
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCrosslinker-modified nucleic acid probes for improved target identification and biomarker detection
Joke
Elskens
 and
Annemieke
Madder
*
and
Annemieke
Madder
*
Department of Organic and Macromolecular Chemistry, Faculty of Sciences, Ghent University, Krijgslaan 281 Building S4, 9000 Ghent, Belgium. E-mail: Annemieke.Madder@ugent.be; Joke.Elskens@ugent.be; Fax: +32-9-264-49-98; Tel: +32-9-264-44-72
First published on 22nd February 2021
Abstract
Understanding the intricate interaction pattern of nucleic acids with other molecules is essential to gain further insight in biological processes and disease mechanisms. To this end, a multitude of hybridization-based assays have been designed that rely on the non-covalent recognition between complementary nucleic acid sequences. However, the ephemeral nature of these interactions complicates straightforward analysis as low efficiency and specificity are rule rather than exception. By covalently locking nucleic acid interactions by means of a crosslinking agent, the overall efficiency, specificity and selectivity of hybridization-based assays could be increased. In this mini-review we highlight methodologies that exploit the use of crosslinker-modified nucleic acid probes for interstrand nucleic acid crosslinking with the objective to study, detect and identify important targets as well as nucleic acid sequences that can be considered relevant biomarkers. We emphasize on the usefulness and advantages of crosslinking agents and elaborate on the chemistry behind the crosslinking reactions they induce.
 Joke Elskens | Joke Elskens studied Chemistry at the University of Ghent and graduated as Master of Science in Chemistry in 2016. She performed her thesis in the research group of Prof. A. Madder. In 2016 she started her PhD project, concerning the design of modified nucleic acid probes for the development of improved RNA target identification strategies. |
 Annemieke Madder | Annemieke Madder studied at Ghent University and obtained her PhD in chemistry in 1997. After postdoctoral stays with Prof. Dr C. Gennari at the University of Milan and with Prof. Dr R. Strömberg at the Karolinska Institute in Stockholm, she returned to Ghent and obtained a position as Lecturer in 2002. In 2014 she was promoted Full Professor at the Department of Organic and Macromolecular Chemistry. Currently she is heading the Organic and Biomimetic Chemistry Research Group specialized in the design and synthesis of modified peptides and nucleic acids and methods for their conjugation and labeling. |
1. Introduction
Nucleic acids such as deoxyribonucleic acid (DNA) and ribonucleic acid (RNA), are key regulators in living organisms. DNA, the holder of the genetic information, directs biological processes via transcription into messenger RNA (mRNA), which serves as template for the translation into proteins. This theory, known as the central dogma of molecular biology, states that information flows from nucleic acids to proteins.1 Originally, RNA was considered a helper molecule, serving the main purpose of assisting and controlling the processes involved in protein synthesis. As only 3% of the total human genome codes for proteins, researchers in the field wondered what the function of the remaining non-coding portion could be. Certain housekeeping RNAs like transfer or ribosomal RNAs were known to be expressed and to be important for normal cell functioning, but they could not account for all non-protein coding sequences in the genome. It was only with the discovery of regulatory non-coding RNAs (ncRNA) that the importance of these non-coding regions started to be revealed.2 These ncRNAs are not constitutively expressed in cells and exert their regulatory function by forming highly ordered dynamic structures capable of interacting with DNA, RNA and proteins. They are subdivided based on their length into small or long ncRNAs. Small regulatory ncRNAs (<200 nucleotides) include the small interfering RNAs (siRNA), the piwi-interacting RNAs and the micro-RNAs (miRNA). These ncRNAs act as key regulators of gene expression (e.g. miRNAs are involved in regulating mRNA levels) and their mode of action is quite well understood.2 The long non-coding RNAs (lncRNA), with a length over 200 nucleotides, are the most numerous but least comprehended regulatory RNAs to date. It is estimated that more than 16![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 lncRNAs exist, while currently only the function of approximately 50 lncRNAs is understood.3 They are involved in transcriptional4- and epigenetic regulation,5 in controlling mRNA stability6 and function as miRNA sponges.7 For many years, the function of RNA as regulatory unit was underestimated and to this day, just a tip of its full potential has been uncovered. Considering their role in regulating biological processes, further insight in ncRNA function is pivotal. To achieve this, identification of the ncRNA interaction partners (e.g. proteins and other nucleic acids) is of major importance. For this purpose, different hybridization-based technologies such as ChiRP (chromatin isolation by RNA purification),8 CHART (capture hybridization analysis of RNA targets),9 immunoprecipitation (IP)10 assays and variants thereof have been developed. These assays use nucleic acid capture probes to isolate and analyse ncRNA complexes and merely differ in buffer conditions and capture probe design.†
000 lncRNAs exist, while currently only the function of approximately 50 lncRNAs is understood.3 They are involved in transcriptional4- and epigenetic regulation,5 in controlling mRNA stability6 and function as miRNA sponges.7 For many years, the function of RNA as regulatory unit was underestimated and to this day, just a tip of its full potential has been uncovered. Considering their role in regulating biological processes, further insight in ncRNA function is pivotal. To achieve this, identification of the ncRNA interaction partners (e.g. proteins and other nucleic acids) is of major importance. For this purpose, different hybridization-based technologies such as ChiRP (chromatin isolation by RNA purification),8 CHART (capture hybridization analysis of RNA targets),9 immunoprecipitation (IP)10 assays and variants thereof have been developed. These assays use nucleic acid capture probes to isolate and analyse ncRNA complexes and merely differ in buffer conditions and capture probe design.†
Besides regulating biological processes, nucleic acids also control pathological pathways and can consequently be considered as relevant biomarkers for e.g. cancer,11,12 neurodegenerative-13,14 and infectious diseases.15 For example, studies indicated that the expression levels of certain mRNAs and ncRNAs correlate with disease manifestation.3,16,17 On the other hand, typical DNA biomarkers arise from variations occurring at the genomic level which include single nucleotide polymorphisms (SNP) and short tandem sequences to name a few.18 Additionally, epigenetically modified nucleic acids containing methylation or hydroxymethylation sites can also serve as biomarkers, as they control gene silencing and gene overexpression.19 Indeed, the methylation pattern in DNA has been correlated with diseases such as cancer.20 In addition to chromosomal DNA, mitochondrial or pathogenic DNA sequences can also be considered as biomarkers.21,22
Nucleic acid biomarker detection and target identification assays such as the above-mentioned ChiRP, CHART and IP assays, are often based on the non-covalent hybridization of an oligonucleotide capture probe recognizing its complementary DNA or RNA target (Fig. 1A).18,23,24 The transient nature of this hybridization-based interaction hampers the overall efficiency as target sequences are often lost during work-up. While a series of bifunctional, externally added crosslinking agents are available and could be used to reinforce the non-covalent interactions for better complex stability during work-up, most of these show little selectivity in terms of the exact site of covalent connection between the strands in a complex. In a very elegant approach, Kool et al. were able to achieve site-specific crosslinking using their so-called BINARI probes (bis-nicotinic azide reversible interaction) for trapping RNA–RNA interactions.25 In this set-up, the inherently reactive bifunctional agent is used for site-selective simultaneous acylation of 2′OH groups of opposing non-complementary RNA sequences located at a specific distance from a short complementary region within the RNA–RNA complex. It is clear that the above-mentioned assays could further benefit from nucleic acid capture probes modified with a crosslinking agent to achieve site-specific crosslinking in a broader context. Upon hybridization of the crosslinker-modified nucleic acid probe with its target oligonucleotide, a covalent linkage is introduced between both, rendering the interaction irreversible (Fig. 1B). This ensures that a maximal amount of target is retained throughout the assay, even when stringent washing conditions are applied. The latter is beneficial for increasing the assay's specificity and selectivity as non-specific interactions can be easily washed away during work-up. This however necessitates careful design of the crosslinker modified capture probes. Indeed, the conditions required for crosslinking and the covalent linkage formed upon reaction may not interfere with downstream processes such as PCR and MS to ensure accurate detection with high sensitivity.
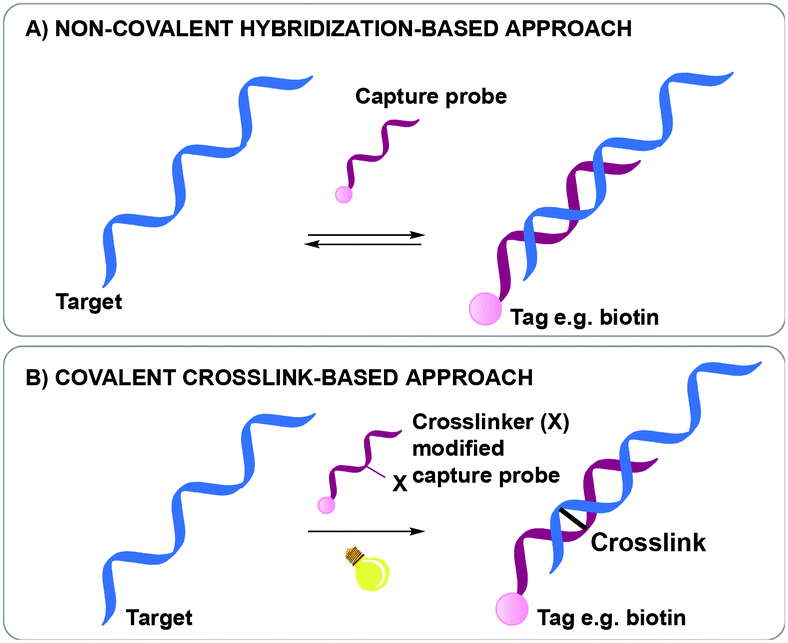 | ||
| Fig. 1 Difference between a non-covalent hybridization assay (A) and a covalent hybridization assay (B). | ||
Considering their importance and contribution in unravelling the biology of cells, we describe in this review a series of methodologies that exploit the use of crosslinker-modified nucleic acid probes for interstrand nucleic acid crosslinking with the objective to study, detect and identify important targets as well as nucleic acid sequences that can be considered relevant biomarkers. Furthermore, we elaborate on the chemistry of the crosslink reactions. The discussed crosslinking agents can be divided into three categories: (1) photoactivatable crosslinking agents; (2) crosslinking probes activated by chemical agents; (3) oligonucleotides containing abasic sites as inherently reactive crosslinking agents. Table 1 provides a summary of the discussed agents and their key features.
| Crosslinker | Activation mode | Reaction type | Reversible | Target | Target base & position | Biological applications | In vivo/in vitro |
|---|---|---|---|---|---|---|---|
| Coumarin | UV, 350 nm | [2+2] | Yes, 254 nm | DNA | T | SNP detection | In vitro |
| Base should oppose the crosslinker | Detection and quantification of viral DNA | ||||||
| DNA methylation | |||||||
| Psoralen | UV, 365 nm | [2+2] | Yes, 254 nm | DNA, RNA | C, T, U | SNP detection | In vitro |
| Base should oppose the crosslinker | Antisense | In vivo | |||||
| Identification of miRNA targets (miR-TRAP, miR-CLIP) | |||||||
| DNA methylation | |||||||
| Vinylcarbazole: | [2+2] | Yes, 312 nm | DNA, RNA | C, T, U | Antisense | In vitro | |
| CNVK | UV, 365 nm | Base should be located at position −1 in the complementary strand | DNA methylation | In vivo | |||
| CNVD | UV, 365 nm | Detection and identification of miRNA targets | |||||
| PCX | VIS, 400 nm | FISH | |||||
| SNP detection | |||||||
| 4-Thiouridines | UV, 365 nm | [2+2] | No | DNA, RNA | C | RNA target identification | In vitro |
| Base should oppose the crosslinker | |||||||
| Diazirines | UV, 365 nm | Carbene | No | DNA, RNA | A, C, G, T | Identification of miRNA targets | In vitro |
| Reaction with nearby nucleophiles, predominantly with base opposing the crosslinker | |||||||
| Phenylselenide | Dual activation mode: | No | DNA | A | SNP detection | In vitro | |
| – UV, 350 nm | Radical | Base should oppose the crosslinker | |||||
| – Chemical (1O2, NaIO4) | Michael type reaction | ||||||
| Furan | Chemical activation: | No | DNA, RNA | A, C | Detection of DNA and RNA targets | In vitro | |
| – 1O2 | [4+2] | Base should oppose the crosslinker | |||||
| – NBS | Nucleophilic addition | ||||||
| Abasic site | Inherently reactive | Nucleophilic addition | No | DNA | A: positioned one nucleotide closer to the 3′ site of the complementary strand | SNP detection | In vitro |
| G: positioned one nucleotide closer to the 5′ site of the complementary strand | |||||||
2. Photo-activatable crosslinking agents for target identification and biomarker detection
2.1 Coumarin
The first class of crosslinking agents requires light to initiate the crosslink reaction. Light activation imposes some advantages over other activation methods as it allows spatiotemporal control of the reaction. If needed, the light beam can be directed to a specific area and irradiation can be halted and resumed at any time. Examples of photo-activatable crosslinking agents include coumarin derivatives, psoralen, vinylcarbazoles, 4-thiouridines and diazirines.Coumarin-based crosslinkers are able to photo-crosslink to nearby thymine bases via a [2+2] cycloaddition reaction upon irradiation at 350 nm, yielding predominantly the syn-cycloaddition adduct. Photo-reversibility is achieved when a wavelength of 254 nm is employed (Fig. 2A). Coumarins have mainly been used as medicines (e.g. warfarin26) or as fluorophores for biomolecule labeling. Therefore, multiple coumarin variants have been designed to date (Fig. 2B).27–32
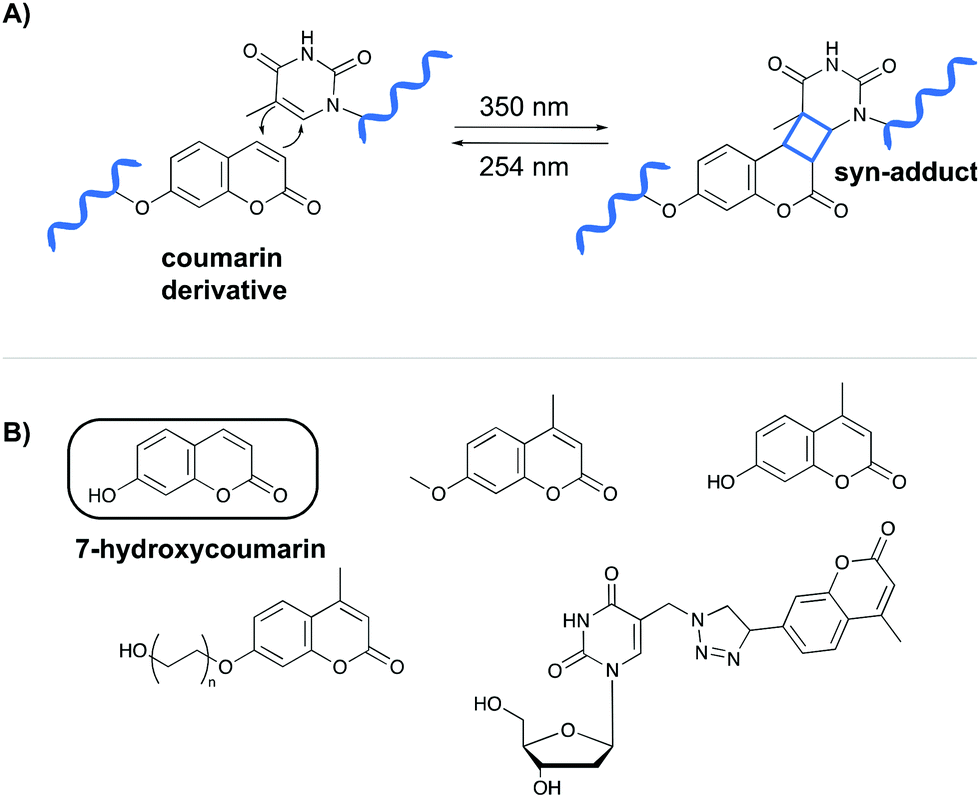 | ||
| Fig. 2 (A) Mechanism of the [2+2] cycloaddition reaction between coumarin and thymine. (B) Examples of coumarin derivatives. 7-Hydroxycoumarin is used as crosslinking agent in the NAXCOR assay. | ||
A specific application of coumarin derivatives in the context of biomarker identification can be found in the NAXCOR assay. This hybridization-based diagnostic test exploits a 7-hydroxycoumarin derivative for photo-crosslinking and is used for the detection of the factor V leiden mutation, a G to A single nucleotide mutation in the factor V gene and the most common inherited risk for thrombosis (Fig. 3A).33 Individuals are either homozygotes (two normal or two mutant factor V alleles) or heterozygotes (one copy of the normal and mutant allele).
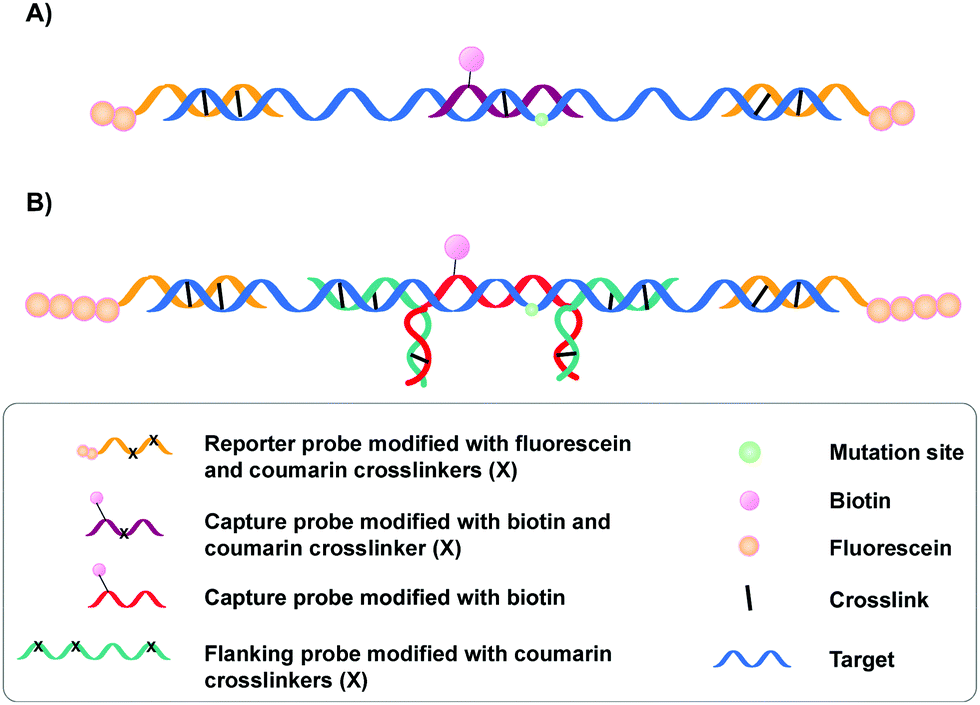 | ||
| Fig. 3 (A) Illustration of the NAXCOR assay. Fluorescein modified reporter probes (yellow) and biotinylated capture probes (purple) are crosslinked to the factor V leiden gene (blue). (B) Variant of the NAXCOR assay in which reporter probes (yellow) are modified with a tail of fluorescein molecules. The biotinylated capture probe (red) is indirectly crosslinked to the factor V leiden gene (blue) by means of crosslinker-modified flanking probes (green). | ||
This diagnostic assay combines the use of coumarin modified reporter probes and capture probes for detection (yellow in Fig. 3A) and recognition (purple in Fig. 3A) respectively. A total of six reporter probes is used, each modified with two coumarin crosslinking moieties and two fluorescein labels. These reporter probes are complementary to non-allele specific regions of exon 10 and intron 10 of the factor V gene, thereby flanking the capture probe binding site. The capture probe is an allele-specific 16-mer oligonucleotide modified with both coumarin and biotin that is able to hybridize at the mutation site in the factor V gene. Upon incubation, the reporter (yellow) and capture probes (purple) hybridize with the factor V gene (blue). This is followed by crosslink formation and streptavidin pull-down, with the latter separating crosslinked probes from their non-crosslinked counterparts. Next, detection of crosslinked product is achieved by incubating the samples with anti-fluorescein antibodies labelled with alkaline phosphatase. Addition of a suitable fluorescent enzyme substrate generates a colored product with an intensity that correlates to the concentration of fluorescein in the sample. Discrimination between normal or mutant alleles (both homo- or heterozygotic) is achieved by using a three-fold analysis system, where each sample is subjected to different probe sets. Each probe set contains the same six fluorescein modified reporter probes but differs in the used capture probe – the capture probe is either fully complementary to the normal gene (set 1) or the mutated gene (set 2). Alternatively, the probe set contains both the normal and the mutant capture probe (set 3). Assay conditions were optimized in such a way that hybridization and crosslinking with mismatched sequences is minimized. Consequently, the intensity of the fluorescent read-out is dependent on the used probe set and the presence or absence of the factor V leiden mutation. For individuals with normal alleles, the fluorescent intensity will be maximal when the normal or mixed capture probe sets are used (set 1 and 3) and the lowest with capture probes complementary to the mutant alleles (set 2). The opposite is true for individuals with the factor V leiden mutation. Hence, single nucleotide polymorphism detection in this NAXCOR assay is solely based on a difference in hybridization and crosslinking efficiency of two distinct capture probes, which in turn results in a change in fluorescent read-out. Besides detection of single nucleotide polymorphisms, the detection and quantification of viral DNA34 as well as the assessment of gene dosage and methylation status has also been described.35
After the initial report, a variant of the NAXCOR assay was published with improved sensitivity and signal to noise ratio (Fig. 3B).36 While the general principle remains the same, the optimized assay differs from the original assay in two distinct features: (1) the reporter probes (yellow) are modified with a polyamine tail allowing the coupling of approximately 10 times more fluorescein moieties. This in turn significantly enhanced the signal generation capacity and enhanced the signal to noise ratio; (2) instead of crosslinking the capture probe directly to the target sequence, an indirect 3-probe approach was opted for. Herein, the capture probe (red) is positioned between, and covalently connected to two adjacent flanking probes (green), by means of crosslink formation between the overhanging partially complementary regions of the capture and flanking probes (Fig. 3B). The flanking probes are further responsible for crosslinking to the target. This indirect crosslinking approach resulted in a 2.8-fold improvement in sensitivity and enhanced the applicability of the NAXCOR assay for SNP detection.
2.2 Psoralen
Psoralen and derivatives thereof belong to the family of furocoumarins and are characterised by a planar tricyclic structure that allows intercalation in any AT or AU region of double stranded DNA and RNA sequences respectively. Upon irradiation with long wavelength UV-A (365 nm), psoralens are able to react via a [2+2] cycloaddition reaction with the C5–C6 bond of pyrimidines (Fig. 4A), either via their furan or pyrone photoreactive site, which results in the formation of a cyclobutane adduct. Hence, one psoralen moiety is able to react with two different pyrimidine residues in a regio- and stereoselective manner as only the cis–syn products are formed. The syn nomenclature refers to the positioning of the N1 amine of the pyrimidine bases relative to the C2 of the pyrone or the O1′ of the furan moiety. In the syn product, these are located on adjacent corners of the cyclobutene ring (Fig. 4A). The cis configuration indicates the position of the psoralen compared to the pyridimine base. After photo-crosslinking, both are located on the same side of the cyclobutane ring.37,38 This photo-reaction can be reversed by controlling the irradiation wavelength.39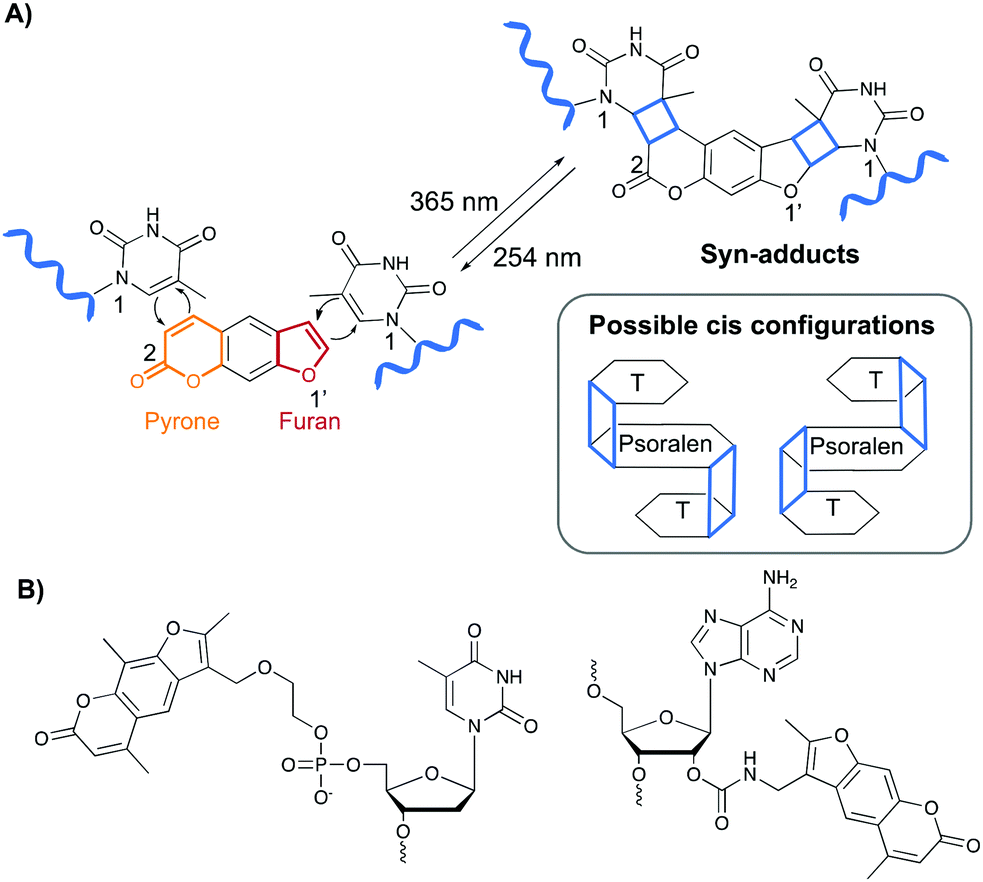 | ||
| Fig. 4 (A) Upon irradiation with UV-A (365 nm), psoralens react via a [2+2] cycloaddition reaction with nearby pyrimidine residues. The reaction can be reversed upon irradiation with a shorter wavelength (254 nm). The mechanism is depicted for the reaction with thymine bases. The cyclobutane product is highlighted in blue and the syn-adduct is shown. A schematic representation of the cis-configuration is also given. (B) Examples of psoralen derivatives incorporated in oligonucleotide sequences. | ||
Psoralens can also be incorporated in oligonucleotide sequences to allow sequence-specific photo-crosslinking. For this purpose, different psoralen-modified nucleosides have been designed (Fig. 4B).40–47 These constructs have been applied for crosslinking triplex forming oligonucleotides to double stranded DNA44,47 and, although not yet translated into therapeutic applications, psoralens have been used in the conceptual design of crosslinking antisense oligonucleotides.40,42,48
Psoralen-modified oligonucleotides are also used in the context of biomarker detection. More specifically, Yamayoshi et al. confirmed, in a proof-of-concept study, the utility of psoralen in the discrimination of C5-methylated cytosine from non-methylated cytosine bases in DNA.41 Cytosine C5-methylation–demethylation is involved in the epigenetic regulation of gene expression and determination of the position and frequency of methylation provides crucial information to understand this process. Psoralen crosslinking proved up to 5-fold higher with C5-methyl cytosine compared to demethylated cytosine, in single as well as in double stranded DNA, thus allowing to discriminate between the two modifications. The same research group also tested the ability of 2′ psoralen modified deoxyadenosine residues to identify a G → T single nucleotide polymorphisms in the H-ras gene.43 Discrimination between the mutant and wild type form is based on the observed pyrimidine selectivity of psoralen crosslinking. As such, crosslinking proceeded in moderate yield with the mutant H-ras gene while almost no crosslinking was observed with the wild type. However, while promising, this method would benefit from improved crosslinking efficiencies, which requires careful design of the linker connecting the psoralen moiety to the sugar ring of deoxyadenosine.
Psoralens have also been intensively used for the in vivo identification of miRNA targets. In cells, processed miRNAs carry out their function by forming a miRNA–protein complex with Argonaute proteins resulting in the RNA induced silencing complex (RISC). This RISC complex directs the miRNA to complementary mRNAs, resulting in degradation of the latter and gene silencing. The miR-TRAP strategy (miRNA-target RNA affinity purification), was developed for the direct identification of miRNA targets, thereby avoiding the use of antibodies typically employed in classical immunoprecipitation strategies.49 The miR-TRAP strategy exploits the recognition of miRNAs by the Argonaute proteins and uses psoralen-modified miRNAs for photo-crosslinking of miRNA–RNA complexes. These miRNAs are 3′-biotinylated and after crosslinking of the biotinylated miRNA probes to their targets, the complexes are isolated via streptavidin pull-down and analysed via reverse transcription quantitative polymerase chain reaction (RT-qPCR). Photo-crosslinking resulted in a 4 to 20-fold enrichment of miRNA targets and facilitated the identification of downregulated or repressed miRNA targets. An alternative strategy for the identification of miRNA targets was developed by Hall et al., and relies on miRNA crosslinking in combination with immunoprecipitation.50 Their strategy, called miR-CLIP (miRNA crosslinking and immunoprecipitation), was tested with the miRNA-106a, which is a psoralen- and biotin-modified miRNA mimic. Upon transfection of the miRNA-106a probe into HeLa cells and subsequent photo-crosslinking, the complexes were purified via Argonaute-based immunoprecipitation. Subsequently, the RNA was isolated and miRNA-target complexes were further purified via streptavidin affinity purification. During this step only miRNA crosslinked targets are isolated. Analysis via deep sequencing identified hundreds of targets, with amongst them the H19 lncRNA which is involved in cell proliferation51 and is regarded as an important cancer biomarker.52 These examples illustrate the benefit of crosslinker-based strategies in applications such as SNP detection and miRNA target identification even if, in the discussed examples, the authors did not provide any comparative data as to the pull-down efficiency of the crosslinker-based strategy versus that of the non-covalent counterpart.
2.3 Vinylcarbazole-based crosslink agents
Psoralens have been demonstrated to be thymine selective photo-crosslinkers (vide supra). However, the necessity for an AT or AU region restricts their general applicability. Moreover, the short wavelength UV-irradiation (254 nm) required for the photo-reversibility results in the formation of pyrimidine dimers, a common type of DNA damage. To circumvent these drawbacks, Fujimoto and co-workers reported on the synthesis of a 3-cyanovinylcarbazole nucleoside (CNVK) as a novel photo-activatable crosslinking agent for site-specific intermolecular nucleic acid crosslinking.53 This agent is selectively activated by short exposure to long-wavelength UV-A light (366 nm) and possesses higher photo-reactivity compared to psoralens. Indeed, the crosslinking reaction with thymidine was reported to proceed at 97% yield upon 1 second of photo-irradiation. Moreover, the reversible nature of the crosslink reaction at 312 nm irradiation was confirmed and shown to proceed without DNA damage. Molecular modelling studies indicated that the vinyl group of the CNVK moiety is stacked onto the C5–C6 double bond of pyrimidine nucleobases located at the −1 position on the complementary strand. This allows the photochemical [2+2] cycloaddition reaction to take place (Fig. 5). Kinetic, thermodynamic and NMR structural analysis indicated that the photo-crosslinking reaction with thymine proceeds via the trans-isomer of CNVK, yielding one single photoadduct.54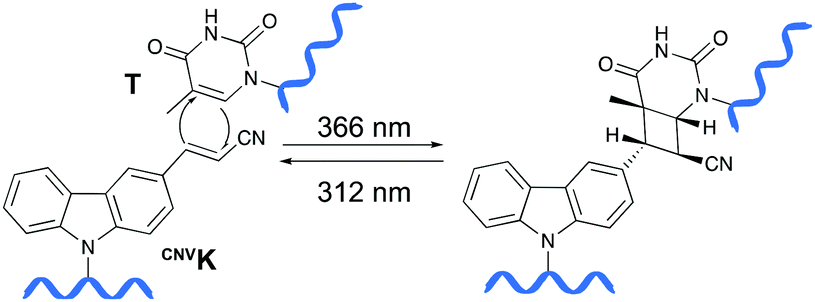 | ||
| Fig. 5 Mechanism of the photo-crosslinking reaction between 3-cyanovinylcarbazole (CNVK) and thymine (T) upon UV-A irradiation (366 nm). Photo-reversibility of the crosslinking reaction can be achieved by irradiating the product at 312 nm. | ||
This ultrafast nucleic acid crosslinking strategy proved to be clean, high-yielding and controllable in terms of selectivity and reversibility. Moreover, its biocompatibility was demonstrated both in vitro and in vivo (vide infra). Throughout the past years, variants of the CNVK moiety have been designed that operate via a similar [2+2] cycloaddition mechanism. For example, the pyranovinylcarbazole derivative PCX was developed (Fig. 6), which could be selectively activated by visible light irradiation (400 nm).55 Substitution of the rigid deoxyribose sugar in CNVK and PCX with a more flexible D-threoninol linker was also explored. The use of this modified building block allows increasing the crosslinking reaction rate substantially as the increased flexibility introduced by the D-threoninol linker minimizes entropic loss during hybridization (Fig. 6).56–58
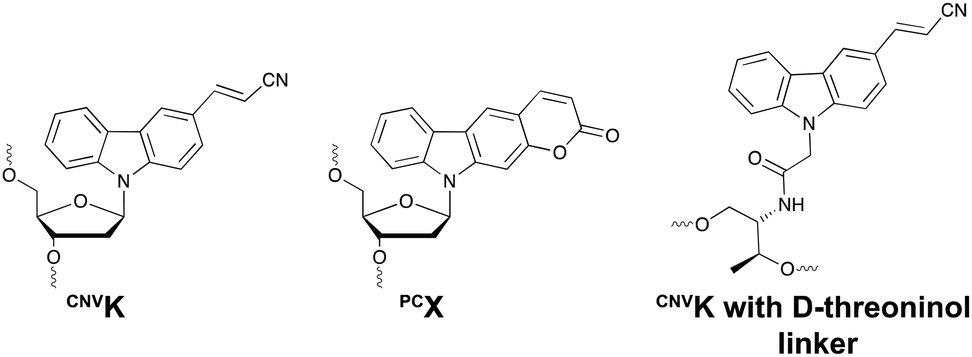 | ||
| Fig. 6 Structure of 3-cyanovinylcarbazole (CNVK), pyranocarbazole (PCX) and 3-cyanovinylcarbazole with D-threoninol linker. | ||
From a practical point of view, CNVK crosslinkers have been used in the design of shielded covalent probes. These are nucleic acid hairpin structures containing a single stranded tail sequence and a crosslinking moiety in their stem region (Fig. 7A).59 In the presence of a complementary DNA or RNA target, hybridization between the target and the tail sequence (step A) triggers the unfolding of the hairpin structure and results in probe-target binding (step B). After complete hybridization (step B), selective crosslinking to the target can be initiated using UV-A irradiation, thereby locking the probe-target interaction (step C). By optimizing the length of the single stranded tail, efficient mismatch discrimination could be achieved, even for SNP detection. The observed high selectivity is a consequence of the conformational change imposed by the probe-target binding event. Indeed, any mismatch destabilizes the probe–target complex and favours the non-target-bound hairpin structure. CNVK crosslinkers were also used in a pull-down experiment for the identification of miR-29b targets, a miRNA involved in e.g. apoptosis and cell differentiation.60 In this assay, miR-29b mimics were employed that contained both biotin and CNVK moieties. Addition of the CNVK modified miR-29b mimics to intact or lysed mouse and human cell lines, successfully induced complex formation with the endogenous Argonoute 2 complex present in these cells, with no observed cytotoxicity. Isolation of the complex then allowed the identification of both known and novel miR-29b targets. By employing the CNVK probes, a 3.9 to 20-fold enrichment was achieved compared to the non-crosslink-based method.
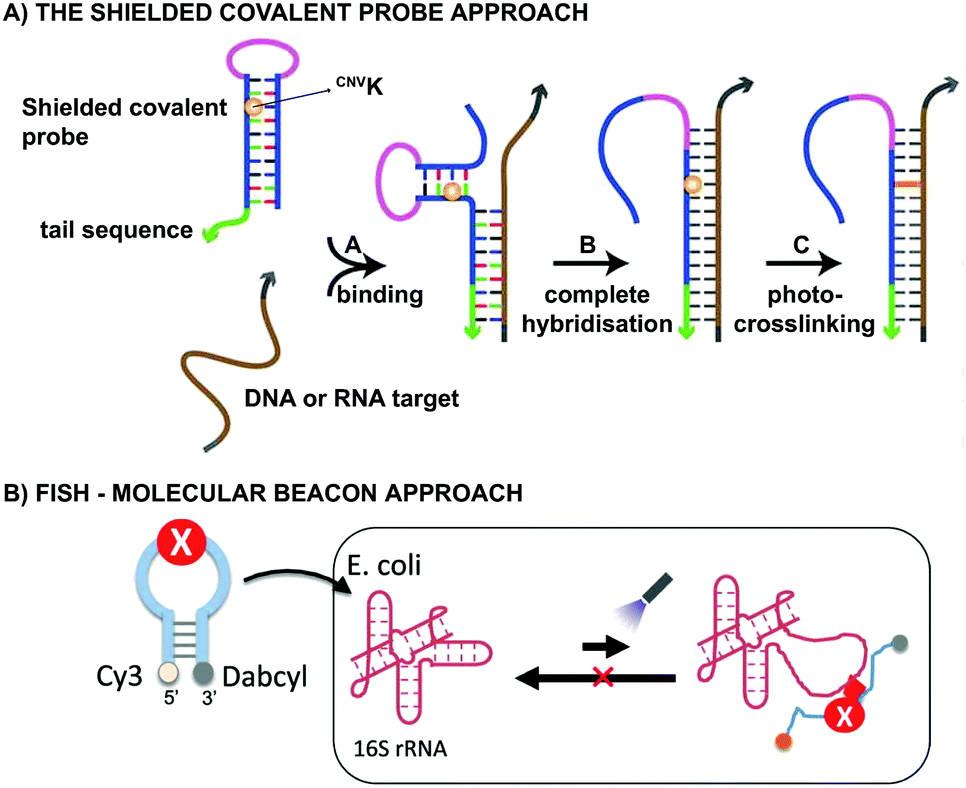 | ||
| Fig. 7 (A) Principle of the shielded covalent probe approach. Upon hybridization of a DNA or RNA target with the probe's tail sequence (step A), the hairpin structure unfolds (step B). After complete hybridization, the probe-target interaction can be locked by initiation of the crosslink reaction (step C). Figure reprinted (adapted) with permission from J. Am. Chem. Soc., 2013, 135, 26, 9691–9699 (https://pubs.acs.org/doi/10.1021/ja4009216), further permission related to the material excerpted should be directed to ACS. (B) Principle of the molecular beacon approach applied in the fluorescence in situ hybridization assays. Cy3 = cyanine fluorophore, Dabcyl = quencher, X = CNVK or PCX crosslinker moiety. Figure reprinted (adapted) from K. Fujimoto, M. Hashimoto, N. Watanabe and S. Nakamura, Bioorganic Med. Chem. Lett., 2019, 29, 2173–2177, Copyright (2020) with permission from Elsevier. | ||
Both CNVK and PCX crosslinkers have been used in wash-free fluorescence in situ hybridization assays (FISH). This methodology is commonly employed for investigating the location of nucleic acids in cells.61,62 In the here described application, a molecular beacon modified at the 5′ end with a fluorophore (e.g. Cy3) and at the 3′ end with a quencher (e.g. dabcyl) is used (Fig. 7B). Prior to target binding, fluorophore and quencher are in close proximity to each other, which suppresses the fluorescent signal. Upon target binding, the beacon unfolds, which in turn increases the distance between the fluorophore and quencher. As a result, the fluorescent signal is switched on. The probe-RNA interaction can subsequently be locked through photo-irradiation of the crosslinker moiety. CNVK and PCX modified molecular beacons have been applied for the visualisation of E. coli 26S rRNA in fixed and living cells respectively (Fig. 7B). The fast photo-crosslinking reaction allowed targeting RNA molecules with complex secondary structures and improved the detection sensitivity of the assay. An additional advantage of the molecular beacon approach is that washing steps can be omitted. Applications of cyanovinyl-based crosslinkers are not limited to the examples given above. Indeed, these fascinating constructs have paved their way in applications such as 19F-NMR based detection of nucleic acid sequences,63 in DNA-peptide photo-crosslinking,64 in DNA assemblies65 and in antisense applications66,67 to name a few.
2.4 4-Thiouridines
Within the realm of photo-activatable crosslinking agents, 4-thiouridines have not received the same attention as vinylcarbazole-based crosslinkers. The photo-reactive properties of these uridine analogues are obtained by replacing the carbonyl oxygen at position 4 by a sulphur atom rendering them capable of forming adducts with nucleobases and amino acids. Upon UV irradiation at 365 nm, 4-thiouridines react with nearby cytosines via a [2+2] cycloaddition reaction (Fig. 8).68,69 The unstable thietane intermediate formed, ring opens with concomitant formation of a stable covalent bond. Contrary to previous photo-activatable crosslinkers, this reaction is irreversible. 4-Thiouridines are easily incorporated into RNA sequences with minimal distortion due to their small size and retained base pairing ability. Consequently, they have been used in the identification of protein–RNA interactions‡ (e.g. in immunoprecipitation approaches70), and RNA–nucleic acid interactions. For example, binding of miRNA-10a to the 5’ untranslated region of ribosomal protein mRNAs was discovered by employing 4-thiouridine crosslinking in combination with streptavidin affinity purification.71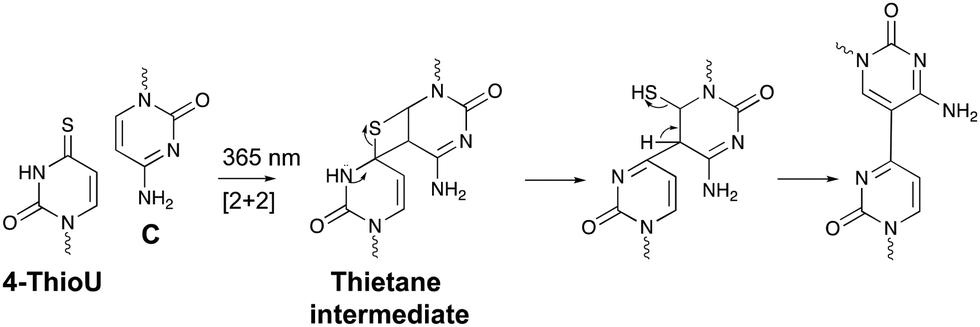 | ||
| Fig. 8 Mechanism for the reaction between 4-thiouridines (4-ThioU) and cytosine (C). | ||
2.5 Diazirines
A last example of photo-activatable crosslinking agents concerns the diazirines. Unlike aforementioned photo-crosslinkers, diazirines do not react via a [2+2] cycloaddition mechanism. Instead, irradiation with UV-A light generates a reactive carbene intermediate, which is capable of reacting with a variety of chemical groups, even unreactive C–H bonds (Fig. 9). | ||
| Fig. 9 Mechanism of diazirine activation shown for a 3-phenyl-3-trifluoromethyl-3H-diazirine derivative. | ||
Diazirine crosslinking has been used for interstrand crosslinking of nucleic acids and to study nucleic acid–protein or protein–protein interactions. It should therefore not come as a surprise that throughout the years a plethora of diazirine analogues have been developed and synthesized for this purpose.72–76 Diazirines possess certain properties which renders them superior compared to other existing (photo)-crosslinking methods in certain applications. For instance, the long wavelength UV-A photo-irradiation minimizes UV-mediated damage in biological systems and although other crosslinkers exist that allow remediating this problem (vide supra), the small nature of diazirines minimizes steric hindrance compared to its contesters. Furthermore, the stable C–C bonds formed upon crosslinking accounts for the robustness of this methodology. The high-reactivity of carbenes and their ability to react with all 4 nucleobases (A, C, G, T/U) makes them ideal for the identification of unknown nucleic acid targets (e.g. miRNA targets).73 Nakamoto et al. investigated the use of RNA probes modified with 3-phenyl-3-trifluoromethyl-3H-diazirines for capturing miRNA targets. In a first proof-of-concept study, they substituted the nucleobases of ribonucleosides with a trifluoromethyl diazirine moiety, which was connected via an acetal linkage to the sugar ring. This was followed by testing the efficiency and gene silencing ability of these analogues.77 Having obtained satisfactory results, the authors assessed the applicability of the most promising photo-reactive diazirine moiety for the labelling of mRNA targets in living cells.78 The model system of choice was the miRNA-145, which targets the known mRNAs FSCN1 and KLF4. Transfection of dual crosslinker- and biotin-modified miRNA probes in DLD-1 cancer cells, followed by subsequent photo-crosslinking, streptavidin purification and quantification of the isolated crosslinked product, showed an enhanced enrichment of the FSCN1 and KLF4 mRNA targets with the crosslinker-modified probes compared to the unmodified control probes. This illustrates the applicability of diazirine crosslinkers for the labelling and targeting of miRNAs targets. In 2018, an alkyne-modified diazirine nucleoside was designed for the development of tag-free miRNA probes.79 Biotinylation of miRNAs hampers efficient RISC loading and consequently interferes with mRNA targeting.72 By introducing the biotin moiety after photo-crosslinking, here achieved via a copper catalysed azide–alkyne cycloaddition reaction (CuAAC) between the alkyne modified diazirine nucleoside and the azide-modified biotin moiety, the degree of RISC-loading was increased. Moreover, these alkyne-modified miRNAs showed comparable gene silencing abilities as their unmodified counterparts. As such, this strategy could be potentially useful for the identification of unknown miRNA targets.
3. Crosslinking probes activated by chemical agents
3.1 Phenyl selenide
In general, light-controlled activation of crosslinking agents represents a green and biocompatible strategy, which is often applied in a biological context. However, when irradiation with short wavelength UV is required, UV-mediated damage is often observed and an undesirable side effect. Additionally, the efficiency of photo-activation depends on the transparency of the sample and the limited penetration depth of UV- or visible light would hamper photo-activation in turbid samples or in tissues. Crosslinking probes activated by chemical agents might offer a solution in this context.A first example of crosslinking agents that are activated by chemical means encompasses phenyl selenides. These intrinsic unreactive probes can be activated via photo-irradiation or through the addition of an oxidant (e.g. singlet oxygen 1O2 or sodium periodate NaIO4).80,81 In the case of oxidative activation, the phenyl selenide is converted into a selenoxide (Fig. 10, step a), which reacts further via a [2,3] sigmatropic rearrangement (step b). The formed methide species acts as a powerful Michael-acceptor and reacts with the N1 amine of purine nucleobases (step c). In the case of adenine alkylation, the formed N1 crosslinked adduct isomerizes further to the more stable N6-adduct (step d).82 Alternatively, when photo-irradiation is employed to activate the phenyl selenide, a radical intermediate is formed (Fig. 10, step a′) which is capable of alkylating the N1 position of purine bases. To explore the utility of phenyl selenides in crosslinking applications, multiple analogues have been developed either as bifunctional phenols83 or as modified pyrimidine nucleobases80,82 (Fig. 10B). The major advantage of the latter is their compatibility with solid phase DNA synthesis. Phenyl selenide analogues have been applied for single nucleotide discrimination in plasmid DNA.84 In this assay, biotinylated oligonucleotide probes containing a phenyl selenide-modified 2′-deoxythymidine moiety (PhSe-dT) were crosslinked to target DNA sequences. The formed adducts were subsequently detected using a fluorescent reporter assay based on an avidin-horseradish peroxidase conjugate. The target sequence was codon 248 in exon 7 of the p53 gene, which is often prone to G → A mutations in human cancer. This methodology allowed the discrimination of single nucleotide mismatches with an efficiency of 193:1. Moreover, the target DNA could be detected in the low nanomolar range without the need for PCR amplification.
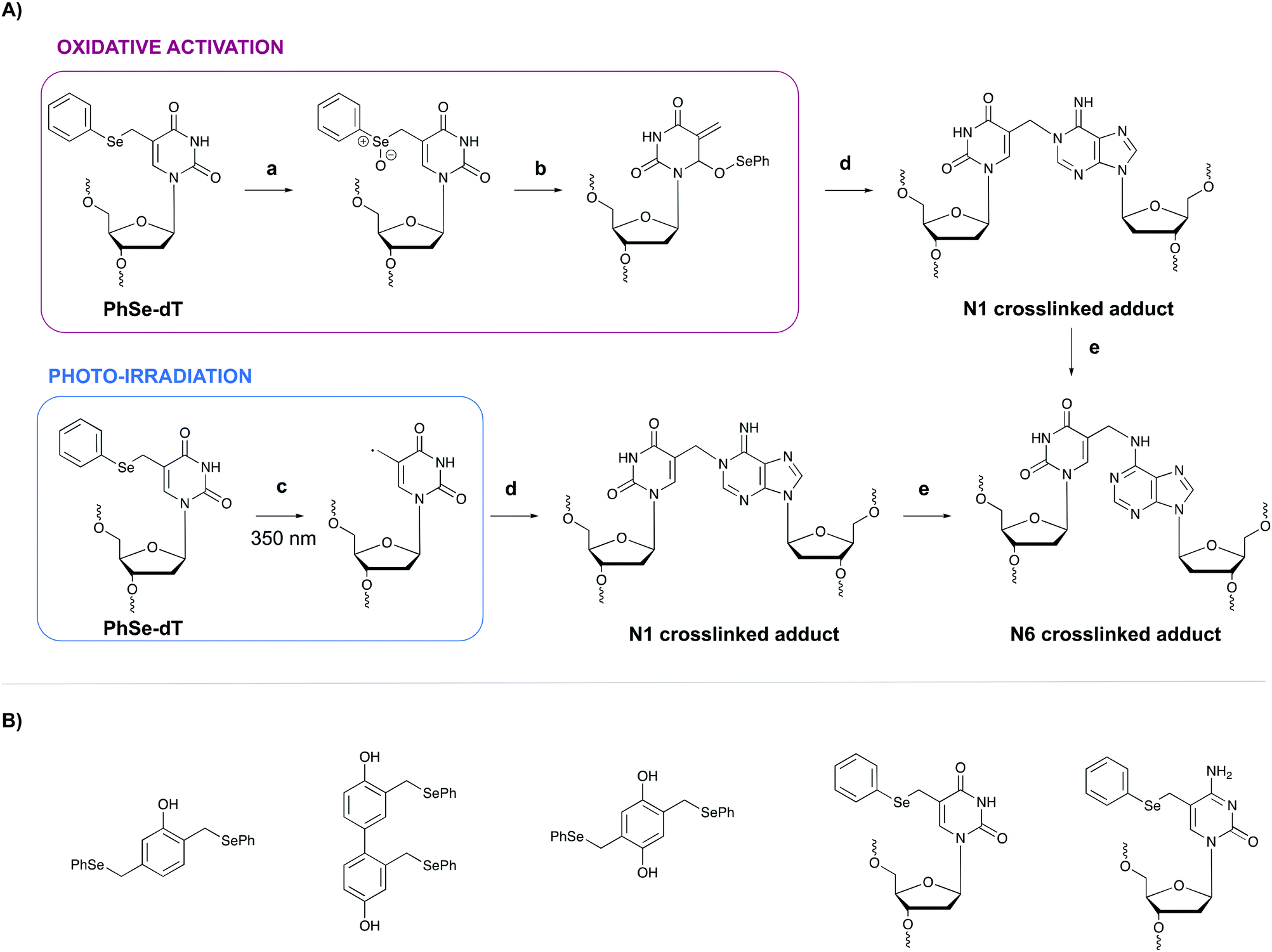 | ||
| Fig. 10 (A) Mechanism of the oxidative activation (top) and photo-irradiation (bottom) of the phenyl selenide-modified thymidine (PhSe-dT) and subsequent reaction with an opposing deoxyadenosine (dA). (a) NaIO4 or 1O2 (b) [2,3]-sigmatropic rearrangement (c) photo-irradiation (d) Reaction with N1 amine of dA (e) isomerisation. (B) Structure of bifunctional phenyl selenide-modified phenol derivatives and phenyl selenide-modified pyrimidine nucleobases. PhSe-dT = phenyl selenide-modified 2′-deoxythymidine; PhSe-5-Me-dC = phenyl selenide-modified 5-methyl-2′-deoxycytidine. | ||
3.2 Furan
The major advantage of phenyl selenides is their dual mode of activation in which one can choose the chemical or photo-irradiation approach depending on the application. In our research group, probes benefitting from a similar dual mode of activation were developed based on furan as a masked reactive moiety. The idea to use furan as reactive group is inspired by the intrinsic reactivity of this molecule in the body. Indeed, oxidation of furan by cytochrome P450 leads to a highly electrophilic di-aldehyde that can react with diverse nucleophiles such as those present on amino acid side chains or nucleic acid bases. From an experimental point of view, furan activation can be achieved chemically via oxidation with N-bromo-succinimide or via a photo-activation strategy. The latter employs singlet oxygen as oxidant, which is produced upon visible light irradiation of a suitable photosensitizer. The generated singlet oxygen induces furan oxidation through a [4+2] cycloaddition reaction with concomitant formation of the corresponding endoperoxide (Fig. 11A). Hydrolysis of this intermediate followed by elimination of the hydrogen peroxide anion yields the 4-oxo-2-enal moiety.85 The natural occurrence of singlet oxygen in biological systems and the possibility to perform singlet oxygen mediated reactions in aqueous solutions renders this approach biocompatible and green. Furthermore, the need for furan activation guarantees spatiotemporal control. Indeed, as the irradiation position, intensity and frequency can be varied, local activation of furan can be achieved without affecting non-target tissue or cells.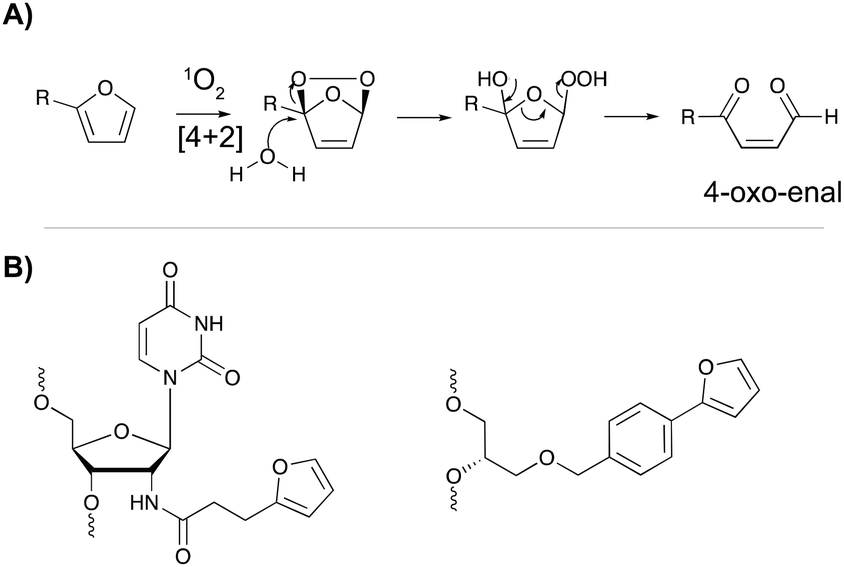 | ||
| Fig. 11 (A) Mechanism of furan activation via reaction with singlet oxygen. (B) Examples of furan crosslinkers incorporated in oligonucleotide sequences. | ||
For nucleic acid interstrand crosslinking, hybridization of a furan-modified oligonucleotide with its complementary strand brings both reaction partners in close proximity. The base-pair complementarity needed for hybridization is responsible for the sequence selectivity of the system. Subsequent activation of the furan by singlet oxygen triggers reaction with an opposing adenine or cytosine base in the complementary strand, resulting in crosslink formation. The proximity-dependence of this reaction and the possibility for visible light activation makes this a selective, attractive and biocompatible crosslinking methodology. Hence, it should not come as a surprise that different furan derivatives have been developed (Fig. 11B) and that the furan-based crosslinking strategy has been successfully implemented for interstrand nucleic acid crosslinking,85–88 PNA crosslinking,89,90 crosslinking of DNA and RNA with proteins91 and crosslinking of peptides with proteins.92 Recently, furan-PNA probes were used in a diagnostic context for the detection of 22-mer DNA or RNA targets. This assay is based on the templated ligation of a furan modified PNA strand, immobilized on a surface, and a PNA strand modified with a photosensitizer and a nucleophilic moiety. The photosensitizer enables local singlet oxygen production, while the nucleophile is capable of reacting with the furan warhead. Ligation between both PNA strands is obtained upon furan activation with visible light, but only occurs in presence of a complementary oligonucleotide target serving as template for both strands (Fig. 12). Only upon ligation, a read-out is obtained. This templated set-up enables selective enzyme-free detection of oligonucleotide sequences, and is potentially valuable for the detection of miRNA targets.90 Moreover, this furan-oxidation based methodology is currently under investigation for the development of an improved ChiRP assay for the in vitro identification of long non-coding RNA targets. Classical ChiRP experiments typically rely on the non-covalent hybridization of a series of biotinylated capture probes to their complementary sequences on a ncRNA target, followed by biotin–streptavidin pull-down. The aim is to isolate the ncRNA and identify its relevant interaction partners. Unfortunately, this strategy suffers from low selectivity and efficiency, partly due to the non-covalent nature of the interaction between capture probe and ncRNA target. By covalently fixing this interaction through activation of a furan crosslinker, more selective and higher yielding in vitro ncRNA target identification should become possible. Recent studies in our group focus on the use of furan crosslinking-enhanced enrichment of lncRNA sequences in vitro.
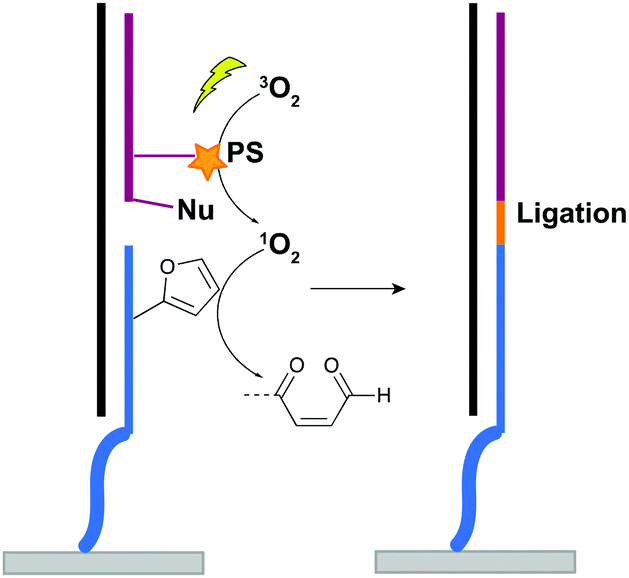 | ||
| Fig. 12 Principle of the templated ligation of two PNA strands: a furan modified PNA strand (blue) and a PNA strand (purple), dually modified with a nucleophile (Nu) and a photosensitizer (PS). Ligation is only observed upon hybridization with an oligonucleotide target (black strand). | ||
4. Oligonucleotides containing abasic sites as inherently reactive crosslinking agents
Among the methodologies at hand, furan-oxidation based crosslinking is not the only one inspired by a naturally occurring process. Abasic sites (AP), also referred to as apurinic or apyrimidinic sites (Fig. 13), are one of the most occurring types of DNA damage. They are generally formed upon hydrolysis of the glycosidic bond connecting the nucleobase with the sugar backbone in nucleotides, but can also be generated upon exposure to radiation, anticancer drugs or mutagens.93 AP sites exist in two forms that are in equilibrium with each other, namely the cyclic hemiacetal form and the ring-opened aldehyde form (Fig. 13). Their reactivity is inherently linked to the electrophilic nature of the aldehyde, which makes them prone to react with nearby nucleophiles. Consequently, AP sites are highly mutagenic as they can introduce crosslinks in duplex DNA, which might hinder replication, transcription or the cellular repair mechanisms. Moreover, abasic sites lack the heterocyclic purine or pyrimidine bases that form stabilizing hydrogen bonding interactions upon base-pairing, which results in destabilization of the DNA duplex.93,94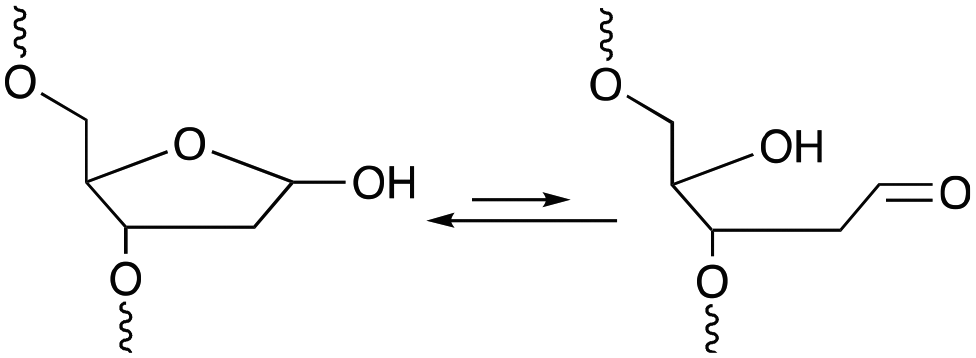 | ||
| Fig. 13 Equilibrium between the cyclic hemiacetal form and the ring-opened aldehyde form of the abasic site. | ||
The inherent reactivity of AP sites has inspired researchers to exploit this moiety for interstrand oligonucleotide crosslinking. Consequently, methods compatible with solid phase DNA synthesis, have been developed for the generation of abasic sites in oligonucleotides. For example, AP sites can be generated upon reaction of 2′-deoxyuridine with the enzyme uracil DNA glycosylase (UDP), through periodate oxidation of a vicinal diol or upon irradiation of a photolabile o-nitrobenzyl acetal.95 Sequences containing abasic sites have been employed as crosslinking agents for the detection of single nucleotide polymorphisms. Gates et al. designed AP probes targeting a T → A mutation at position 1799 in the human BRAF kinase gene.96 The AP sites haven proven to be reactive towards the N6 exocyclic amino group of 2′-deoxyadenosine, when positioned one nucleotide closer to the 3′ site of the opposing strand (Fig. 14A).97 Probes fully complementary to the wildtype sequence and modified with an abasic site, successfully discriminated between the wildtype and the mutant gene, with crosslinking yields up to 83% for the latter and no detectable yields for the wildtype. The flexibility induced by the mismatch in the mutant gene is most likely responsible for the selectivity of the reaction and for the observed yield. Detection of the crosslinked product and hence the SNP, has been performed via a variety of methods such as fluorescent, colorimetric or electrochemical detection. Additionally, a nanopore detection strategy was developed based on an α-hemolysin ion channel which is capable of capturing nucleic acid structures when subjected to an electric potential.96 The measured read-out is significantly different for crosslinked duplexes compared to their non-crosslinked counterparts, thus allowing the efficient detection of the mutant genes (in this case the BRAF gene).
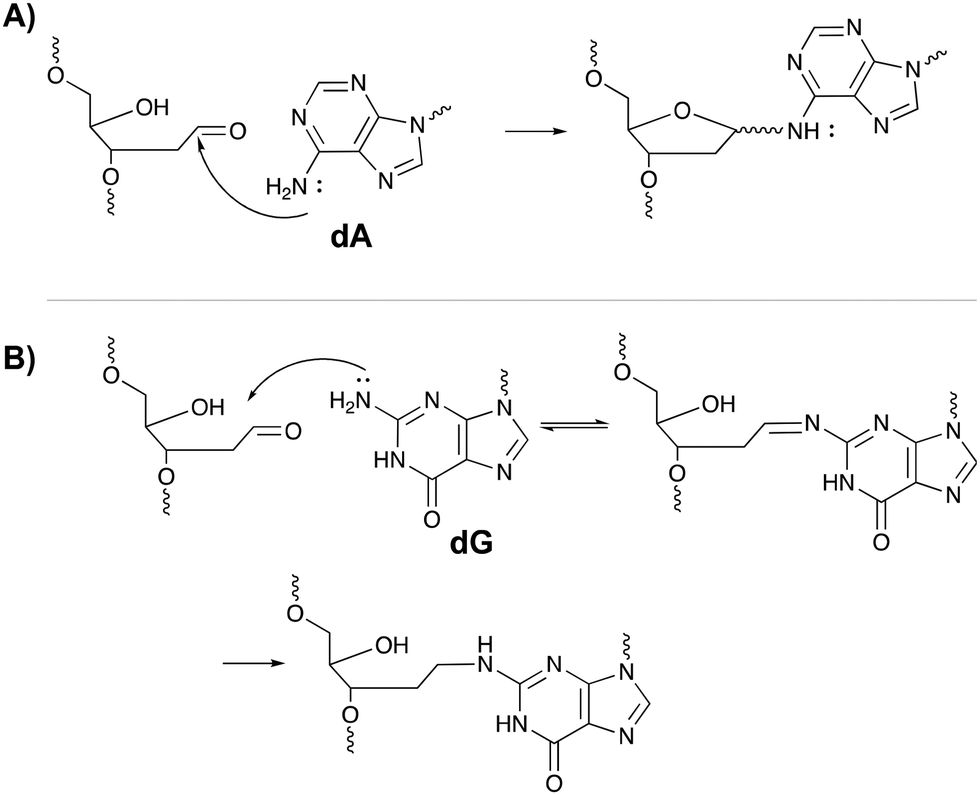 | ||
| Fig. 14 Mechanism of crosslink formation between the abasic site on one oligonucleotide strand and the N6-exocyclic amine of 2′-deoxyadenosine (A) or the N2-exocyclic amine of 2-deoxyguanosine (B) on the opposite strand. | ||
A similar strategy was employed for targeting the cancer driven C → G mutation at position 35 in the human KRAS gene.98 Reaction between the N2-exocyclic amine of 2-deoxyguanosine and the abasic site proceeds via the formation of an imine intermediate, which can be reduced with NaCNBH3 to form a stable covalent linkage (Fig. 14B). In contrast to 2′-deoxyadenosine crosslinking, the AP site should be located one nucleotide closer to the 5′ site of the opposing strand. This discrepancy is a consequence of the position of the exocyclic amine within a DNA duplex (major groove for adenine vs. minor groove for guanine).97 Interestingly, AP probes introducing non-canonical structures such as loops and bulges drastically increase the yield of the crosslinking reaction, most likely due to the increased flexibility.98
Overall, abasic sites offer elegant and efficient possibilities for interstrand nucleic acid crosslinking and have proven useful for SNP detection. The ease of incorporating AP precursors in DNA sequences via solid phase synthesis, the efficiency of abasic site generation and the compatibility of the resulting AP probes with different detection strategies, have paved the way for further exploration of such crosslinking agents in biology-related applications.
5. Conclusion
Hybridization-based assays are standard practice when it comes to the study and detection of nucleic acids and their interaction partners. These assays are crucial for gaining insight in biological processes but suffer from limited affinity and selectivity. The transient nature of nucleic acid interactions and the reversibility of hybridization often complicates straightforward analysis. Crosslinker-modified nucleic acid probes can offer an elegant way to circumvent the pitfalls of standard hybridization-based methods as probe-target interactions are covalently locked. However, whilst a plethora of crosslinking agents has been designed, only few have reached the cellular biology labs. This is mainly due to the need for such reactions to be fast, high yielding, biocompatible and to occur with high specificity leading to stable conjugates. Furthermore, the crosslinking reactions should proceed in highly diluted conditions, in presence of other biomolecules that might impede binding of the crosslinking probe to the nucleic acid target. The occurrence of secondary and tertiary DNA/RNA structure might further complicate reaction. To date, no crosslinking agent exists that meets all these criteria. Nevertheless, it is clear from the applications discussed in this review that hybridization-based assays can indeed benefit from the use of crosslinking agents. Further optimization of existing agents or the design of novel crosslinking methodologies can help improving the general applicability of crosslinking agents in a biological context and may eventually result in the common use of crosslinkers in the study and detection of nucleic acids. In this prospect, there is room for improvement in the design of new crosslinking moieties. Indeed, the ideal crosslinker should combine the various features of the existing agents: (1) a small size which ensures minimal sterical hindrance in analogy with diazirines; (2) a fast reaction which proceeds in high yield such as the vinylcarbazole derivatives; (3) reversible in nature like coumarin, psoralen and vinycarbazole crosslinkers; (4) a pro-reactive moiety to ensure selective reaction and (5) based on a moiety that can be incorporated chemically or enzymatically in oligonucleotide sequences in analogy to thiouridines.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Vincent Ornelis is gratefully acknowledged for proofreading and correcting the manuscript. Joke Elskens is indebted to the Fonds Wetenschappelijk Onderzoek (FWO)-Vlaanderen (FWO18-SBB-073) for a position as SB PhD fellow.Notes and references
- C. Francis, Nature, 1970, 227, 561–563 CrossRef.
- S. Hombach and M. Kretz, Adv. Exp. Med. Biol., 2016, 937, 53–69 CrossRef.
- I. Tsagakis, K. Douka, I. Birds and J. L. Aspden, J. Pathol., 2020, 250, 480–495 CrossRef CAS.
- I. Martianov, A. Ramadass, A. Serra Barros, N. Chow and A. Akoulitchev, Nature, 2007, 445, 666–670 CrossRef CAS.
- P. Grote and B. G. Herrmann, RNA Biol., 2013, 10, 1579–1585 CrossRef CAS.
- M. Kretz, Z. Siprashvili, C. Chu, D. E. Webster, A. Zehnder, K. Qu, C. S. Lee, R. J. Flockhart, A. F. Groff, J. Chow, D. Johnston, G. E. Kim, R. C. Spitale, R. A. Flynn, G. X. Y. Zheng, S. Aiyer, A. Raj, J. L. Rinn, H. Y. Chang and P. A. Khavari, Nature, 2013, 493, 231–235 CrossRef CAS.
- Y. Wang, Z. Xu, J. Jiang, C. Xu, J. Kang, L. Xiao, M. Wu, J. Xiong, X. Guo and H. Liu, Dev. Cell, 2013, 25, 69–80 CrossRef CAS.
- H. Y. C. Ci Chu, Methods Mol. Biol., 2016, 1480, 115–123 CrossRef.
- M. D. Simon, C. I. Wang, P. V. Kharchenko, J. A. West, B. A. Chapman, A. A. Alekseyenko, M. L. Borowsky, M. I. Kuroda and R. E. Kingston, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 20497–20502 CrossRef CAS.
- F. C. Y. Lee and J. Ule, Mol. Cell, 2018, 69, 354–369 CrossRef CAS.
- A. M. Schmitt and H. Y. Chang, Cancer Cell, 2016, 29, 452–463 CrossRef CAS.
- H. Schwarzenbach, D. S. B. Hoon and K. Pantel, Nat. Rev. Cancer, 2011, 11, 426–437 CrossRef CAS.
- M. Grasso, P. Piscopo, A. Confaloni and M. A. Denti, Molecules, 2014, 19, 6891–6910 CrossRef.
- P. Wan, W. Su and Y. Zhuo, Mol. Neurobiol., 2017, 54, 2012–2021 CrossRef CAS.
- L. Tribolet, E. Kerr, C. Cowled, A. G. D. Bean, C. R. Stewart, M. Dearnley and R. J. Farr, Front. Microbiol., 2020, 11, 1–15 CrossRef.
- R. A. Sunde, J. Nutr. Biochem., 2010, 21, 665–670 CrossRef CAS.
- I. Gareev, O. Beylerli, G. Yang, J. Sun, V. Pavlov, A. Izmailov, H. Shi and S. Zhao, Clin. Exp. Med., 2020, 20, 349–359 CrossRef CAS.
- T. Gilboa, P. M. Garden and L. Cohen, Anal. Chim. Acta, 2020, 1115, 61–85 CrossRef CAS.
- E. Olkhov-Mitsel and B. Bapat, Cancer Med., 2012, 1, 237–260 CrossRef CAS.
- M. Kulis and M. Esteller, Adv. Genet., 2010, 70, 27–56 Search PubMed.
- P. K. Periyannan Rajeswari, L. M. Soderberg, A. Yacoub, M. Leijon, H. Andersson Svahn and H. N. Joensson, J. Microbiol. Methods, 2017, 139, 22–28 CrossRef CAS.
- J. P. Jakupciak, W. Wang, M. E. Markowitz, D. Ally, M. Coble, S. Srivastava, A. Maitra, P. E. Barker, D. Sidransky and C. D. O’Connell, J. Mol. Diag., 2005, 7, 258–267 CrossRef CAS.
- S. Nainar, C. Feng and R. C. Spitale, ACS Chem. Biol., 2016, 11, 2091–2100 CrossRef CAS.
- M. D. Simon and M. Machyna, Cold Spring Harb. Perspect. Biol., 2019, 11 DOI:10.1101/cshperspect.a032276.
- W. A. Velema, H. S. Park, A. Kadina, L. Orbai and E. T. Kool, Angew. Chem., Int. Ed., 2020, 59, 22017–22022 CrossRef CAS.
- S. Shapiro, Angiology, 1953, 4, 380–390 CrossRef CAS.
- H. Sun, H. Fan, H. Eom and X. Peng, ChemBioChem, 2016, 17, 2046–2053 CrossRef CAS.
- M. M. Haque, H. Sun, S. Liu, Y. Wang and X. Peng, Angew. Chem., Int. Ed., 2014, 53, 7001–7005 CrossRef CAS.
- H. Sun, H. Fan and X. Peng, J. Org. Chem., 2014, 79, 11359–11369 CrossRef CAS.
- M. Shadmehr, G. J. Davis, B. T. Mehari, S. M. Jensen and J. C. Jewett, ChemBioChem, 2018, 19, 2550–2552 CrossRef CAS.
- Y. Chen, C. M. Clouthier, K. Tsao, M. Strmiskova, H. Lachance and J. W. Keillor, Angew. Chem., Int. Ed., 2014, 53, 13785–13788 CrossRef CAS.
- R. M. Franzini and E. T. Kool, ChemBioChem, 2008, 9, 2981–2988 CrossRef CAS.
- J. Zehnder, R. Van Atta, C. Jones, H. Sussman and M. Wood, Clin. Chem., 1997, 43, 1703–1708 CrossRef CAS.
- V. C. H. Lai, R. Guan, M. L. Wood, S. K. Lo, M. F. Yuen and C. L. Lai, J. Clin. Microbiol., 1999, 37, 161–164 CrossRef CAS.
- R. Peoples, H. Weltman, R. Van Atta, J. Wang, M. Wood, M. Ferrante-Raimondi, P. Cheng and B. Huan, Clin. Chem., 2002, 48, 1844–1850 CrossRef CAS.
- C. French, C. Li, C. Strom, W. Sun, R. Van Atta, B. Gonzalez and M. Wood, Clin. Chem., 2004, 50, 296–305 CrossRef CAS.
- J. E. Hearst, Chem. Res. Toxicol., 1989, 2, 69–75 Search PubMed.
- S. Peckler, B. Graves, D. Kanne, H. Rapoport, J. E. Hearst and S. H. Kim, J. Mol. Biol., 1982, 162, 157–172 CrossRef CAS.
- P. S. Song and K. J. J. Tapley, Photochem. Photobiol., 1979, 29, 1177–1197 CrossRef CAS.
- J. M. Kean, A. Murakami, K. R. Blake, C. D. Cushman and P. S. Miller, Biochemistry, 1988, 27, 9113–9121 CrossRef CAS.
- A. Yamayoshi, Y. Matsuyama, M. Kushida, A. Kobori and A. Murakami, Photochem. Photobiol., 2014, 90, 716–722 CrossRef CAS.
- A. Yamayoshi, R. Iwase, T. Yamaoka and A. Murakami, Chem. Comm., 2003, 3, 1370–1371 RSC.
- A. Yamayoshi, M. Higuchi, Y. Sakai, A. Kobori, T. Yamamoto, T. Shibata and A. Murakami, Nucleosides, Nucleotides and Nucleic Acids, 2019, 0, 1–12 Search PubMed.
- H. Li, V. J. Broughton-Head, G. Peng, V. E. C. Powers, M. J. Ovens, K. R. Fox and T. Brown, Bioconjug. Chem., 2006, 17, 1561–1567 CrossRef CAS.
- M. Higuchi, A. Yamayoshi, T. Yamaguchi, R. Iwase, T. Yamaoka, A. Kobori and A. Murakami, Nucleosides, Nucleotides and Nucleic Acids, 2007, 26, 277–290 CrossRef CAS.
- M. Higuchi, A. Kobori, A. Yamayoshi and A. Murakami, Bioorg. Med. Chem., 2009, 17, 475–483 CrossRef CAS.
- R. A. Cassidy, N. S. Kondo and P. S. Miller, Biochemistry, 2000, 39, 8683–8691 CrossRef CAS.
- A. Murakami, A. Yamayoshi, R. Iwase, J. Nishida, T. Yamaoka and N. Wake, Eur. J. Pharm. Sci., 2001, 13, 25–34 CrossRef CAS.
- H. Baigude, Ahsanullah, Z. Li, Y. Zhou and T. M. Rana, Angew. Chem., Int. Ed., 2012, 51, 5880–5883 CrossRef CAS.
- J. Imig, A. Brunschweiger, A. Brümmer, B. Guennewig, N. Mittal, S. Kishore, P. Tsikrika, A. P. Gerber, M. Zavolan and J. Hall, Nat. Chem. Biol., 2015, 11, 107–114 CrossRef CAS.
- T. Iempridee, Exp. Biol. Med., 2017, 242, 184–193 CrossRef CAS.
- Y. Liu, A. He, B. Liu, Z. Huang and H. Mei, BioMed Res. Int., 2019, 2019, 9056458 Search PubMed.
- Y. Yoshimura and K. Fujimoto, Org. Lett., 2008, 10, 3227–3230 CrossRef CAS.
- K. Fujimoto, A. Yamada, Y. Yoshimura, T. Tsukaguchi and T. Sakamoto, J. Am. Chem. Soc., 2013, 135, 16161–16167 CrossRef CAS.
- K. Fujimoto, S. Sasago, J. Mihara and S. Nakamura, Org. Lett., 2018, 20, 2802–2805 CrossRef CAS.
- S. Nakamura, R. Nakajima and K. Fujimoto, Chem. Lett., 2018, 47, 875–877 CrossRef CAS.
- K. Fujimoto, T. Yamaguchi, T. Inatsugi, M. Takamura, I. Ishimaru, A. Koto and S. Nakamura, RSC Adv., 2019, 9, 30693–30697 RSC.
- T. Sakamoto, Y. Tanaka and K. Fujimoto, Org. Lett., 2015, 17, 936–939 CrossRef CAS.
- J. R. Vieregg, H. M. Nelson, B. M. Stoltz and N. A. Pierce, J. Am. Chem. Soc., 2013, 135, 9691–9699 CrossRef CAS.
- A. Choudhary, D. P. Vanichkina, C. Ender, J. Crawford, G. J. Baillie, A. D. Calcino, K. Ru and R. J. Taft, RNA, 2018, 24, 597–608 CrossRef CAS.
- K. Fujimoto, M. Hashimoto, N. Watanabe and S. Nakamura, Bioorg. Med. Chem. Lett., 2019, 29, 2173–2177 CrossRef CAS.
- S. Nakamura, C. Kano and K. Fujimoto, Chem. Lett., 2017, 46, 1711–1713 CrossRef CAS.
- S. Nakamura, Y. Takashima and K. Fujimoto, Org. Biomol. Chem., 2018, 16, 891–894 RSC.
- Z. Qiu, S. Nakamura and K. Fujimoto, Org. Biomol. Chem., 2019, 17, 6277–6283 RSC.
- T. Gerling and H. Dietz, Angew. Chem., Int. Ed., 2019, 58, 2680–2684 CrossRef CAS.
- T. Sakamoto, A. Shigeno, Y. Ohtaki and K. Fujimoto, Biomater. Sci., 2014, 2, 1154–1157 RSC.
- K. Fujimoto, H. Yang-Chun and S. Nakamura, Chem. – Asian J., 2019, 1292, 1912–1916 CrossRef.
- A. Favre, C. Saintomé, J. L. Fourrey, P. Clivio and P. Laugâa, J. Photochem. Photobiol., B, 1998, 42, 109–124 CrossRef CAS.
- E. J. Sontheimer, Mol. Biol. Rep., 1994, 20, 35–44 CrossRef CAS.
- M. Hafner, M. Landthaler, L. Burger, M. Khorshid, J. Hausser, P. Berninger, A. Rothballer, M. Ascano, A. C. Jungkamp, M. Munschauer, A. Ulrich, G. S. Wardle, S. Dewell, M. Zavolan and T. Tuschl, Cell, 2010, 141, 129–141 CrossRef CAS.
- U. A. Ørom, F. C. Nielsen and A. H. Lund, Mol. Cell, 2008, 30, 460–471 CrossRef.
- M. Winnacker, S. Breeger, R. Strasser and T. Carell, ChemBioChem, 2009, 10, 109–118 CrossRef CAS.
- Z. Qiu, L. Lu, X. Jian and C. He, J. Am. Chem. Soc., 2008, 130, 14398–14399 CrossRef CAS.
- L. Dubinsky, B. P. Krom and M. M. Meijler, Bioorg. Med. Chem., 2012, 20, 554–570 CrossRef CAS.
- M. Liebmann, F. Di Pasquale and A. Marx, ChemBioChem, 2006, 7, 1965–1969 CrossRef CAS.
- Y. Sugihara, S. Tatsumi and A. Kobori, Chem. Lett., 2017, 46, 236–239 CrossRef CAS.
- K. Nakamoto and Y. Ueno, J. Org. Chem., 2014, 79, 2463–2472 CrossRef CAS.
- K. Nakamoto, K. Minami, Y. Akao and Y. Ueno, Chem. Comm., 2016, 52, 6720–6722 RSC.
- K. Nakamoto, Y. Akao and Y. Ueno, Bioorg. Med. Chem. Lett., 2018, 28, 2906–2909 CrossRef CAS.
- I. S. Hong and M. M. Greenberg, J. Am. Chem. Soc., 2005, 127, 10510–10511 CrossRef CAS.
- I. S. Hong, H. Ding and M. M. Greenberg, J. Am. Chem. Soc., 2006, 128, 485–491 CrossRef CAS.
- X. Peng, S. H. In, H. Li, M. M. Seidman and M. M. Greenberg, J. Am. Chem. Soc., 2008, 130, 10299–10306 CrossRef CAS.
- X. Weng, L. Ren, L. Weng, J. Huang, S. Zhu, X. Zhou and L. Weng, Angew. Chem., Int. Ed., 2007, 46, 8020–8023 CrossRef CAS.
- X. Peng and M. M. Greenberg, Nucleic Acids Res., 2008, 36, 5–11 CrossRef.
- M. Op de Beeck and A. Madder, J. Am. Chem. Soc., 2012, 134, 10737–10740 CrossRef CAS.
- S. Halila, T. Velasco, P. De Clercq and A. Madder, Chem. Comm., 2005, 936–938 RSC.
- M. O. De Beeck and A. Madder, J. Am. Chem. Soc., 2011, 133, 796–807 CrossRef.
- N. De Laet and A. Madder, J. Photochem. Photobiol. A Chem., 2016, 318, 64–70 CrossRef CAS.
- J. Elskens, A. Manicardi, V. Costi, A. Madder and R. Corradini, Molecules, 2017, 22, 2010 CrossRef.
- A. Manicardi, E. Cadoni and A. Madder, Chem. Sci., 2020, 11729–11739 RSC.
- L. L. G. Carrette, E. Gyssels, N. De Laet and A. Madder, Chem. Comm., 2016, 52, 1539–1554 RSC.
- W. Vannecke, C. Ampe, M. Van Troys, M. Beltramo and A. Madder, ACS Chem. Biol., 2017, 12, 2191–2200 CrossRef CAS.
- S. Dutta, G. Chowdhury and K. S. Gates, J. Am. Chem. Soc., 2007, 129, 1852–1853 CrossRef CAS.
- E. Gyssels, N. De Laet, E. Lumley and A. Madder, in Modified Nucleic Acids in Biology and Medicine, ed. S. Jurga, V. A. Erdmann (Deceased) and J. Barciszewski, Springer International Publishing, Cham, 2016, pp. 339–369 Search PubMed.
- M. M. Greenberg, Acc. Chem. Res., 2014, 47, 646–655 CrossRef CAS.
- M. I. Nejad, R. Shi, X. Zhang, L. Q. Gu and K. S. Gates, ChemBioChem, 2017, 18, 1383–1386 CrossRef CAS.
- N. E. Price, K. M. Johnson, J. Wang, M. I. Fekry, Y. Wang and K. S. Gates, J. Am. Chem. Soc., 2014, 136, 3483–3490 CrossRef CAS.
- X. Guo, M. I. Nejad, L. Q. Gu and K. S. Gates, RSC Adv., 2019, 9, 32804–32810 RSC.
Footnotes |
| † The term crosslinking is often used in the context of these hybridization-based assays but refers to the cell fixation in which oligonucleotide sequences and their interaction partners are covalently locked in a random fashion. In the current review, the term crosslinking refers to the introduction of a site-specific covalent link between a capture probe and its complementary nucleic acid target. |
| ‡ Thiouridines are expected to interact with lysine, tyrosine, tryptophane, methionine and cysteine residues. Although thiouridines are commonly applied for locking nucleic acid–protein interactions, the exact nature of the formed photoadducts and the mechanism of the photo-reaction is not very-well understood. |
| This journal is © The Royal Society of Chemistry 2021 |