
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
DNase I functional microgels for neutrophil extracellular trap disruption†
Aisa
Hosseinnejad
a,
Nadine
Ludwig
 b,
Ann-Katrin
Wienkamp
b,
Rahul
Rimal
a,
Christian
Bleilevens
c,
Rolf
Rossaint
c,
Jan
Rossaint
b and
Smriti
Singh
*ad
b,
Ann-Katrin
Wienkamp
b,
Rahul
Rimal
a,
Christian
Bleilevens
c,
Rolf
Rossaint
c,
Jan
Rossaint
b and
Smriti
Singh
*ad
aDWI—Leibniz-Institute for Interactive Materials e.V., Forckenbeckstr. 50, 52056 Aachen, Germany
bDepartment of Anesthesiology, Intensive Care and Pain Medicine, University Hospital Münster, Albert-Schweitzer-Campus 1, Bldg. A1, 48149 Münster, Germany
cDepartment of Anesthesiology, University Hospital RWTH Aachen, Pauwelsstraße 30, 52074 Aachen, Germany
dMax-Planck-Institut für medizinische Forschung, Jahnstraße 29, 69120 Heidelberg, Germany. E-mail: smriti.singh@mr.mpg.de
First published on 23rd November 2021
Abstract
Neutrophil extracellular traps (NETs) are web-like chromatin structures produced and liberated by neutrophils under inflammatory conditions which also promote the activation of the coagulation cascade and thrombus formation. The formation of NETs is quite prominent when blood comes in contact with artificial surfaces like extracorporeal circuits, oxygenator membranes, or intravascular grafts. DNase I as a factor of the host defense system, digests the DNA backbone of NETs, which points out its treatment potential for NET-mediated thrombosis. However, the low serum stability of DNase I restricts its clinical/therapeutic applications. To improve the bioavailability of the enzyme, DNase I was conjugated to the microgels (DNase I MG) synthesized from highly hydrophilic N-(2-hydroxypropyl) methacrylamide (HPMA) and zwitterionic carboxybetaine methacrylamide (CBMAA). The enzyme was successfully conjugated to the microgels without any alternation to its secondary structure. The Km value representing the enzymatic activity of the conjugated DNase I was calculated to be 0.063 μM demonstrating a high enzyme–substrate affinity. The DNase I MGs were protein repellant and were able to digest NETs more efficiently compared to free DNase in a biological media, remarkably even after long-term exposure to the stimulated neutrophils continuously releasing NETs. Overall, the conjugation of DNase I to a non-fouling microgel provides a novel biohybrid platform that can be exploited as non-thrombogenic active microgel-based coatings for blood-contacting surfaces to reduce the NET-mediated inflammation and microthrombi formation.
1. Introduction
The morbidity arising from the material-induced thrombosis along with the risk of bacterial colonization and subsequent infections limits the biomedical applications of blood-contacting materials such as extracorporeal membrane oxygenators (ECMOs) used in artificial lungs-supporting systems.1,2 The lack of active thrombosis- and pathogenesis-resistance on the artificial surfaces promotes adverse interactions with blood components. The most important of these is the adsorption of proteins on the surfaces followed by the activation of platelets and subsequent inflammation, which triggers thrombus formation and complement activation.3–7 The complement system is a major part of the innate immunity for early effective recognition and lysis of pathogens. The activation of this system is mainly mediated by adsorbed inflammatory complement peptides at sites of thrombo-inflammation including C3b and C5b to activate and recruit leukocytes particularly neutrophils.5,8,9 Additionally, activated platelets also play a significant role in neutrophil recruitment and thereby the pathogenesis of the inflammatory response.10,11 Neutrophils adhere to activated platelets mainly via the binding of Mac1 (integrin αMβ2) and P-selectin glycoprotein ligand-1 (PSGL-1) to the platelet adhesion molecules glycoprotein Ib (GPIb) and P-selectin (CD62P) expressed on the platelet plasma membrane. These platelet-neutrophil aggregates exacerbate the functions bilaterally resulting in a vicious cycle of thrombo-inflammation. Thereby the activated platelets and platelet-derived agonists/proteins initiate outside-in signaling in neutrophils and stimulate them to release neutrophil extracellular traps (NETs).11–17 NETs are web-like chromatin structures decorated with histones and antimicrobial enzymes expelled from neutrophils as a part of the host defense system to trap and kill pathogens at sites of inflammation.18,19 However, uncontrolled or dysregulated NET formation (NETosis) may intensify inflammation and the NET-induced activation of the coagulation cascade. NETs and granular proteins released by the stimulated neutrophils including neutrophil elastase (NE), serine proteases, and cathepsin G contribute to procoagulant reactions in different ways such as the proteolysis of tissue factor pathway inhibitor (TFPI), thereby activating the intrinsic coagulation cascade.20 Moreover, the interaction of NET components like histones H3 and H4 with platelets via Toll-like-receptors (TLRs) or fibrinogen enhances the activation and aggregation of platelets and thus induces the local thrombin generation.14,18,19,21,22 These adverse effects of NETosis dysregulation lead to the formation of clots and vessel occlusion,23 host tissue injuries24,25 and chronic human inflammatory diseases/disorders, e.g. pulmonary fibrosis or acute respiratory distress syndrome (ARDS) recently observed as a common clinical symptom of COVID-19 during the current SARS-CoV-2 pandemic.17,26–30The recent findings have also shown that NETs are sufficient to generate all the aforementioned life-threatening complications even in the absence of platelets. As we recently demonstrated, the platelet reduction in an in vitro ECMO circuit inhibits platelet activation but it is not sufficient to prevent clot formation, possibly due to the generation of NETs.31
As antiplatelet and antithrombotic drugs are ineffective to disrupt NET formation, finding ways to degrade NETs has been a matter of intense research and may lead to great therapeutic opportunities. Physiologically, NETs are degraded by serum DNases such as DNase I.32–34 DNase I is an endonuclease predominant in plasma. It is a member of the DNase protection family expressed by non-hematopoietic cells, which preferentially cleaves/hydrolyzes protein-free DNA strands in a non-sequence-specific manner.24,35 Apart from being used as a molecular biology tool, DNase I is U.S. Food and Drug Administration (FDA)-approved and is commonly used in therapeutic methods of modern medicine for cystic fibrosis36 to clear extracellular DNA fibers in the lungs and systemic lupus erythematosus.37 Recent findings have also shown the effectiveness of DNase I in the digestion of NETs.38–40 However, the low serum stability, and fast deactivation by environmental stimuli have been considered as the limiting factors for clinical applications of DNase I.41
In this work, we show the synthesis of biohybrid, highly hydrophilic microgels conjugated to the DNase I (DNase I MGs) for the digestion of NETs. The systematically studied conjugation of the enzyme to the microgel abstains any influence on the structural stability of the protein. In this regard, comparing with the native enzyme, the enzymatic activity of conjugated DNase I was measured over time using variable concentrations of a fluorescently labeled DNA probe as a substrate. The DNase I MG showed negligible fouling when challenged with bovine serum albumin (BSA) used as a model foulant in both static and flow conditions. To show the efficiency of the DNase I MGs to digest NETs in biological media, the conjugated microgels were incubated with stimulated polymorphonuclear neutrophils continuously secreting NETs. Using live-cell imaging and the specific citrullinated histone H3 based ELISA, the fast and more efficient digestion of NETs by the DNase I MGs compared to the native enzyme was demonstrated for a long duration of time. We envision that these novel biohybrid microgels can be used as a coating material for blood-contacting surfaces42 to digest the formed NETs.
2. Experimental
2.1. Materials
Anhydrous dichloromethane (DCM), Span 80 (sorbitan monooleate), Tween 80 (polyethylene glycol sorbitan monooleate), n-hexadecane 99%, anhydrous sodium sulfate, N-[3-(dimethylamino)propyl]methacrylamide, 3-hydroxypropionic acid lactone (β-propiolactone 97%), anhydrous tetrahydrofuran (THF), diethyl ether (DEE), N,N′-methylenebis(acrylamide) (MBAA) 99%, purified DNase I from the bovine pancreas and Phosphate-buffered saline (PBS) (1× pH 5.0) were purchased from Sigma-Aldrich. 2-Methylprop-2-enoyl chloride (methacryloyl chloride 97%) and 2,2′-azobis(2-methylpropionamidine) dihydrochloride (AMPA 97%) were purchased from Acros organics. 1-Aminopropan-2-ol 98% and liquid chromatography-mass spectrometry (LC-MS) grade water, PBS (1× pH 7.4), sodium carbonate and blotting-grade sodium dodecyl sulfate (SDS ≥ 99.5%) were purchased from Merck, Lonza AG, ChemPur and Carl Roth GmbH respectively. 1-Ethyl-3-(3-dimethylaminopropyl) carbodiimide hydrochloride (EDC) and sulfo-N-hydroxysuccinimide (sulfo-NHS) crosslinkers were purchased from ThermoFischer Scientific. The fluorometric DNase I activity assay kit (K-429) was purchased from BioVision containing a vial of 25 μM DNA probe and 25 mL molecular biology grade water. The chemicals used for the NET formation assays were included 0.5 M ethylenediaminetetraacetic acid (EDTA) solution, 1 μg mL−1 Hoechst 33342 staining dye solution, 100 nM phorbol 12-myristate 13-acetate (PMA) from Sigma-Aldrich, discontinuous 1077/1119 Pancoll gradient separating solution and imaging medium (RPMI-1640) plus 0.5% fetal calf serum (FCS) from PAN Biotech, SYTOX™ orange and micrococcal nuclease solution from Thermo Fischer Scientific and Poly-L-Lysine (PLL) coated micro-slides from Ibidi, Anti-Histone H3 (citrulline R2 + R8 + R17) capture antibody from abcam and Anti-DNA-POD detection antibody from Roche. All chemicals and reagents were of analytical grade and used as received without further purification.2.2. Methods
![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CH2), 5.7 ppm (s, 1H, CCH2).
CH2), 5.8 (m, 1H, CCH2).
CH2), 5.7 ppm (s, 1H, CCH2).
CH2), 5.8 (m, 1H, CCH2).
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 used as the continuous phase. HPMA (122 mg, 0.80 mmol, 75 mmol%), CBMAA (36 mg, 0.14 mmol, 13 mmol%), and different amounts of MBAA were dissolved in 0.5 mL of PBS followed by addition to the organic phase. The mixture was vigorously stirred for 5 min at room temperature, transferred to a rosette cooling cell (Sonic materials™, Fischer Scientific GmbH, NRW, Germany) and subjected to ultrasonication. The pre-emulsion in the cooling cell was firmly closed to the clamp stand immersed in/under the water bath. The mixture was sonicated for 5 min using a microtip of 6.4 mm in diameter adjusted closely to the bottom of the cell. The sonication parameters were kept at 50% amplitude in a set pulse regime (0.9 s sonication, 0.1 s pause) using a Branson 250 W sonifier. AMPA (25.5 mg, 0.094 mmol, 9 mmol%) dissolved in 0.5 mL PBS was added dropwise to the mixture along with the ultrasonication treatment for a further 5 min. The mixture was sonicated once more for 5 min and then purged with N2 gas. The polymerization was initiated upon heating the emulsion to 70 °C under constant stirring (700 rpm) and inert atmosphere for 45 min. Separation of the microgels was achieved after removing the organic phase by centrifuging the emulsion at 10000 rpm for 5 min. The microgels were further washed with hexadecane (3 × 10 mL) and purified by dialysis (MWCO: 50 kDa; reg. cellulose) against distilled water for 7 days. The obtained microgels were freeze-dried and kept at 2–8 °C (yield 50 mg, 31%).
:20) using an ACE 600 sputter coater (Leica, Germany).
:1 used as the continuous phase. HPMA (122 mg, 0.80 mmol, 75 mmol%), CBMAA (36 mg, 0.14 mmol, 13 mmol%), and different amounts of MBAA were dissolved in 0.5 mL of PBS followed by addition to the organic phase. The mixture was vigorously stirred for 5 min at room temperature, transferred to a rosette cooling cell (Sonic materials™, Fischer Scientific GmbH, NRW, Germany) and subjected to ultrasonication. The pre-emulsion in the cooling cell was firmly closed to the clamp stand immersed in/under the water bath. The mixture was sonicated for 5 min using a microtip of 6.4 mm in diameter adjusted closely to the bottom of the cell. The sonication parameters were kept at 50% amplitude in a set pulse regime (0.9 s sonication, 0.1 s pause) using a Branson 250 W sonifier. AMPA (25.5 mg, 0.094 mmol, 9 mmol%) dissolved in 0.5 mL PBS was added dropwise to the mixture along with the ultrasonication treatment for a further 5 min. The mixture was sonicated once more for 5 min and then purged with N2 gas. The polymerization was initiated upon heating the emulsion to 70 °C under constant stirring (700 rpm) and inert atmosphere for 45 min. Separation of the microgels was achieved after removing the organic phase by centrifuging the emulsion at 10000 rpm for 5 min. The microgels were further washed with hexadecane (3 × 10 mL) and purified by dialysis (MWCO: 50 kDa; reg. cellulose) against distilled water for 7 days. The obtained microgels were freeze-dried and kept at 2–8 °C (yield 50 mg, 31%).
:20) using an ACE 600 sputter coater (Leica, Germany).
The particle size and distribution were analyzed from the obtained SEM micrographs using ImageJ analysis software (ImageJ/Fiji).43 It facilitates measuring the diameter of an adequate number of particles (N) for statistical significance of each preparation of microgels with different amounts of crosslinker (MBAA) used. The frequency distribution of particle radius was further plotted with Origin data analysis software (OriginLab 2018b).
:20) using an ACE 600 sputter coater (Leica, Germany). The microgel structure was further characterized by a Fourier Transform Infrared Spectroscopy with Thermo Nicolet Nexus 470 FT-IR spectrometer (Thermo Fisher Scientific Inc., MA, USA) using a KBr tablet. To determine the elemental composition of the microgel, X-ray photoelectron spectroscopy (XPS) was conducted with a Kratos Ultra Axis (Kratos Analytical Ltd, Manchester, UK). The samples were excited with monochromatic Al-Kα1,2 radiation (1486.6 eV) and the resulting spectra were analyzed with CasaXPS processing software (Casa Software Ltd, United Kingdom). The binding energy (BE, eV) was corrected with C 1s (285.0 eV) as the standard. The conformational structure of DNase I modified microgels was evaluated by CD spectroscopy. The cuvette and the holding chamber were flushed with a constant stream of dry N2 gas to avoid water condensation on the cuvette exterior. Data were collected from 280 to 190 nm with a 1 s response time and a 1 nm bandwidth using a Jasco V-780 spectrophotometer (Jasco, Tokyo, Japan) in a 0.1 cm quartz cuvette. The UV-CD measurements for 2 μM free DNase I and 5 μM conjugated DNase I corresponded to 0.6 mg mL−1 DNase I MGs (the concentration of immobilized enzyme will be discussed in the following section of enzymatic activity) were carried out in PBS (pH 7.4) at 25 °C. Each spectrum shown is the average of three individual scans and is corrected according to the baseline spectrum of the buffer.
000 rpm for 5 min and analyzed by XPS (Kratos Analytical Ltd, Manchester, UK) before and after contact with the protein solution. Additionally, the antifouling activity of the microgels was evaluated by a QCM-D measurement performed on a QSense® Explorer device (Biolin Scientific, Västra Frölunda, Sweden) and an IPC 8 multichannel peristaltic pump (Ismatec, Wertheim, Germany). For all measurements, gold-coated QCM-D sensors (QSX 301 QSensor®, Biolin Scientific, Västra Frölunda, Sweden) have been used as supports for microgel adsorption. Before use, each sensor has been exposed to UV light for 15 min, immersed in a 2 wt% solution of blotting-grade SDS for 45 min, then rinsed with Milli-Q water, dried under a mild stream of N2 flow, and exposed again to UV light for another 15 min. 100 μL of the microgels were spin-coated at 2000 rpm with an initial acceleration of 800 rpm for a min on a clean surface of a gold sensor previously treated by 300 s of air plasma at a pressure of 0.2 mbar, which activates the surface and further improves the microgel adsorption. An additional sensor without any coating was used as a reference.
In each experiment, microgel-coated sensors have been allocated in a standard QSense flow module at a constant temperature of 25 °C and the medium has been pumped at a constant flow rate of 20 μL min−1. After initial equilibration in PBS (pH 5.0), the measurement was restarted to obtain a baseline value for the frequency and the dissipation curves around 0 Hz and 0 ppm, respectively followed by monitoring study of protein fouling by alternating use of PBS (pH 5.0) as a medium and BSA (in PBS pH 5.0) as a model foulant. The experimental data have been collected using software QSoft401® and processed by Dfind software (QSense AB)44 and Origin (OriginLab 2018b). The mass change on the surface of the QCM-D sensor before and after washing step was calculated using Sauerbery equation, Δm = C·n−1·Δf, with C mass sensitivity constant (17.7 ng cm−2 Hz for 5 MHz quartz crystal sensors used in this study); n number of harmonics and Δf variation of frequency.
To have a more specific measurement of NET formation and to exclude the false positive signals, the impact of the DNase I MGs on the persistence of NETs was analyzed in a H3Cit-DNA complex ELISA. The NET formation was induced by stimulating isolated human PMNs with PMA in a 24-well plate for 6 h at 37 °C. DNase I MGs was supplemented in the same way as shown in the SYTOX™ assay. Appropriate controls (unstimulated, DNase I treatment, DNase I MGs only) were added to the well plates. The supernatants were collected after treatment with a micrococcal nuclease to detach NETs from surfaces and cell remnants. Samples were analyzed in the H3Cit-DNA complex ELISA using the Anti-Histone H3 (citrulline R2 + R8 + R17) antibody as capture and the Anti-DNA-POD antibody as a detection antibody.
3. Results and discussion
3.1. Synthesis of p(HPMA-co-CBMAA) microgels
The microgels were synthesized using N-(2-hydroxypropyl)methacrylamide (HPMA) and zwitterionic carboxybetaine methacrylamide (CBMAA). The synthesis of HPMA was through the Schotten–Baumann condensation of methacryloyl chloride and 1-aminopropan-2-ol as reported in the literature.45,46 The zwitterionic CBMAA was synthesized by the ring opening of β-propiolactone in reaction with N-[3-(dimethylamino)propyl]methacrylamide.47,48 The synthesis route of monomers and the 1H NMR evaluation of their chemical structure are shown in ESI Fig. S1 and S2.† The characteristic peaks of HPMA and CBMAA are evident at 4.0 and 3.6 ppm respectively. The typical methacryloyl peaks observable at 5.7–5.8, 5.5 and 2.0 ppm confirm the successful formation of the methacrylamide in both monomers.The microgels were synthesized using HPMA and CBMAA in a molar ratio of 17:3 with different molar ratios of N,N′-methylenebis(acrylamide) (MBAA) as a crosslinker, using a free-radical emulsion polymerization in an inverse miniemulsion (Scheme 1A). The emulsion was prepared by ultrasonically dispersing a 1:10 volume of monomeric phase in hexadecane using 2,2′-azobis(2-methylpropionamidine) dihydrochloride (AMPA) as an initiator. A mixture of non-ionic Span 80 and Tween 80 was applied as a surfactant in a weight ratio of 3:1, which provided a hydrophilic–lipophilic balance (HLB) of 7 required for stability of the inverse emulsions. Further optimized parameters to achieve a stable colloidal system are summarized in Table S1.† On completion of the reaction, microgels were centrifuged, dialyzed, and freeze-dried.
 | ||
| Scheme 1 (A) Schematic representation of the w/o inverse miniemulsion method used for fabrication of microgels from HPMA and CBMAA: (1) the aqueous phase consists of the monomers and the crosslinker in PBS (blue) while the organic phase consists of n-hexadecane and the surfactants Span 80 and Tween 80 in the ratio of 3:1 (yellow); (2) the initiator AMPA dissolved in PBS was added along with ultrasonic dispersion of the phases; (3) the polymerization was initiated within the stabilized droplets upon heating the miniemulsion to 70 °C under an inert atmosphere which leads to the formation of microgels after 45 min. (B) Different molar ratios of the crosslinker were used to optimize the size of p(HPMA-co-CBMAA) microgels. | ||
HPMA and CBMAA were used as monomers for the synthesis of microgels due to their excellent non-fouling properties. Fig. S3† shows the NMR spectrum of the p(HPMA-co-CBMAA) microgels. The integral ratio of the characteristic peaks of HPMA and CBMAA at 3.8 and 3.5 ppm respectively, are attributed to the molar ratio of the corresponding protons (17 × 1H):(3 × 2H), which is in agreement with the feed composition ratio of copolymerized HPMA and CBMAA (17:3). The relevance of this ratio is later discussed in section 3.5 the microgels were further tuned for their size by varying the crosslinker amounts (Scheme 1B). The morphology of the prepared microgels with different crosslink densities is shown in Fig. S4A–D.† As can be seen from the SEM images in the dry state the microgels tend to flatten which is an inherent nature of the microgels due to the soft open structure. The particle size and distribution were evaluated from the given SEM micrograph for each microgel sample without prior filtration. The analysis was conducted using ImageJ analysis software (ImageJ/Fiji)43 analyzing each particle under the Feret parameter. This allows calculating the average diameter of the particles in each micrograph considering the distance between two parallel tangents to the projected silhouette/outline of a particle at any angle.49 As shown in Fig. S4A–D,† the diameter of approximately 100 particles (N) was measured from the SEM micrographs for statistical significance of each preparation of microgels with varying amounts of crosslinker (MBAA) used. The frequency distribution of particle radius was plotted with Origin data analysis software (OriginLab 2018b). The results are summarized in Table S2† indicating the obtained particle radius in the range of 7 μm to 300 nm with the increasing amount of MBAA, however, it leads to a decline of the monodispersity of particles. The particles size and the mechanical properties (e.g. stiffness) of micro-/nanogels can be fine-tuned by varying the crosslinking density. This inevitably induces a change in the surface properties of microgels as the microgel composition varies and therefore, it might influence their interaction with enzymes on conjugation.50 The extent of enzyme–substrate interaction is a predominant parameter impacting the enzymatic activity of enzyme–microgel conjugates and it is thought to be enhanced due to the high surface area offering by the microgelic support. However, it significantly depends on the morphology and the pore size of the microgels tuned by crosslink density. Highly crosslinked microgels impose physical restrictions in accessing the available surface area for enzyme conjugation and the substrate (and solvent) diffusion due to their compact structure, while ultra-low crosslinked microgels with an extremely open swollen structure suffer from the lack of available surface area.51,52 This implies that the tunability of crosslinking density facilitates control over the average particle and pore size, swelling capacity, and mechanical properties of microgels, which further regulates the modality of their interaction with both enzyme and substrate and later on the local density of immobilized enzyme as well as the diffusion rate of the substrate.
3.2. Conjugation of DNase I with p(HPMA-co-CBMAA) microgels
The bovine pancreatic DNase I was chosen as a model nuclease for conjugation with the microgels, due to its potential clinical and pharmacological relevance.41,53 The conjugation was carried out through amide crosslinks formed between the carboxylic group of CBMAA and lysine residues of the enzyme in a molar ratio of 5:1. The reaction was mediated by the zero-length 1-ethyl-3-(3-dimethylaminopropyl) carbodiimide hydrochloride/sulfo-N-hydroxysuccinimide (EDC/sulfo-NHS) crosslinkers (Fig. 1A). The use of sulfo-NHS stabilizes the ester intermediate, prevents its fast hydrolysis, and facilitates a nucleophilic addition with amines.54,55 The EDC/sulfo-NHS mediated conjugation was carried out in PBS at a physiological pH of 7.4. At this pH the DNase I is negatively charged (isoelectric point ≈ 5),56 while the microgels have a net positive charge due to the EDC/sulfo-NHS driven conversion of acid groups of CBMAA to active esters. This facilitates the electrostatic attraction of the DNase I to the highly hydrophilic microgels followed by a covalent conjugation over sufficient time. The electrical neutrality of the microgels is re-established once the reaction is complete.55,57,58
 | ||
| Fig. 1 (A) Schematic illustration of the DNase I conjugation with p(HPMA-co-CBMAA) microgels mediated by EDC/sulfo-NHS at pH 7.4. (B) FESEM micrograph of the DNase I MGs revealing the spherical morphology of the synthesized microgels. (C) The size distribution of 100 particles (N = 100) of DNase I MGs from the given SEM micrograph. (D) CD spectra of the conjugated DNase I compared to the free enzyme revealing the preservation of the secondary structure of the enzyme after a successful conjugation. | ||
3.3. Characterization of DNase I conjugated p(HPMA-co-CBMAA) microgels
The FESEM was used to ascertain the morphology of the DNase I MGs. Fig. 1B shows a FESEM micrograph of monodisperse spherical microgels. The particle size and distribution were evaluated from the given SEM micrograph indicating the microgel size of 7.3 ± 1.4 μm in radius with 3% of MBAA (Fig. 1C). The enzyme conjugation and its structural stability in an enzyme–microgel biohybrid were assessed by circular dichroism (CD) spectroscopy. Fig. 1D demonstrates the CD spectrum of the DNase I MG compared to the free DNase I at 25 °C. DNase I is an alpha, beta-protein with two 6-stranded beta-pleated sheets packed against each other which forms the core of a ‘sandwich-type structure’. The anti-parallel beta-sheets are flanked by three longer alpha-helices and extensive loop regions making the alpha helix the dominant structure.59 Any change in the conformational stability of the enzyme will reduce the dominant alpha-helix structure while increasing the beta-sheet. The CD spectrum of DNase I MG shows a positive band around 192 nm and a strong negative band around 205 nm in the far UV region as it can be seen in the spectrum of free DNase I due to the dominant alpha-helical structure of DNase I60–63 (Fig. 1D). This reveals the absence of any alteration in the secondary structure of the enzyme after being successfully conjugated to the microgels.The high-resolution XPS further ascertained the microgel composition after the incorporation of DNase I (Fig. 2). The C 1s spectrum of the DNase I MG (Fig. 2B) reveals the signals of C–C (284.9 eV), O–CO (286.2 eV), and N–CO (287.9 eV) mostly related to the amide and carboxylic groups, which are also detectable in the structure of unconjugated microgels (Fig. 2A), however, an increase in the peak intensity of N–CO and O–CO is evident after conjugation. The peaks of C–NH–C (400.1 eV) and C–N (402.6 eV) are invariant in the N 1s spectrum of the microgels before (Fig. 2C) and after conjugation (Fig. 2D) with DNase I, but the C–N peak in DNase I MGs shows a broadening.
 | ||
| Fig. 2 High-resolution C 1s and N 1s XPS spectra of the p(HPMA-co-CBMAA) microgels (A and C) before and (B and D) after conjugation with DNase I demonstrating the characteristic structural peaks. | ||
According to the XPS data before and after conjugation (n = 3) summarized in Table 1, the relative carbon/nitrogen ratio (C/N) shows an obvious decrease after conjugation (C/N = 5.4 ± 0.9) compared to the composition ratio of the unconjugated microgels (C/N = 7.2 ± 0.5). This is due to the significant 1.3 fold increase of nitrogen atomic percentage after the enzyme incorporation, thus it can justify the observed increase in peak intensity of N–CO and O–CO and confirm the successful biofunctionalization of microgels with DNase I. Additionally, the broadening of the C–N peak after conjugation corroborates the incorporation/distribution of DNase I throughout the microgel structure, as the same broad peak for C–N can be also seen in the native DNase I (Fig. S5†).
| C 1s | N 1s | O 1s | C/N | |
|---|---|---|---|---|
| Before conjugation | 53.8 ± 1.2% | 7.6 ± 0.6% | 38.6 ± 1.4% | 7.2 ± 0.5 |
| After conjugation | 51.5 ± 1.3% | 9.8 ± 1.4% | 38.7 ± 0.3% | 5.4 ± 0.9 |
| DNase I | 34.9 ± 0.2% | 8.4 ± 0.2% | 56.7 ± 0.2% | 4.2 ± 0.1 |
3.4. DNase I conjugation to microgel in flow
To further monitor the successful conjugation of the DNase I to the EDC/sulfo-NHS activated microgels QCM-D was used. The freshly EDC/NHS activated microgel-coated sensor was allocated in a standard QSense flow module at a constant temperature of 25 °C and the media (PBS or different concentrations of DNase) was pumped at a constant flow rate of 20 μL min−1. After initial equilibration in PBS (pH 7.4), the measurement was followed by monitoring the conjugation of DNase I (PBS pH 7.4) with alternate cycles of PBS (pH 7.4) washing. The change in thickness and conjugated mass on the surface of the QCM-D sensor before and after washing steps were extrapolated from the collected experimental QCM-D response using the Voigt model embedded in Smartfit algorithm of Dfind software assigned to the soft viscoelastic surfaces. Three overtones (third, fifth, seventh) were used to fit the layer thickness and mass.As shown in Fig. S6A,† upon injection of varying concentrations of DNase (0.1, 0.5, 1.0 wt% in PBS pH 7.4), a damping of the resonance frequency due to the mass adsorption on the coated surface was evident, while the energy dissipation increased concerning the adhered film/mass on a surface. The dampening effect on the frequency is directly related to the DNase concentration. With increasing DNase concentration from 0.1 to 1.0 wt% with intermittent washing by PBS a total of 28.8 pmol cm−2 of DNase was conjugated at the microgel surface (Fig. S6B†). This gives a clear indication of the efficiency of conjugation and that the microgel coating on the QCM-D sensor is thin and sensitive enough to observe any changes in mass owing to either conjugation or fouling.
3.5 Antifouling microgels
Effective biofunctionalization of polymer without impairing antifouling properties poses a challenge. To study the antifouling behavior of DNase I MGs, the microgels were incubated in 1 mg mL−1 bovine serum albumin (0.1% BSA) for 60 min at room temperature and subsequently centrifuged and washed. Since, the temporal evaluation of adsorbed protein corona on the DNase I MG can indicate the non-fouling nature of the microgels, the change in the chemical nature of microgels due to protein adsorption was in-depth analyzed by XPS (Fig. S7†). The adsorption of the protein layer is reflected by an increase in C–O and CO components in C 1s peak and C–NH–C in N 1s spectra of the microgels after incubation with the protein. Fig. S7† shows no changes in the characteristic peaks of C 1s and N 1s spectra of DNase I MG after BSA treatment. Additionally, the persistent atomic contents of C (51.5 ± 1.3%) and N (9.8 ± 1.4%) calculated from XPS analysis before the BSA incubation corresponded to the percentages after incubation, C (51.3 ± 0.4%) and N (10.1 ± 0.9%). This corroborates the antifouling nature of the DNase I MGs (Table S3†).
The antifouling property of the microgels was also evaluated using quartz crystal microbalance with dissipation (QCM-D). This method provides the possibility to probe binding and interactions under dynamic conditions and in real-time by monitoring the variations in resonance frequency (Δf) resulting from changes in the adsorbed mass on the surface. To confirm the antifouling property of the DNase I MGs under flow conditions, the microgels were spin-coated on a QCM-D gold chip and a bare gold chip was used as a reference surface. In a typical adsorption experiment by QCM-D, with an increase of adsorbed mass on the surface, the Δf decreases.64 With the injection of 0.1% BSA (in PBS pH 5.0) used as a model foulant onto the DNase I MG coated QCM-D chip, the frequency of the crystal sensor slightly decreased (Fig. S8-1†) due to the mass adsorption on the coated surface. Using, Sauerbery equation the adsorbed protein was calculated to be 3.54 ng cm−2. However, the surface was almost completely regenerated on washing with PBS (pH 5.0) (Fig. S8-2†), which implies the efficient removal of adsorbed proteins from the surface. This shows the excellent protein resistance of the DNase I MG coating in comparison to the control surface, where the frequency drastically dropped from 0 to −6 Hz upon BSA exposure (Fig. S8-1†) and the surface could barely be regenerated even after prolonged washing (Fig. S8-3†). The amount of adsorbed protein on the control after washing was 97.35 ng cm−2.
Protein adsorption on blood-contacting surfaces leads to the initiation of coagulation cascade or in the case of nanoparticle influences the immune response in the body. To overcome this, microgels were synthesized with HPMA and CBMAA. The high hydrophilicity and the low surface charge of HPMA supplies a strong hydration shell surrounding its hydroxyl or amide groups, which enthalpically hinders protein fouling.65,66 Additionally, the electroneutrality and the potent electrostatically induced hydrophilicity due to the zwitterionic nature of CBMAA give rise to a high surface wettability (ionic solvation) and non-specific protein fouling resistance.48,67–69 The random or statistical copolymerization of high carboxyl-functional low-fouling zwitterionic CBMAA with the high fouling resistant HPMA, gives a novel enhanced antifouling functional polymer that performs significantly better than individual homopolymers.70,71 It has been reported in the literature that commonly used non-fouling homopolymers like CBMAA during functionalization often encounter the reaction of too many functional groups, loss of neutrality and/or crosslinking of the chains that leads to antifouling deterioration.72 Since in this work, the attainment of a biofunctionalized and antifouling microgel was intended, the approach of HPMA and CBMAA combination enabled the efficient biofunctionalization of the p[HPMA-co-CBMAA] based microgels while preserving antifouling properties owing to the HPMA units. It has been shown that different ratios of HMPA:CBMAA particularly with an optimized CBMAA molar content (15%) (≈HPMA:CBMAA 17:3) provided a higher conjugation capacity and resistance to plasma fouling that was comparable or much better than recently developed ultra-low-fouling CB polymers or conventional antifouling carboxyl-functional polymeric coatings.73
Until now, different types of hydrophilic and/or zwitterionic polymeric materials are commonly used in form of linear or brush-like polymers,74,75 hydrogels76 or nano-/microgels77,78 have been extensively studied for different purposes particularly in the fields of antifouling surface modifications.79 However, the success achieved so far mostly relies on zwitterionic or hydrophilic homopolymers. In this work, we aimed to show a novel functional antifouling microgel system from a particular combination of both highly fouling resistant HPMA and functional low-fouling zwitterionic CBMAA promoting the antifouling properties. These microgels can be biofunctionalized and can be tuned for a variety of applications including drug delivery or functional surface coating materials.
3.6. Enzymatic activity of conjugated DNase I
The enzymatic activity of DNase I MGs was quantitatively evaluated by a fluorometric assay, by measuring the ability of the conjugated DNase I to cleave the quenched fluorescently labeled DNA probe.80 The successful cleavage results in fluorescent DNA fragments detected at an excitation/emission (Ex/Em) wavelength of 651/681 nm (Fig. 3A). For this, 1 mg mL−1 of DNase I MG was incubated with model DNA as a substrate, and changes in fluorescence intensity were recorded for 5 h at 37 °C. With increasing time and concentration of the substrate the formation of the product was increased. A plateau was attained with a substrate concentration of 0.8 μM making the substrate concentration rate-limiting (Fig. 3B). The velocity of the enzyme (RFU min−1) catalyzing the rate of reaction as a function of the substrate concentration was plotted (Fig. 3C). The plot of rate against substrate concentration is a rectangular parabola as shown by Michaelis–Menten kinetics. The Km calculated from Lineweaver–Burk was 0.06275 ± 0.010 μM. The low Km value of the DNase I MG compared to the KHill of the free enzyme, 0.15945 ± 0.006 μM (Fig. S9†), indicates its much higher affinity for the substrate. The higher substrate–enzyme interaction of DNase I MGs is attributed to the increased localized density of enzymes in a given volume and enhanced mass transport due to the open structure of highly swollen microgels with a diffused outer boundary and well solvated inner chain segments allowing high mobility of solvent and solute molecules.81,82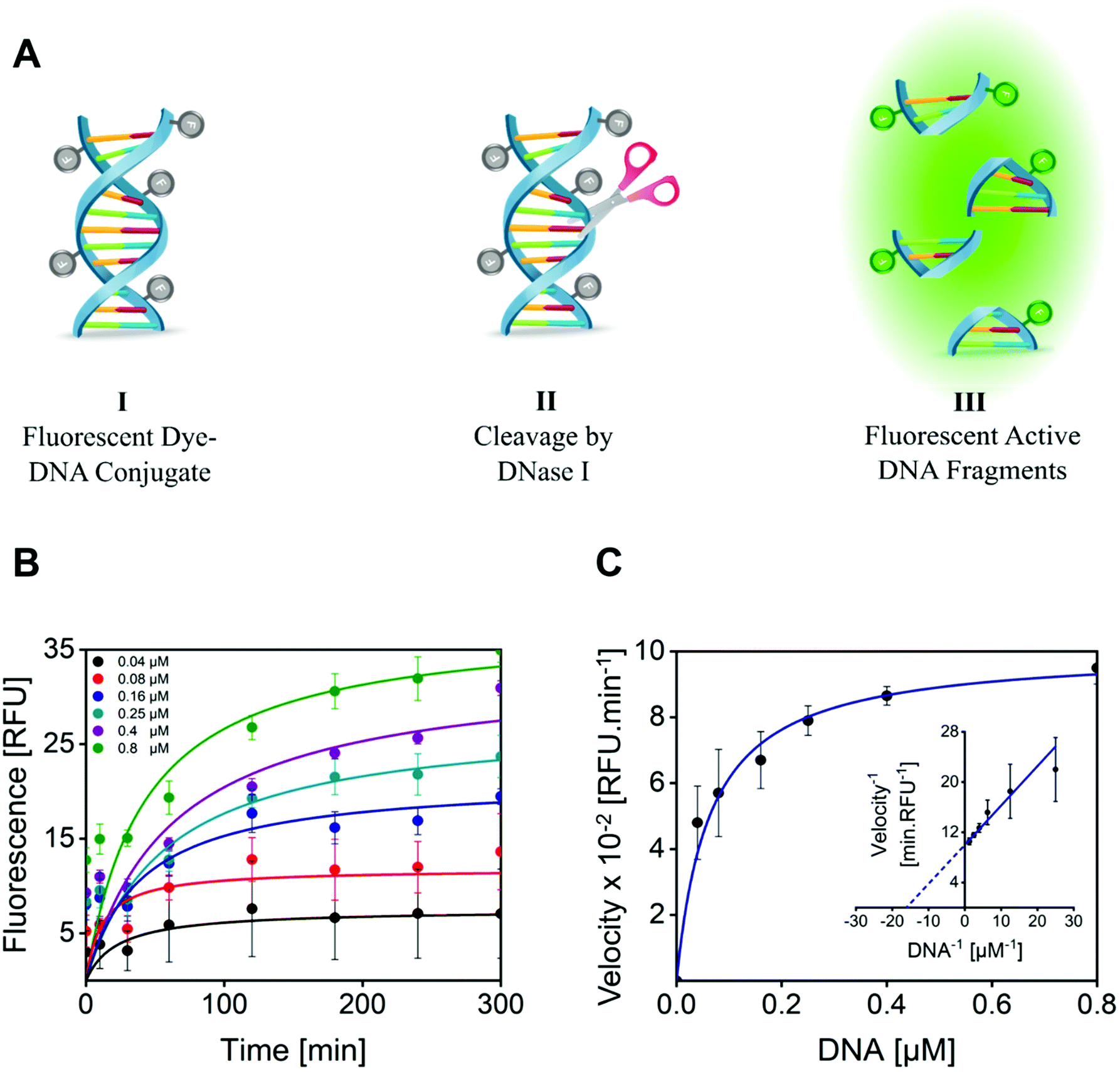 | ||
| Fig. 3 (A) The fluorometric DNase I activity assay: (I) Self-quenched covalent fluorescent dye–DNA conjugates (II) undergoes cleavage by DNase I leading to (III) unquenching of fluorescence tags on formed DNA fragments. The fluorescently activated DNAs are detectable at Ex/Em wavelength of 651/681 nm as a result of the enzymatic activity of DNase I. (B) Quantitative analysis of DNase I MG kinetics by measuring the fluorescence over time using different substrate concentrations. Reactions contained 8 μM of the incorporated enzyme. Fluorescence was normalized by subtraction of background fluorescence observed in the absence of enzyme. (C) The velocity data were fitted to the Michaelis–Menten equation by non-linear regression. The inset shows the Lineweaver–Burk plot of the fluorometric kinetic data. The error bars represent the standard error of the regression (n = 3). | ||
3.7. Enzymatic degradation of NETs exposed to DNase I conjugated microgels
To ascertain the bioactivity of the DNase I MGs, their ability to degrade NETs was measured. To determine this, a time course measurement was performed at 37 °C for 6 h which is the limit of DNase half-life in serum.83,84 The DNase I MGs were incubated with polymorphonuclear neutrophils (PMNs) in RPMI-1640 media with 0.5% fetal calf serum (FCS) and visualized using time-lapse microscopy. To produce NETs, PMNs were stimulated with phorbol 12-myristate13-acetate (PMA). The cell impermeable DNA dye SYTOX™ orange was added to the stimulated PMNs to visualize the extracellular DNA like NETs. On stimulation, the PMNs commence the formation of NETs after an hour of incubation and add up significantly during the measurement (ESI Video S1†). Fig. 4A above shows fluorescent microscopy images of stimulated PMNs with Hoechst staining (blue) of the nuclei and SYTOX™ orange staining of the NETs. In presence of DNase I MGs the stimulated PMNs started to form NETs after 2 h, however no sooner than the NETs were formed they were degraded by the DNase I MGs. (Fig. 4A below, ESI Video S2†). In presence of DNase I MGs the unstimulated PMNs did not show any significant release of NETs compared to the stimulated PMNs. This shows that the microgel itself did not contribute towards the amplification of NETs (Video 3†). Unstimulated PMNs were used as baseline control (Video 4†). | ||
| Fig. 4 (A) The live-cell imaging of neutrophils with stimulated neutrophils (PMA) and stimulated neutrophils in presence of DNase I MGs (PMA + DNase I MGs). The results are recorded for 6 h at 37 °C revealing the interaction of DNase I MGs with neutrophils and its NETosis degradation effect in presence of NETs released upon neutrophils stimulation. (B) The quantitative expression of live-cell imaging results presents the density of NETs with time. In presence of free DNase I (purple) and DNase I MGs (green) the NETs are disrupted over time in the same way while an intense accumulation of NETs is measured after 6 h in the absence of any inhibitors (red). As expected the unstimulated neutrophils do not show significant production of NETs in presence of DNase I MGs (blue). (C) The analysis of NET formation by H3Cit-DNA complex ELISA. The NET formation was induced by PMA stimulation for 6 h. The addition of DNase I MGs disrupts NET integrity thereby reducing the measurable NET fragments (green) to a greater extent compared to free DNase I (purple). Moreover, the addition of DNase I MGs also decreases the amount of spontaneously formed NETs (compare Ctrl (gray) and DNase I MGs (blue)). Mean ± SD, n = 5, *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001. | ||
The results were further quantified as NET density over time by plotting the SYTOX™ orange area per cell shown in Fig. 4B. As mentioned before, the unstimulated neutrophils (black) represent the baseline control which does not show significant production of NETs in presence of the DNase I MGs (blue). The PMA-induced formed NETs are disrupted overtime in front of the DNase I MGs (green) while an intense accumulation of NET is measured after 6 h in the negative control group excluded any inhibitory enzymes (red). The significant anti-NET performance of the DNase I MGs was similar to the free DNase (purple) considered as a positive control in the same concentration with the conjugated enzyme. It shows the successful conjugation of DNase I to form a microgel-based biohybrid as well as the significantly preserved activity of anti-NETosis in DNase I MGs.
However, since the SYTOX™ can also measure apoptotic and necrotic events that may or may not be associated with NET formation, a new set of the assay was performed. The presence of a typical NET-related protein, such as myeloperoxidase (MPO), neutrophil elastase (NE), or citrullinated histone H3 (H3Cit) would exclude false-positive signals originating from other types of cell death and would distinctly prove the formation of NETs. Several publications demonstrate that the citrullination of histone tails (histones H3, H4 and H2A) (H3Cit) plays a crucial role in the development of NETs. Furthermore, H3Cit is a positive marker for NET formation similar to NE and MPO85–88 Hence, the influence of the DNase I MGs on the persistence of NETs was additionally analyzed using H3Cit-DNA complex ELISA. The NET formation after stimulating isolated human PMNs with PMA was evaluated for 6 h at 37 °C. DNase I MGs and the appropriate controls were prepared and supplemented in the same way as was done in the SYTOX™ assay. The collected supernatants were analyzed in the H3Cit-DNA complex ELISA, using the Anti-Histone H3 antibody as capture and the Anti-DNA-POD antibody as a detection antibody. As shown in Fig. 4C, the addition of DNase I MGs destroys PMA-induced NETs more efficiently compared to free DNase I which was used as a positive control. These promising results are attributed to the increased localized density of enzymes in the DNase I MG in a given volume which results in fast and effective disruption of the NETs.
NETosis is a process that leads to an explosive release of NETs by neutrophils mainly causing programmed death of the cell. This process is accompanied by granulation of cell membrane and nucleus rupture which leads to chromatin decondensation and the release of NETs into extracellular space.20 DNase I as a host defense system is known to ameliorate the dissociation of NETs and somewhat alters the proteolytic activity of NET-related proteins.89 There are a significant number of studies reporting the beneficial aspects of DNase I treatment ranging from the recovery of disease indications90 to the suppression or the reduction of inflammatory40 and coagulation91 responses to the immunomodulation.92 However, the use of DNase I by systemic administration to tackle NETs is limited by its low stability in serum.20,83,84 To improve the serum stability of DNase I, the conjugation of the DNase I to non-fouling microgels has been shown that can improve the stability with preservation of enzymatic activity in the biological milieu for a significantly longer duration of time.
4. Conclusion
Non-fouling microgels were synthesized from highly hydrophilic HPMA and zwitterionic CBMAA through a free-radical emulsion polymerization in an inverse miniemulsion. The obtained microgels were functionalized with DNase I to yield active biohybrid DNase I MGs. The enzyme-conjugated microgels were systematically evaluated for successful conjugation, structural characteristics, conformational stability, fouling resistance and enzymatic activity. The conjugation of DNase I on the microgels enhanced the bioactive surface area for enzyme–substrate interaction without alteration in its secondary structure and antifouling property of the microgels. The biohybrid microgels when challenged by stimulated neutrophils explosively expelling NETs, showed fast disruption of NETs and the more efficient clearance of NET accumulation in comparison to free DNase I throughout the 6 h measurements in serum. This corroborates the significant enzymatic bioactivity of DNase I MGs even in complex biological solutions. Altogether, the biohybrid microgel shows a high potential for use as a bioactive coating material for blood-contacting artificial surfaces like in membrane oxygenators to reduce the NET-mediated inflammation and microthrombi formation.93,94Ethical statement
All experiments were performed in accordance with the Helsinki declaration and were approved by the ethics committee at the University of Münster. Informed consents were obtained from human participants of this study.Conflicts of interest
Authors declare no conflict of interest.Acknowledgements
The authors acknowledge the financial support by the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation) via Schwerpunktprogramm “Towards an Implantable Lung” Project Number: SI 2164/2-1, RO 2000/25-1, RO4537/3-1, RO4537/4-1 and SFB1450C07B to J. R. The authors would like to thank Prof. Martin Möller for constructive discussions. A.W. is a member of the CiM-IMPRS graduate school. Open Access funding is provided by the Max Planck Society.References
- S. Doymaz, J. Intensive Crit. Care, 2018, 4(02), 1–6 CrossRef.
- M. Weber, H. Steinle, S. Golombek, L. Hann, C. Schlensak, H. P. Wendel and M. Avci-Adali, Front. Bioeng. Biotechnol., 2018, 6, 99 CrossRef PubMed.
- X. Liu, L. Yuan, D. Li, Z. Tang, Y. Wang, G. Chen, H. Chen and J. L. Brash, J. Mater. Chem. B, 2014, 2, 5718 RSC.
- M. Mulder, I. Fawzy and M. Lancé, Neth. J. Crit. Care, 2018, 26, 6–13 Search PubMed.
- I. Jaffer, J. Fredenburgh, J. Hirsh and J. Weitz, J. Thromb. Haemostasis, 2015, 13, S72–S81 CrossRef.
- L. Brewster, E. M. Brey and H. P. Greisler, in Principles of Tissue Engineering, Elsevier, 2014, pp. 793–812 Search PubMed.
- B. Shen, M. K. Delaney and X. Du, Curr. Opin. Cell Biol., 2012, 24, 600–606 CrossRef CAS PubMed.
- C. A. Janeway Jr., P. Travers, M. Walport and M. J. Shlomchik, in Immunobiology: The Immune System in Health and Disease. 5th edition, Garland Science, 2001 Search PubMed.
- T. M. Maul, M. P. Massicotte and P. D. Wearden, Extracorporeal Membrane Oxygenation: Advances in Therapy, 2016, 27 Search PubMed.
- M. Mezger, H. Nording, R. Sauter, T. Graf, C. Heim, N. von Bubnoff, S. M. Ensminger and H. F. Langer, Front. Immunol., 2019, 10, 1731 CrossRef CAS.
- T. Lisman, Cell Tissue Res., 2018, 371, 567–576 CrossRef CAS.
- L. K. Jennings, Thromb. Haemostasis, 2009, 102, 248–257 CrossRef CAS PubMed.
- J. Rossaint and A. Zarbock, Cardiovasc. Res., 2015, 107, 386–395 CrossRef.
- A. Z. Zucoloto and C. N. Jenne, Front. Cardiovasc. Med., 2019, 6, 85 CrossRef CAS PubMed.
- K. Stark, HemaSphere, 2019, 3, 89–91 CrossRef.
- C. Sperling, M. Fischer, M. F. Maitz and C. Werner, Biomater. Sci., 2017, 5, 1998–2008 RSC.
- J. Rossaint, A. Margraf and A. Zarbock, Front. Immunol., 2018, 9, 2712 CrossRef.
- G. Sollberger, D. O. Tilley and A. Zychlinsky, Dev. Cell, 2018, 44, 542–553 CrossRef CAS.
- B. Shah, N. Burg and M. H. Pillinger, in Kelley and Firestein's textbook of rheumatology, Elsevier, 2017, pp. 169–188 Search PubMed.
- V. Papayannopoulos, Nat. Rev. Immunol., 2018, 18, 134 CrossRef CAS PubMed.
- D. Stakos, P. Skendros, S. Konstantinides and K. Ritis, Thromb. Haemostasis, 2020, 120(03), 373–383 CrossRef.
- D. Puhr-Westerheide, S. J. Schink, M. Fabritius, L. Mittmann, M. E. Hessenauer, J. Pircher, G. Zuchtriegel, B. Uhl, M. Holzer and S. Massberg, Sci. Rep., 2019, 9, 1–13 CAS.
- M. Leppkes, J. Knopf, E. Naschberger, A. Lindemann, J. Singh, I. Herrmann, M. Stürzl, L. Staats, A. Mahajan and C. Schauer, EBioMedicine, 2020, 58, 102925 CrossRef.
- M. Jiménez-Alcázar, C. Rangaswamy, R. Panda, J. Bitterling, Y. J. Simsek, A. T. Long, R. Bilyy, V. Krenn, C. Renné and T. Renné, Science, 2017, 358, 1202–1206 CrossRef.
- D. Nakazawa, S. Kumar, J. Desai and H.-J. Anders, Histol. Histopathol., 2017, 32, 203–213 CAS.
- N. Ruparelia, J. T. Chai, E. A. Fisher and R. P. Choudhury, Nat. Rev. Cardiol., 2017, 14, 133–144 CrossRef CAS PubMed.
- A. S. Kimball, A. T. Obi, J. A. Diaz and P. K. Henke, Front. Immunol., 2016, 7, 236 Search PubMed.
- M. Jimenez-Alcazar, M. Napirei, R. Panda, E. Köhler, J. A. Kremer Hovinga, H. Mannherz, S. Peine, T. Renné, B. Lämmle and T. Fuchs, J. Thromb. Haemostasis, 2015, 13, 732–742 CrossRef CAS PubMed.
- J. Perdomo, H. H. Leung, Z. Ahmadi, F. Yan, J. J. Chong, F. H. Passam and B. H. Chong, Nat. Commun., 2019, 10, 1–14 CrossRef CAS.
- E. A. Middleton, X.-Y. He, F. Denorme, R. A. Campbell, D. Ng, S. P. Salvatore, M. Mostyka, A. Baxter-Stoltzfus, A. C. Borczuk and M. Loda, Blood, 2020, 136, 1169–1179 CrossRef CAS.
- C. Bleilevens, J. Lölsberg, A. Cinar, M. Knoben, O. Grottke, R. Rossaint and M. Wessling, Sci. Rep., 2018, 8, 1–9 CAS.
- T. Mohanty, J. Fisher, A. Bakochi, A. Neumann, J. F. P. Cardoso, C. A. Karlsson, C. Pavan, I. Lundgaard, B. Nilson and P. Reinstrup, Nat. Commun., 2019, 10, 1–13 CrossRef CAS.
- M. Wilton, T. W. Halverson, L. Charron-Mazenod, M. D. Parkins and S. Lewenza, Infect. Immun., 2018, 86(9), e00403-18 CrossRef PubMed.
- P. Sumby, K. D. Barbian, D. J. Gardner, A. R. Whitney, D. M. Welty, R. D. Long, J. R. Bailey, M. J. Parnell, N. P. Hoe and G. G. Adams, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 1679–1684 CrossRef CAS.
- J. J. Swartjes, T. Das, S. Sharifi, G. Subbiahdoss, P. K. Sharma, B. P. Krom, H. J. Busscher and H. C. van der Mei, Adv. Funct. Mater., 2013, 23, 2843–2849 CrossRef CAS.
- A. Durward, V. Forte and S. D. Shemie, Crit. Care Med., 2000, 28, 560–562 CrossRef CAS.
- W. S. Prince, D. L. Baker, A. H. Dodge, A. E. Ahmed, R. W. Chestnut and D. V. Sinicropi, Clin. Exp. Immunol., 1998, 113, 289–296 CrossRef CAS.
- J. T. Buchanan, A. J. Simpson, R. K. Aziz, G. Y. Liu, S. A. Kristian, M. Kotb, J. Feramisco and V. Nizet, Curr. Biol., 2006, 16, 396–400 CrossRef CAS PubMed.
- Z. Konsoula, Mater. Methods, 2019, 9, 2714 Search PubMed.
- W. Meng, A. Paunel-Görgülü, S. Flohé, I. Witte, M. Schädel-Höpfner, J. Windolf and T. T. Lögters, Mediators Inflammation, 2012, 2012, 149560 CAS.
- M. Kovaliov, D. Cohen-Karni, K. A. Burridge, D. Mambelli, S. Sloane, N. Daman, C. Xu, J. Guth, J. K. Wickiser and N. Tomycz, Eur. Polym. J., 2018, 107, 15–24 CrossRef CAS.
- A. Hosseinnejad, T. Fischer, P. Jain, C. Bleilevens, F. Jakob, U. Schwaneberg, R. Rossaint and S. Singh, J. Colloid Interface Sci., 2021, 601, 604–616 CrossRef CAS PubMed.
- C. A. Schneider, W. S. Rasband and K. W. Eliceiri, Nat. Methods, 2012, 9, 671–675 CrossRef CAS.
- B. Scientific, 2021, https://www.biolinscientific.com/qsense/software/.
- M. F. Ebbesen, D. H. Schaffert, M. L. Crowley, D. Oupický and K. A. Howard, J. Polym. Sci., Part A: Polym. Chem., 2013, 51, 5091–5099 CrossRef CAS.
- J. Kopeček and H. Baẑilová, Eur. Polym. J., 1973, 9, 7–14 CrossRef.
- T. Gresham, J. Jansen, F. Shaver, R. Bankert and F. Fiedorek, J. Am. Chem. Soc., 1951, 73, 3168–3171 CrossRef CAS.
- S.-P. Tao, J. Zheng and Y. Sun, J. Chromatogr., A, 2015, 1389, 104–111 CrossRef CAS PubMed.
- Y. Galindez, E. Correa, A. Zuleta, A. Valencia-Escobar, D. Calderon, L. Toro and P. Chacón, Met. Mater. Int., 2019, 1–18 Search PubMed.
- L. Zhang, Z. Cao, Y. Li, J.-R. Ella-Menye, T. Bai and S. Jiang, ACS Nano, 2012, 6, 6681–6686 CrossRef CAS PubMed.
- S. Reinicke, T. Fischer, J. Bramski, J. Pietruszka and A. Böker, RSC Adv., 2019, 9, 28377–28386 RSC.
- L. Bayne, R. V. Ulijn and P. J. Halling, Chem. Soc. Rev., 2013, 42, 9000–9010 RSC.
- W.-J. Chen and T.-H. Liao, Protein Pept. Lett., 2006, 13, 447–453 CrossRef CAS PubMed.
- G. T. Hermanson, Bioconjugate techniques, Academic press, 2013 Search PubMed.
- M. P. Wickramathilaka and B. Y. Tao, J. Biol. Eng., 2019, 13, 63 CrossRef PubMed.
- A. Funakoshi, Y. Tsubota, K. Fujii, H. Ibayashi and Y. Takagi, J. Biochem., 1980, 88, 1113–1118 CrossRef CAS.
- M. A. Gauthier and H.-A. Klok, Polym. Chem., 2010, 1, 1352–1373 RSC.
- T. Riedel, F. Surman, S. Hageneder, O. Pop-Georgievski, C. Noehammer, M. Hofner, E. Brynda, C. Rodriguez-Emmenegger and J. Dostalek, Biosens. Bioelectron., 2016, 85, 272–279 CrossRef CAS PubMed.
- D. Suck, C. Oefner and W. Kabsch, EMBO J., 1984, 3, 2423–2430 CrossRef CAS PubMed.
- D. Suck, C. Oefner and W. Kabsch, EMBO J., 1984, 3, 2423–2430 CrossRef CAS PubMed.
- K. Ajtai and S. Y. Venyaminov, FEBS Lett., 1983, 151, 94–96 CrossRef CAS.
- Y. Sasaki, D. Miyoshi and N. Sugimoto, Nucleic Acids Res., 2007, 35, 4086–4093 CrossRef CAS PubMed.
- Y. Liu, J. Zhai, J. Dong and M. Zhao, Mol. Imprinting, 2015, 1, 47–54 Search PubMed.
- P. Saha, M. Santi, M. Emondts, H. Roth, K. Rahimi, J. Großkurth, R. Ganguly, M. Wessling, N. K. Singha and A. Pich, ACS Appl. Mater. Interfaces, 2020, 12(52), 58223–58238 CrossRef CAS PubMed.
- M. Vorobii, A. de los Santos Pereira, O. Pop-Georgievski, N. Y. Kostina, C. Rodriguez-Emmenegger and V. Percec, Polym. Chem., 2015, 6, 4210–4220 RSC.
- H. Yuan, B. Yu, L.-H. Fan, M. Wang, Y. Zhu, X. Ding and F.-J. Xu, Polym. Chem., 2016, 7, 5709–5718 RSC.
- N. Y. Kostina, C. Rodriguez-Emmenegger, M. Houska, E. Brynda and J. Michalek, Biomacromolecules, 2012, 13, 4164–4170 CrossRef CAS PubMed.
- A. Laschewsky, Polymers, 2014, 6, 1544–1601 CrossRef.
- A. Sinclair, M. B. O'Kelly, T. Bai, H. C. Hung, P. Jain and S. Jiang, Adv. Mater., 2018, 30, 1803087 CrossRef PubMed.
- M. Vorobii, N. Y. Kostina, K. Rahimi, S. Grama, D. Söder, O. Pop-Georgievski, A. Sturcova, D. Horak, O. Grottke and S. Singh, Biomacromolecules, 2019, 20, 959–968 CrossRef CAS PubMed.
- H. Vaisocherová-Lísalová, F. e. Surman, I. Víšová, M. Vala, T. s. Špringer, M. L. Ermini, H. Šípová, P. Šedivák, M. Houska and T. s. Riedel, Anal. Chem., 2016, 88, 10533–10539 CrossRef PubMed.
- T. Riedel, S. Hageneder, F. Surman, O. Pop-Georgievski, C. Noehammer, M. Hofner, E. Brynda, C. Rodriguez-Emmenegger and J. Dostalek, Anal. Chem., 2017, 89, 2972–2977 CrossRef CAS.
- H. Lisalova, E. Brynda, M. Houska, I. Visova, K. Mrkvova, X. C. Song, E. Gedeonova, F. Surman, T. Riedel and O. Pop-Georgievski, Anal. Chem., 2017, 89, 3524–3531 CrossRef CAS.
- B. Li, Z. Yuan, P. Jain, H.-C. Hung, Y. He, X. Lin, P. McMullen and S. Jiang, Sci. Adv., 2020, 6, eaba0754 CrossRef CAS PubMed.
- E. van Andel, S. C. Lange, S. P. Pujari, E. J. Tijhaar, M. M. Smulders, H. F. Savelkoul and H. Zuilhof, Langmuir, 2018, 35, 1181–1191 CrossRef PubMed.
- P. Zhang, F. Sun, C. Tsao, S. Liu, P. Jain, A. Sinclair, H.-C. Hung, T. Bai, K. Wu and S. Jiang, Proc. Natl. Acad. Sci. U. S. A., 2015, 112, 12046–12051 CrossRef CAS.
- B. Li, Z. Yuan, Y. He, H.-C. Hung and S. Jiang, Nano Lett., 2020, 20, 4693–4699 CrossRef CAS PubMed.
- G. Cheng, L. Mi, Z. Cao, H. Xue, Q. Yu, L. Carr and S. Jiang, Langmuir, 2010, 26, 6883–6886 CrossRef CAS PubMed.
- P. Zhang, B. D. Ratner, A. S. Hoffman and S. Jiang, in Biomaterials Science, Elsevier, 2020, pp. 507–513 Search PubMed.
- S. Sato and S. Takenaka, Sensors, 2014, 14, 12437–12450 CrossRef CAS PubMed.
- S. Ding, A. A. Cargill, I. L. Medintz and J. C. Claussen, Curr. Opin. Biotechnol., 2015, 34, 242–250 CrossRef CAS PubMed.
- F. A. Plamper and W. Richtering, Acc. Chem. Res., 2017, 50, 131–140 CrossRef CAS PubMed.
- W. Prince, D. Baker, A. Dodge, A. Ahmed, R. Chestnut and D. Sinicropi, Clin. Exp. Immunol., 1998, 113, 289 CrossRef CAS PubMed.
- B. M. McGrath and G. Walsh, Directory of therapeutic enzymes, CRC Press, 2005 Search PubMed.
- Y. Wang, M. Li, S. Stadler, S. Correll, P. Li, D. Wang, R. Hayama, L. Leonelli, H. Han and S. A. Grigoryev, J. Cell Biol., 2009, 184, 205–213 CrossRef CAS.
- M. Leshner, S. Wang, C. Lewis, H. Zheng, X. A. Chen, L. Santy and Y. Wang, Front. Immunol., 2012, 3, 307 Search PubMed.
- T. A. Claushuis, L. E. van der Donk, A. L. Luitse, H. A. van Veen, N. N. van der Wel, L. A. van Vught, J. J. Roelofs, O. J. de Boer, J. M. Lankelma and L. Boon, J. Immunol., 2018, 201, 1241–1252 CrossRef CAS.
- C. Thålin, S. Lundström, C. Seignez, M. Daleskog, A. Lundström, P. Henriksson, T. Helleday, M. Phillipson, H. Wallén and M. Demers, PLoS One, 2018, 13, e0191231 CrossRef.
- E. Kolaczkowska, C. N. Jenne, B. G. Surewaard, A. Thanabalasuriar, W.-Y. Lee, M.-J. Sanz, K. Mowen, G. Opdenakker and P. Kubes, Nat. Commun., 2015, 6, 1–13 CrossRef PubMed.
- H. H. Park, W. Park, Y. Y. Lee, H. Kim, H. S. Seo, D. W. Choi, H. K. Kwon, D. H. Na, T. H. Kim and Y. B. Choy, Adv. Sci., 2020, 7, 2001940 CrossRef CAS.
- H. Albadawi, R. Oklu, R. E. R. Malley, R. M. O’Keefe, T. P. Uong, N. R. Cormier and M. T. Watkins, J. Cardiovasc. Surg., 2016, 64, 484–493 Search PubMed.
- M. Macanovic, D. Sinicropi, S. Shak, S. Baughman, S. Thiru and P. Lachmann, Clin. Exp. Immunol., 1996, 106, 243–252 CrossRef CAS.
- J. Cedervall, Y. Zhang, H. Huang, L. Zhang, J. Femel, A. Dimberg and A.-K. Olsson, Cancer Res., 2015, 75, 2653–2662 CrossRef CAS.
- S. F. De Meyer, G. L. Suidan, T. A. Fuchs, M. Monestier and D. D. Wagner, Arterioscler. Thromb. Vasc. Biol., 2012, 32, 1884–1891 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1bm01591e |
| This journal is © The Royal Society of Chemistry 2022 |