) simulations were run with the Amber 18 suite,32,33 using the ff14SB34 and GLYCAM 06j-135 force fields for the proteins and carbohydrate ligands, respectively. Binding histidine residues (H65 and H229) were modelled in their Nδ1–H tautomeric state (reside name HID in Amber). Initial structures were neutralized with either Na+ or Cl− ions and set at the centre of a cubic TIP3P water36 box with a buffering distance between solute and box of 10 Å. For each system, we followed a two-stage geometry optimization approach: the first stage minimizes only the positions of solvent molecules and ions, and the second stage is an unrestrained minimization of all the atoms in the simulation cell. The systems were then heated by incrementing the temperature from 0 to 300 K under a constant pressure of 1 atm and periodic boundary conditions. Harmonic restraints of 10 kcal mol−1 were applied to the solute, under the Andersen temperature coupling scheme.37,38 The time step was kept at 1 fs during the heating stages, allowing potential inhomogeneities to self-adjust. Water molecules were treated with the SHAKE algorithm39 such that the angle between the hydrogen atoms is kept fixed through the simulations. Long-range electrostatic effects were modelled using the particle mesh Ewald method.40 An 8 Å cut-off was applied to Lennard-Jones interactions. Each system was equilibrated for 2 ns with a 2 fs time step at a constant volume and temperature of 300 K. Ten independent production trajectories were then run for additional 2.0 μs under the same simulation conditions, leading to accumulated simulation times of 20.0 μs for each system (hGal-8, hGal-8_N-term:LacNAc_C-term:LacNAc, hGal-8_N-term:3-sialyl-Galβ1–3GalNAc, hGal-8_C-term:A typeII, and hGal-8_N-term:3-sialyl-Galβ1–3GalNAc_C-term:AtypeII). For the complex used as a negative control (hGal-8-_N-term:AtypeII_C-term:3-sialyl-Galβ1–3GalNAc), five independent 1.0 μs production trajectories were run. Additionally, five 200 ns production trajectories were run for the isolated N- and C-terminal domains in complex with 3-sialyl-Galβ1–3GalNAc and A type II respectively.
Author contributions
Marcos Gómez-Redondo and Ana Gimeno performed and analysed the NMR and ITC investigations. Sandra Delgado expressed and purified the lectin domains in the regular and 15N-labelled forms. Reyes Núñez-Franco and Gonzalo Jiménez-Osés performed and analysed the MD simulations. Gonzalo Jiménez-Oses, Ana Ardá, Jesús Jiménez-Barbero, and Ana Gimeno conceptualized the research and supervised all the activity planning and execution. All authors critically commented the results throughout the project. Marcos Gómez-Redondo and Reyes Núñez-Franco (MD part) wrote the original draft. Ana Gimeno and Gonzalo Jiménez-Osés (MD part) reviewed the text. Jesús Jiménez-Barbero wrote the final manuscript. Gonzalo Jiménez-Oses (for the MD part), Ana Ardá, and Jesús Jiménez-Barbero provided the financial support for this Project to be carried out.
Conflicts of interest
The authors state there are no conflicts to declare.
Acknowledgements
This research was funded by European Research Council for financial support (ERC-2017-AdG, project number 788143-RECGLYCANMR). We also thank Agencia Estatal de Investigación (Spain) for projects RTI2018-094751-B-C21, RTI2018-099592-B-C22 and the Severo Ochoa Excellence Accreditation (SEV-2016-0644) and the Mizutani Foundation for Glycoscience (200077 to GJO). We thank Dr S. T. D. Hsu (Institute of Biological Chemistry, Academia Simica, Taipei, Taiwan) for kindly providing the 15N backbone assignment of Gal-8N.
References
- L. Johannes, R. Jacob and H. Leffler, J. Cell Sci., 2018, 131, jcs208884 CrossRef PubMed.
- S. H. Barondes, D. N. W. Cooper, M. A. Gitt and H. Leffler, J. Biol. Chem., 1994, 269, 20807 CrossRef CAS.
- J. P. Cerliani, A. G. Blidner, M. A. Toscano, D. O. Croci and G. A. Rabinovich, Trends Biochem. Sci., 2017, 42, 255 CrossRef CAS PubMed.
- V. C. Martínez Allo, M. A. Toscano, N. Pinto and G. A. Rabinovich, Trends Glycosci. Glycotechnol., 2018, 30, SE97 CrossRef.
- R. Y. Yang, G. A. Rabinovich and F. T. Liu, Expert Rev. Mol. Med., 2008, 10, e17 CrossRef PubMed.
- J. Nieminen, A. Kuno, J. Hirabayashi and S. Sato, J. Biol. Chem., 2007, 282, 1374 CrossRef CAS PubMed.
- S. Bertuzzi, J. I. Quintana, A. Ardá, A. Gimeno and J. Jiménez-Barbero, Front. Chem., 2020, 8, 593 CrossRef CAS PubMed.
- Y. R. Hadari, K. Paz, R. Dekel, T. Mestrovic, D. Accili and Y. Zick, J. Biol. Chem., 1995, 270, 3447 CrossRef CAS PubMed.
- B. W. Kim, S. Beom Hong, J. Hoe Kim, D. Hoon Kwon and H. K. Song, Nat. Commun., 2013, 4, 1613 CrossRef PubMed.
- S. Kumar, M. Frank and R. Schwartz-Albiez, PLoS One, 2013, 8, e59761 CrossRef CAS PubMed.
- J. Hirabayashi, T. Hashidate, Y. Arata, N. Nishi, T. Nakamura, M. Hirashima, T. Urashima, T. Oka, M. Futai, W. E. G. Muller, F. Yagi and K. Kasai, Biochim. Biophys. Acta, 2002, 1572, 232 CrossRef CAS.
- H. Ideo, T. Matsuzaka, T. Nonaka, A. Seko and K. Yamashita, J. Biol. Chem., 2011, 286, 11346 CrossRef CAS PubMed.
- S. R. Stowell, C. M. Arthur, K. A. Slanina, J. R. Horton, D. F. Smith and R. D. Cummings, J. Biol. Chem., 2008, 283, 20547 CrossRef CAS PubMed.
- M. N. Schroeder, M. V. Tribulatti, J. Carabelli, G. André-Leroux, J. J. Caramelo, V. Cattaneo and O. Campetella, Biochem. J., 2016, 473, 887 CrossRef CAS PubMed.
- S. Carlsson, C. T. Öberg, M. C. Carlsson, A. Sundin, U. J. Nilsson, D. Smith, R. D. Cummings, J. Almkvist, A. Karlsson and H. Leffer, Glycobiology, 2007, 17, 663 CrossRef CAS PubMed.
- A. J. Cagnoni, M. F. Troncoso, G. A. Rabinovich, K. V. Mariño and M. T. Ebola, Biochem. Soc. Trans., 2020, 48, 1255 CrossRef CAS PubMed.
- H. Yoshida, S. Yamashita, M. Teraoka, A. Itoh, S. Nakakita, N. Nishi and S. Kamitori, FEBS J., 2012, 279, 3937 CrossRef CAS PubMed.
- T. Wiseman, S. Williston, J. F. Brandts and L. N. Lin, Anal. Biochem., 1989, 179, 131 CrossRef CAS PubMed.
- Y. Si, Y. Wang, J. Gao, C. Song, S. Feng, Y. Zhou, G. Tai and J. Su, Int. J. Mol. Sci., 2016, 17, 2088 CrossRef PubMed.
- A. Ardá and J. Jiménez-Barbero, Chem. Commun., 2018, 54, 4761 RSC.
- J. M. Bardelli, E. Livoti, L. Simonelli, M. Pedotti, A. Moraes, A. P. Valente and L. Vareli, J. Mol. Recognit., 2015, 28, 393 CrossRef PubMed.
- M. Mayer and B. Meyer, J. Am. Chem. Soc., 2001, 123, 6108 CrossRef CAS PubMed.
- A. Gimeno, S. Delgado, P. Valverde, S. Bertuzzi, M. A. Berbís, J. Echavarren, A. Lacetera, S. Martín-Santamaría, A. Surolia, F. J. Cañada and J. Jiménez-Barbero, Angew. Chem., Int. Ed., 2019, 58, 7268 CrossRef CAS PubMed.
- W. B. Turnbull and A. H. Daranas, J. Am. Chem. Soc., 2003, 125, 14859 CrossRef CAS PubMed.
- J. L. Asensio, F. J. Cañada and J. Jiménez-Barbero, Eur. J. Biochem., 1995, 233, 618 CrossRef CAS PubMed.
- A. Gimeno, P. Valverde, A. Ardá and J. Jiménez-Barbero, Curr. Opin. Struct. Biol., 2020, 62, 22 CrossRef CAS PubMed.
- E. R. P. Zuiderweg, Biochemistry, 2002, 41, 1 CrossRef CAS PubMed.
- M. P. Lenza, I. Oyenarte, T. Diercks, J. I. Quintana, A. Gimeno, H. Coelho, A. Diniz, F. Pecatti, S. Delgado, A. Bosch, M. Valle, O. Millet, N. G. A. Abrescia, A. Palazón, F. Marcelo, G. Jiménez-Osés, J. Jiménez-Barbero, A. Ardá and J. Ereño-Orbea, Angew. Chem., Int. Ed., 2020, 59, 23763 CrossRef CAS PubMed.
- P. Valverde, J. I. Quintana, J. I. Santos, A. Ardá and J. Jiménez-Barbero, ACS Omega, 2019, 4, 13618 CrossRef CAS PubMed.
- S. Bertuzzi, A. Gimeno, R. Nuñez, G. Bernardo-Seisdedos, S. Delgado, G. Jimenez-Oses, O. Millet, J. Jiménez-Barbero and A. Ardá, Chem. – Eur. J., 2020, 26, 15643 CrossRef CAS PubMed.
- C.-H. G. Liu, C.-T. H. Chien, C.-H. Lin and S. T. D. Hsu, Biomol. NMR Assign., 2015, 9, 427 CrossRef CAS PubMed.
-
D. A. Case, et al., Amber 2018, University of California, San Francisco, 2018 Search PubMed.
- T. S. Lee, J. Chem. Inf. Model., 2018, 58, 2043 CrossRef CAS PubMed.
- J. A. Maier,
et al.
, J. Chem. Theory Comput., 2015, 11, 3696 CrossRef CAS PubMed.
- K. N. Kirschner,
et al.
, J. Comput. Chem., 2008, 29, 622 CrossRef CAS PubMed.
- W. L. Jorgensen, J. Chandrasekhar, J. D. Madura, R. W. Impey and M. L. Klein, J. Chem. Phys., 1983, 79, 926 CrossRef CAS.
- H. C. Andersen, J. Chem. Phys., 1980, 72, 2384 CrossRef CAS.
- T. A. Andrea, W. C. Swope and H. C. Andersen, J. Chem. Phys., 1983, 79, 4576 CrossRef CAS.
- S. Miyamoto and P. A. Kollman, J. Comput. Chem., 1992, 13, 952 CrossRef CAS.
- T. Darden, D. York and L. Pedersen, J. Chem. Phys., 1993, 98, 10089 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1cb00051a |
|
| This journal is © The Royal Society of Chemistry 2021 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence ab,
Ana
Ardá
*ab,
Jesús
Jiménez-Barbero
ab,
Ana
Ardá
*ab,
Jesús
Jiménez-Barbero