
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Local delivery to malignant brain tumors: potential biomaterial-based therapeutic/adjuvant strategies
Majed
Alghamdi
ab,
Mark
Gumbleton
a and
Ben
Newland
 *ac
*ac
aSchool of Pharmacy and Pharmaceutical Sciences, Cardiff University, King Edward VII Avenue, Cardiff, CF10 3NB, UK. E-mail: newlandb@cardiff.ac.uk
bFaculty of Pharmacy, King Abdulaziz University, Jeddah, 22522, Kingdom of Saudi Arabia
cLeibniz-Institut für Polymerforschung Dresden, Max Bergmann Center of Biomaterials Dresden, Hohe Straße 6, D-01069 Dresden, Germany
First published on 29th July 2021
Abstract
Glioblastoma (GBM) is the most aggressive malignant brain tumor and is associated with a very poor prognosis. The standard treatment for newly diagnosed patients involves total tumor surgical resection (if possible), plus irradiation and adjuvant chemotherapy. Despite treatment, the prognosis is still poor, and the tumor often recurs within two centimeters of the original tumor. A promising approach to improving the efficacy of GBM therapeutics is to utilize biomaterials to deliver them locally at the tumor site. Local delivery to GBM offers several advantages over systemic administration, such as bypassing the blood–brain barrier and increasing the bioavailability of the therapeutic at the tumor site without causing systemic toxicity. Local delivery may also combat tumor recurrence by maintaining sufficient drug concentrations at and surrounding the original tumor area. Herein, we critically appraised the literature on local delivery systems based within the following categories: polymer-based implantable devices, polymeric injectable systems, and hydrogel drug delivery systems. We also discussed the negative effect of hypoxia on treatment strategies and how one might utilize local implantation of oxygen-generating biomaterials as an adjuvant to enhance current therapeutic strategies.
 Majed Alghamdi | Majed Alghamdi is currently pursuing a Ph.D. in pharmaceutical sciences at the School of Pharmacy and Pharmaceutical Sciences, Cardiff University. He graduated with a Pharm.D. degree at King Abdulaziz University, Jeddah, Saudi Arabia. He then graduated with an MSc in pharmaceutics at the Massachusetts College of Pharmacy and Health Sciences (MSCPHS), Boston, USA before beginning his Ph.D. research that focuses on the use of biomaterials for GBM therapy and how oxygen generation may improve radiotherapeutic efficacy in glioblastoma treatment. |
 Mark Gumbleton | Mark Gumbleton is a Professor of Experimental Therapeutics and is the Head of School at the School of Pharmacy and Pharmaceutical Sciences, Cardiff University. His research activities include understanding at the whole body, tissue, cellular and molecular levels the interaction of experimental therapeutics with biological barriers, particularly the blood-brain-barrier. In experimental therapeutics has a particular interest in cancer therapeutics with respect to brain tumours. |
 Ben Newland | Ben Newland joined the School of Pharmacy and Pharmaceutical Sciences at Cardiff University in 2017 to set up his highly interdisciplinary research team focussing on nano, micro and macroscale materials for use in neuroscience research. His group have developed microscale spherical cryogel scaffolds for cell, growth factor, cytokine, and drug delivery via local implantation in the brain. Together with collaboration partners, he has developed a new ex vivo model of multiple sclerosis. His team are interested in the role that soft biomaterials can play in glioblastoma therapeutics and how oxygen generation may improve radiotherapeutic efficacy in glioblastoma treatment. |
1. Introduction
1.1 Glioblastoma
Malignant brain tumors are one of the most lethal solid tumors leading to very poor prognoses.1 They can originate from the brain (primary brain tumors) or be metastasized from non-brain tumors to the brain (secondary brain tumors). Among the primary brain tumors, glioblastoma (GBM) is the most aggressive and most commonly occurring malignant brain tumor and is classified as a grade IV glioma tumor by the World Health Organization (WHO).2,3 GBM can start de novo (primary GBM) or develop from lower grades of gliomas (secondary GBM). Primary and secondary GBMs create distinctive subtypes affecting patients of different ages and are formed by distinct genetic pathways and vary in the prognosis.4 Primary GBM comprises 90% of the GBM cases, which usually affect elderly patients and have a worse prognosis than secondary GBM.5 The global incidence rate of GBM is equal to 10 per 100![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 population every year,6 with an average age of onset between 65 to 75 years.7 Despite the combined regimen of surgery, radiotherapy, and systemic chemotherapy, the prognosis of GBM remains poor.8 New therapeutics and new therapeutic strategies are therefore needed to improve the outcome for patients. To this end, in this review, we have focused on the potential emerging strategy of utilizing biomaterials for local delivery of chemotherapeutics or adjuvant treatments to malignant brain tumors.
000 population every year,6 with an average age of onset between 65 to 75 years.7 Despite the combined regimen of surgery, radiotherapy, and systemic chemotherapy, the prognosis of GBM remains poor.8 New therapeutics and new therapeutic strategies are therefore needed to improve the outcome for patients. To this end, in this review, we have focused on the potential emerging strategy of utilizing biomaterials for local delivery of chemotherapeutics or adjuvant treatments to malignant brain tumors.
1.2 Current standard treatment for newly diagnosed patients
The current standard treatment for newly diagnosed patients with GBM includes total safe surgical resection of the tumor followed by focal external beam radiotherapy to the surgical cavity with concomitant and adjuvant chemotherapy with temozolomide (TMZ).9 Surgery alone results in a median survival equal to 6 months, while the addition of radiation extends the survival to 12.1 months. The combined surgical resection, radiation, and chemotherapy of (TMZ) results in median survival of 14.6 months.9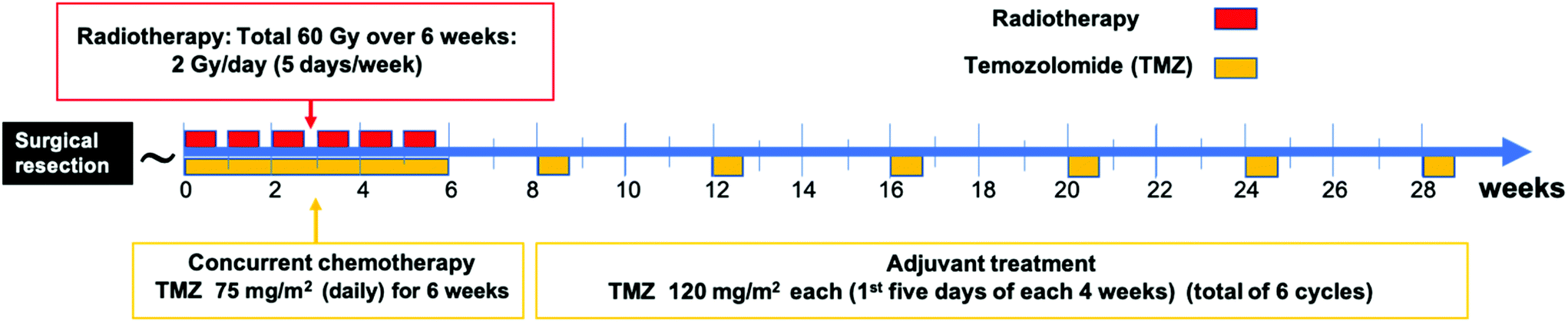 | ||
| Fig. 1 A schematic timeline of the standard treatment for newly diagnosed GBM patients (Stupp protocol9). Patients undergo surgical resection of the tumor followed by six weeks of radiation and concurrent TMZ which are then followed by six cycles of adjuvant TMZ. | ||
The standard radiotherapy regimen in GBM includes focal external beam radiotherapy (EBRT) to the tumor resection cavity plus two centimeters of the surrounding brain tissue. Radiotherapy is typically given as 60 Gy divided over six weeks as 2 Gy per day for five days a week.9 Radiotherapy utilizes ionizing radiation to produce free radicals that induce single-strand and double-strand breaks in the targeted cells’ DNA.15 Other radiation regimens are more recently being considered such as hypofractionation, which uses a lower total dose over a shorter period of time to treat GBM patients with advanced age or poor performance status.16
TMZ is an orally administered alkylating agent that causes DNA damage that eventually leads to cell apoptosis.17 In 2005, Stupp et al. reported on a clinical trial that showed that adding TMZ to radiation increased the median survival from 12.1 to 14.6 months; (P < 0.001) and the 5-year overall survival from 1.9% to 9.8%; (P < 0.001).9 These findings established the current standard treatment of GBM which is known as the Stupp protocol. In this protocol, TMZ is given with radiation (concurrently) with a dosage of 75 mg m−2 daily for the entire six weeks of radiation, then TMZ is given afterward for six cycles as an adjuvant.18
1.3 The rationale for new local therapeutic strategies
Unfortunately, the current standard treatment still results in a poor prognosis for newly diagnosed patients. Hence, other treatment strategies and novel interventions should be developed. Local delivery of chemotherapeutics and other adjuvant agents poses an interesting route to potentially improve GBM therapeutic outcomes.19 A number of factors make local drug delivery strategies particularly well-suited to applications in GBM therapies. Firstly, 65–75 percent of GBM patients undergo surgical resection,12,20 leaving behind a cavity into which drug delivery systems can be implanted.21Additionally, GBM recurrence usually occurs within two centimeters of the original tumor site22 meaning close proximity to the drug delivery system. Moreover, local delivery of therapeutics bypasses the blood–brain barrier (BBB) opening up possibilities of re-purposing chemotherapeutics that cannot cross the BBB. Furthermore, implants or intratumoral delivery could allow modulation/disruption of local microenvironmental factors that increase chemo/radio-resistance such as hypoxia. For example, biomaterials that produce or deliver molecular oxygen could increase the tumor susceptibility to chemo/radiotherapy. Finally, local delivery of chemotherapeutics increases their concentration locally, thus potentially avoiding dose-limiting system-wide adverse effects. For these reasons, local delivery of therapeutics poses several potential advantages for treating GBM over systemic delivery.
1.4 Biomaterials for controlled release of therapeutics
Local delivery of therapeutic agents to GBM is generally introduced through invasive routes such as brain surgery or stereotactic injection. However, the administered agents usually exhibit a short half-life as they are cleared from the brain tissue rapidly.23 To maintain a therapeutic concentration at the target site, without repeating the invasive administration, delivery systems are required to control drug release over a prolonged period of time.24 These systems can be achieved using biomaterials that carry the therapeutic payload and control its release. They can be designed for implantation/injection into the tumor surgical cavity or injection directly into the tumor.Biomaterials for controlled release of therapeutics can comprise of natural or synthetic biocompatible materials that encapsulate or load the therapeutic agents and limit their release by different mechanisms such as diffusion, dissociation, and dissolution.25 Such biomaterials include implantable colloidal carriers, hydrogels, and polymeric-based delivery systems (such as films, rods, disks, and wafers).19
Ideally, local chemotherapeutic drug delivery systems for GBM can be introduced directly inside the surgical cavity for newly diagnosed patients. Patients that undergo surgical resection of the tumor have to wait for the wound to heal before commencing radiotherapy. Local delivery could bridge this time gap to prevent any tumor proliferation before starting the radiation.26 For inoperable GBM tumors, biomaterials could be injected into the tumor directly therefore have to be in a size range that is easy to inject, such as microscale material. Herein, we provide an overview of such biomaterial systems that have been investigated for delivering chemotherapeutic drugs to a variety of gliomas. However, we also propose the use of local delivery systems to manipulate the tumor microenvironment for enhancing the efficacy of current therapeutics. Specifically, we discuss how oxygen-generating biomaterials could offer a means of modulating the hypoxic GBM microenvironment to improve radiotherapeutic outcomes.
2. Biomaterials for local delivery of chemotherapeutics to GBM
Biomaterials for local delivery of chemotherapy can be divided into three main categories: implantable polymeric drug delivery devices, injectable polymeric-based delivery systems, and hydrogel-based delivery systems.2.1 Implantable polymeric drug delivery devices
Implantable drug delivery devices for GBM generally use polymeric biomaterials designed to control the release of chemotherapeutic drugs into the surgical cavity. The basic principle applies only to operable tumors, where the devices are introduced into the surgical cavity during the same operation as the surgical resection. Gliadel, a carmustine filled polymer wafer, is currently the only local delivery device that has been approved by the FDA for use in GBM.27 Gliadel® wafers are white-to-pale, with dimensions of 14.5 mm in diameter and 1 mm in thickness (Fig. 3A). Each wafer contains 7.7 mg of carmustine in a biodegradable co-polymer formed of 1,3-bis-(p-carboxyphenoxy)propane (pCPP) and sebacic acid (SA) in a 20:80 molar ratio.28 The recommended amount of Gliadel® wafers is up to eight wafers (a total dose of 61.6 mg) which are placed to fill the tumor surgical cavity after the surgical resection. The co-polymer is hydrophobic and when in contact with aqueous fluids, it is hydrolyzed slowly into its monomeric contents releasing the carmustine into the surrounding tissue.29 The approval of carmustine wafers came after preclinical and clinical studies showed that local delivery of carmustine was superior to the placebo in terms of efficacy for newly diagnosed and recurrent GBM.30–32 Several other chemotherapeutic agents have also been encapsulated within pCPP and SA co-polymer wafers and have been tested in preclinical brain tumor models, including paclitaxel (PTX),33 carboplatin,34 camptothecin,35 minocycline,36 mitoxantrone,37 acriflavine38 and doxorubicin.39
Despite initial evidence of the efficacy of carmustine wafers as an adjuvant to radiation, only a modest improvement in survival has been achieved (1.7 months in comparison to the placebo wafer).40,41 This might be attributed to the drug release profile, where an initial burst release from the wafer occurs and the majority of carmustine is released in the first week.42,43 Moreover, due to the rigidity of the wafers, a physical and mechanical mismatch between the stiff device and the soft brain tissue can result in severe adverse reactions such as convulsions, confusion, cerebral edema, infection, and peri-cavity necrosis.40 These adverse reactions may last until complete degradation of the polymeric wafers occurs (up to 6 months) even though the carmustine has already been released.23,44,45 Therefore, ideal local drug delivery systems should solve both the problem of the rapid drug release and the toxicities resulted from the stiffness of the device.
Fig. 2 represents a graphical illustration comparing the release profile, efficacy, and toxicity of carmustine wafers and systemic TMZ administration against an ideal local drug delivery system.23 As mentioned before, a local delivery system can give the advantage of a high drug concentration in the tumor (Fig. 2a). However, an ideal local delivery system should maintain a high local concentration of the drug for a more extended period to increase the exposure of the drug to the tumor cells. Indeed, it has been reported that doubling the systemic TMZ cycles could improve survival by 8.4 months but at the expense of increasing the systemic toxicity.46 Moreover, the ideal local drug delivery system should be made of a soft material to avoid the toxicity associated with rigid materials such as Gliadel® wafers (Fig. 2c).
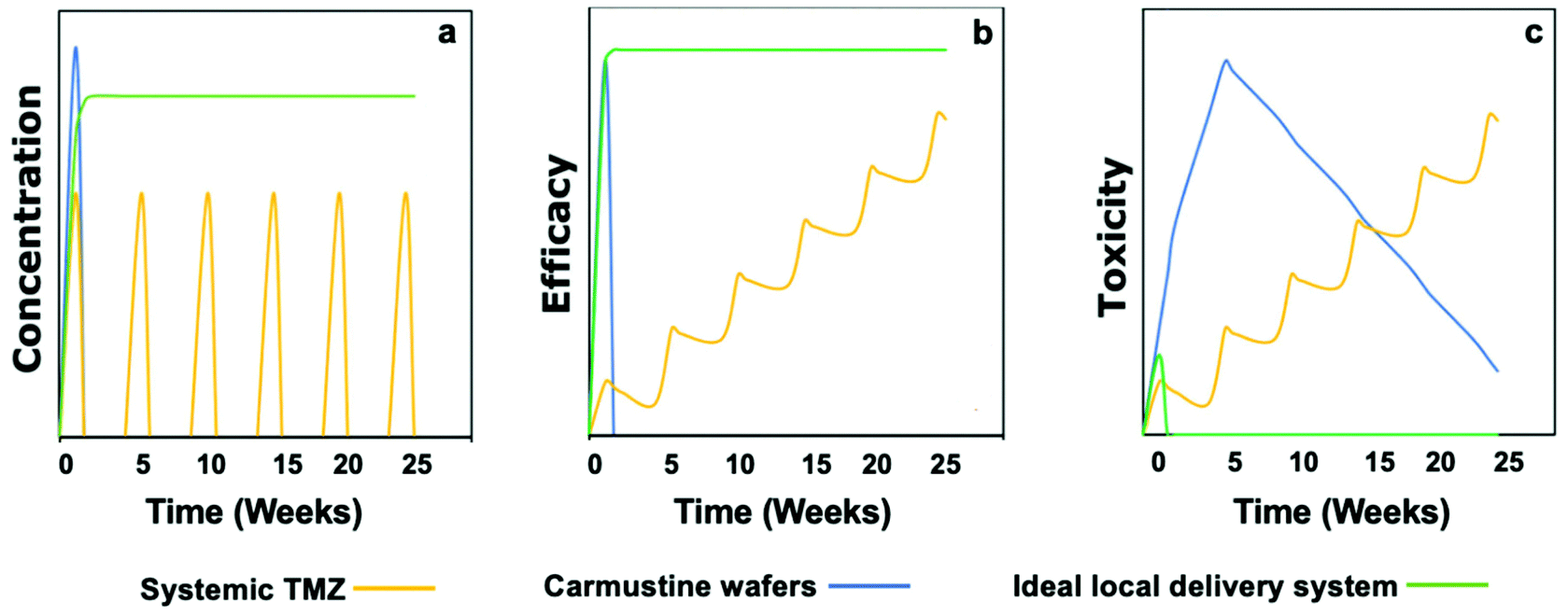 | ||
| Fig. 2 Graphical depiction of concentration, efficacy, and toxicity profiles of the adjuvant systemic TMZ (Stupp protocol), carmustine wafers, and an ideal local delivery system. (a) Local delivery can achieve higher drug concentration than systemic TMZ, the doses of which are limited by its systemic concentration. An ideal local delivery system will exhibit sustained release with zero or 1st order release kinetics. (b) An ideal local delivery system that achieves a long-term high concentration locally will show long-term efficacy in reducing tumor burden and preventing relapse. (c) The toxicity of carmustine wafers due to the rigidity of the device can be overcome in the ideal local delivery system by creating soft materials. In contrast, the toxicity of systemic TMZ limits the prolongation of the current regimen. Reproduced from ref. 23 with permission from Wiley, copyright 2019. | ||
Many approaches have been investigated to reduce the burst release and help sustain the release of the chemotherapeutic drug. Ranganath et al. developed PLGA micron- and submicron-fiber discs using electrospinning techniques, resulting in a high encapsulation efficiency of paclitaxel.47,48 These implants demonstrated a sustained release of paclitaxel over 80 days with only a small initial burst. Moreover, Rahman et al. explored the application of drug encapsulated in PLGA-microparticles delivered via a paste spread into the surgical cavity (Fig. 3B).49 Furthermore, Lee et al. designed a flexible sticky wireless device for drug delivery to brain tumors (Fig. 3C and D).50 The device contained doxorubicin-conjugated to oxidized starch and an electronic wireless heater and sensor that controlled the release and enhanced penetration of doxorubicin into the surrounding tissue.50 Doxorubicin was covalently bound to the oxidized starch which resulted in a decreased burst effect and prolonged doxorubicin release in comparison to the non-conjugated control. Implantation of this electronic device into an in vivo human xenograft GBM mouse model resulted in a significantly reduced GBM tumor volume and increased the survival rate compared to a BCNU wafer control group.
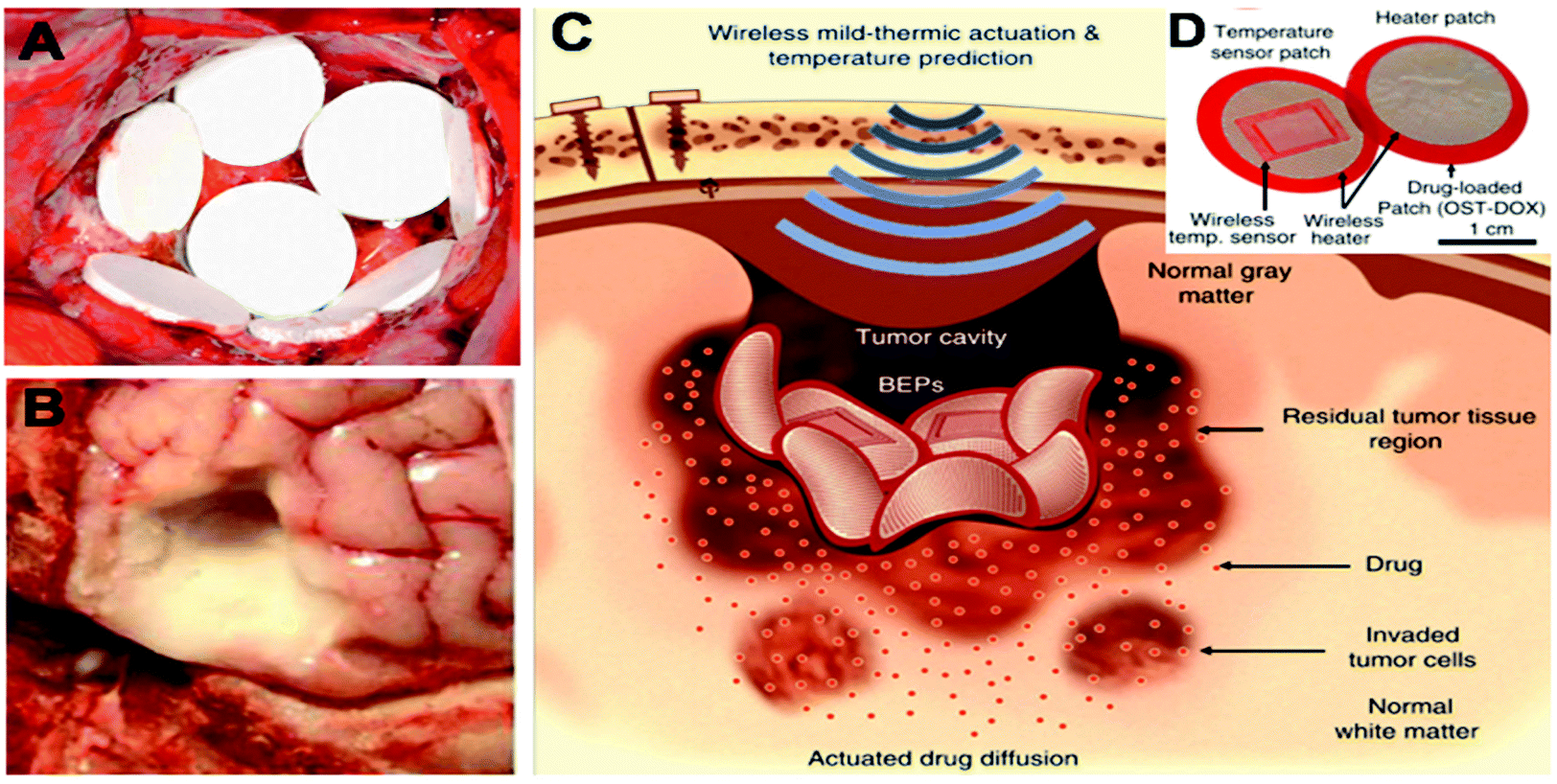 | ||
| Fig. 3 Examples of implantable chemotherapeutic drug devices for tumor resection cavity. (A) Biodegradable drug-loaded wafers (Gliadel® wafers) implanted into a brain tumor resection cavity. Reproduced from ref. 151 with permission from Elsevier, Copyrights (2011). (B) Drug-loaded paste applied to an ex vivo tumor resection cavity Reproduced from ref. 49, licensed under CC-BY, copyright 2013. (C) Schematic illustration of localized and penetrative drug delivery to deep GBM tissues by the bioresorbable electronic patch (BEP) with wireless mild-thermic actuation. (D) Image of the BEP, which includes a bioresorbable wireless heater and a temperature sensor on an oxidized starch (OST) patch containing doxorubicin (DOX). (C) and (D) Reproduced from ref. 50, licensed under CC-BY, copyright 2019. | ||
2.2 Hydrogel drug delivery systems
Hydrogels are three-dimensional (3D) polymeric networks formed through crosslinking of hydrophilic polymers within an aqueous medium.51 Due to the physical or chemical crosslinking of hydrophilic polymers, hydrogels are able to imbibe a large volume of water without dissolution.52 The high-water content of hydrogels can typically resemble biological tissue and can contribute to their excellent biocompatibility through matching the stiffness of their surroundings.53Hydrogels are excellent candidates for local and controlled delivery of chemotherapeutic drugs to GBM.19 Firstly, they can be used to encapsulate both hydrophilic and hydrophobic drugs.54 Secondly, they can be introduced to the brain by implantation post-surgically to the resection cavity. In addition, hydrogels capable of in situ gelation offer the possibility of easy administration by direct injection into either the surgical cavity or directly into a tumor. This in situ gelation can be achieved by various mechanisms such as photo-induced polymerization, forming irreversible covalent bonds, or via self-assembly by chemical reactions in response to specific stimuli such as pH and temperature.55 Furthermore, the water content of hydrogels contributes to their soft and flexible consistency which avoids a mechanical mismatch between the drug device and the soft brain tissue and the associated adverse reactions.56
Hydrogels also offer an attractive means of delivering biomacromolecules such as proteins and DNA, as such cargoes can be loaded to hydrogels without denaturing hydrophobic interactions.52 Lastly, hydrogels can be used to deliver chemotherapeutic drugs to GBM by either direct incorporation of the free drug57–63 or incorporation of drug-loaded nano/micro carriers56,64–66 (Fig. 4B).
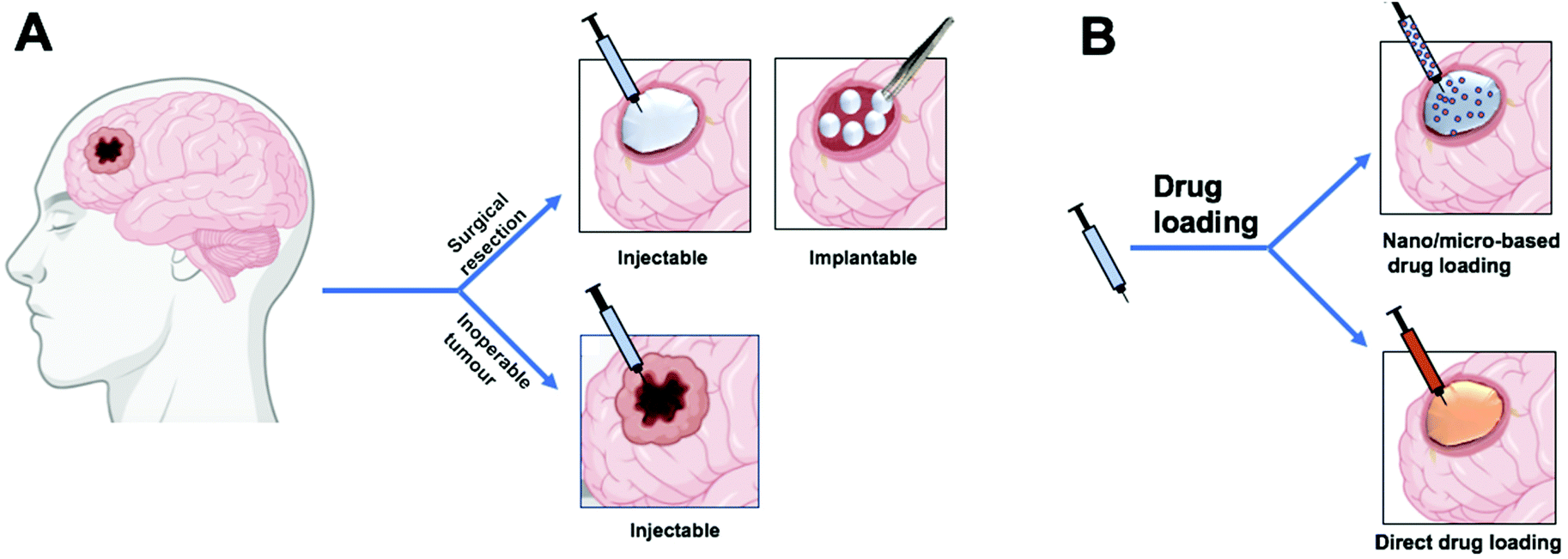 | ||
| Fig. 4 Schematic illustration of the use of hydrogel drug delivery systems. (A) A hydrogel drug delivery system can be injected or implanted into the tumor resection cavity or can be injected directly into the tumor for inoperable tumors. (B) Drug incorporation into the hydrogel can be direct loading of the drug or incorporation of drug-loaded nano/micro carriers. Figure created with BioRender.com. | ||
Several studies have explored the application of hydrogels for delivering chemotherapeutic agents locally to GBM.56,58–60,63,65,67–69 Akbar et al. developed a biodegradable PLGA based hydrogel comprised of a mixture of PLGA: plasticizer with a ratio of (40:60) to load and deliver TMZ directly into the resection cavity.57 Plasticizers with different hydrophilicity and hydrophobicity such as polyethylene glycol (PEG), N-methyl pyrrolidone (NMP), and acetyl triethyl citrate (ATEC) were studied which showed good biocompatibility.57 ATEC, a hydrophobic plasticizer, showed favorable drug release, with an initial burst followed by a constant release over a period of 15 days post-injection. The TMZ containing hydrogel system was tested for safety and efficacy in an in vivo C6-GFP glioma model and showed a significant reduction in tumor load compared to an empty hydrogel.57 Oncogel™ is another PLGA based hydrogel with PEG as a plasticizer that has been used to deliver paclitaxel (PTX) intratumorally.58,59
Oncogel™ is a thermo-sensitive hydrogel which has a low viscosity at lower temperatures and becomes a biodegradable hydrogel at body temperature.19 Oncogel™ was tested as an adjuvant to radiation and in combination with oral TMZ in the rodent 9L sarcoma model. The hydrogel sustained the drug release over six weeks and appeared to be safe, effective, and synergistic in the treatment.59
Achieving sustained release of locally delivered chemotherapeutic drugs is essential for maintaining drug exposure to the tumor site and accomplishing high therapeutic efficacy.23 To prolong the drug release, it can be incorporated first into a hydrophobic polymer-based nano/micro carrier, and then the system can be embedded into hydrogels. For instance, an alginate hydrogel was utilized to incorporate PTX-PLGA microspheres to form monodisperse spherical gel beads for implantation into the resection cavity.48 This resulted in enhancing the control over PTX release which lasted more than 60 days at a near-constant rate and a low initial burst.48 Another example includes the utility of photo polymerizable hydrogels for local delivery of chemotherapy-loaded nanoparticles to GBM.66,68 The photopolymerizable hydrogels enhanced the sustained release, provided easy injectability into the resection cavity, and provided in situ gelation using ultraviolet (UV) light. Zhao et al. incorporated PTX-PLGA nanoparticles into photo-polymerizable polyethylene glycol dimethyl acrylate which could be injected into the resection cavity.66 Using the U87 cell in vivo mouse model, this hydrogel/nanoparticle system allowed long-term survival (<150 days) and the empty hydrogel was well-tolerated in healthy mice for up to four months. These studies suggest that hydrogels can be used to retain nano/micro carriers at an injection site and extend the release profile of the chemotherapeutic payload.
Recently, Schiapparelli et al. synthesized a camptothecin-based self-assembled prodrug that can be injected into the surgical cavity.61 The prodrug consists of camptothecin covalently bound to peptide filaments which spontaneously assemble into a hydrogel in an aqueous solution i.e., within the surgical cavity. The in vitro release profile showed a near zero-order release over a period of 30 days and in vivo analysis showed significantly improved survival of a post-resection GBM murine model in comparison to the placebo hydrogel.
2.3 Injectable polymer-based drug delivery systems
Injectable polymer-based delivery systems involve drug-loaded nano/micro-carriers intended for injection into malignant brain tumors that cannot be resected. They can be administered by direct stereotactic injection into the tumor (intratumoral injection) or via the convection enhanced delivery (CED) injection. CED is a route of administration where a microcatheter is implanted into the tumor to deliver drugs by controlled-rate infusion which creates a high bulk flow, instead of simple diffusion, to maximize the distribution of the drug within the tumor.70–73 The use of nano/micro-polymeric carriers tends to improve drug penetration and retention into the tumor. Indeed, injection of free drug often results in poor drug penetration into, and retention within, the tumor due to its high interstitial pressure.74,75 An ideal intratumoral delivery system would result in equal drug distribution throughout the GBM, hence the use of CED, and the drug should be retained at the tumor site for a sufficient time.Several studies have explored the utility of intratumoral injections of injectable polymeric delivery systems into malignant brain tumors.76–81 PLGA based injectable nano/micro-carriers are one of the most heavily investigated because of the excellent biocompatibility and biodegradability of PLGA.82 Emerich et al. examined the efficacy of intratumoral injection of carboplatin- or BCNU-loaded PLGA microspheres into an in vivo rat glioma model which showed controlled drug release and superior efficacy than unloaded microspheres or bolus injection of the free drugs.81 However, multiple injections at the perimeter of the large tumors were needed to ensure good drug distribution and better efficacy.81 Other studies have also explored the utility of injectable polymer-based delivery systems containing different chemotherapeutic agents such as imatinib,76 curcumin,77 TMZ78 and docetaxel.80
In addition to direct intratumoral injections, drug-loaded nanoparticles can also be administered via CED. The drug distribution achieved by CED depends on a number of factors including catheter design, infusion rate and volume, the physiochemical properties of the injected drug or delivery system and the affinity towards the targeted tissue.83–85 Drug-loaded nanoparticles can offer an enhancement of the depth of penetration and therefore better volume of distribution across the tumor.70,86 Moreover, drug-loaded nanoparticles provide a controlled release of the drug as well as decrease the drug clearance rate.70,86 PLGA nanoparticles have been investigated for delivery of chemotherapeutic agents, such as camptothecin,87 dithiazanine iodide,88 TMZ89 and carboplatin,90 to be delivered via CED. Sawyer et al. synthesized camptothecin-loaded PLGA nanoparticles with an average particle size of 100 nm for CED.87 These loaded-PLGA nanoparticles were tested in vivo in a 9L tumor model via CED administration and showed a significant improvement in survival in comparison to CED injection of either unloaded nanoparticles or the free drug.87 To improve the drug penetration and distribution, Zhou et al. synthesized a smaller size of PLGA nanoparticles (average size of 74 nm) intended for CED.88 Formulating a smaller nanoparticle size enabled deeper penetration through the interstitial spaces which were reported to be 38–64 nm for healthy tissue and 7–100 nm for GBM tumor.91 Therefore, with careful design, drug-loaded nanoparticles, capable of sustained drug release, may increase the therapeutic efficacy of CED.
3. Biomaterials for radio-sensitization
3.1 The problem of tumor hypoxia
Tumor hypoxia, in which the partial oxygen pressure (pO2) can be defined as below 2%,92 occurs in solid tumors due to fast-growing tumor cells increasing oxygen demands, which are exacerbated by a disorganized tumor vascular supply.93 In gliomas, a larger degree of hypoxia is associated with a higher WHO grade and a poorer prognosis.94,95 This is a consequence of cellular responses to hypoxia which enable their survival under hypoxic conditions and results in a more aggressive tumor phenotype.95,96 In addition, a hypoxic environment is well-recognized to increase radiation resistance.15,97 GBM is associated with moderate hypoxia (pO2 0.5–0.1%) to severe hypoxia (pO2 < 0.1%), which occurs in ∼40% of GBM cases.94,95The cellular response of tumor cells to hypoxia depends on the expression of multiple genes that are mainly controlled by hypoxia-inducible transcription factors (HIFs). Many of these genes are linked to tumor progression by promoting survival activity such as angiogenesis, invasion/metastasis, glycolysis and drug resistance.98 Such genes include vascular endothelial growth factor-A (VEGF-A) which acts as a potent proangiogenic and is overexpressed in many tumors including GBM.99 Furthermore, HIFs induce a change in metabolism, shifting from oxidative phosphorylation to the glycolytic pathway (known as the Warburg effect), via the expression of genes regulating glucose transporters, glycolytic enzymes (causing lactate production), and pyruvate metabolism.100 Moreover, HIFs activation is linked to metastasis and migration through the regulation of key factors that control cell adhesion, extracellular matrix formation, and cell migration such as E-cadherin, lysyl oxidase, and CXCR4.99,101 Furthermore, HIFs activity is also associated with an increased expression of multiple drug resistance gene (MDR1) responsible for drug resistance.102 Lastly, hypoxia is found to promote the formation of a subpopulation of cells known as glioblastoma stem cells which are associated with self-renewal properties and are also highly resistant to chemo/radiotherapy.103
In addition to the cellular responses to hypoxia, a hypoxic environment also has a direct negative effect on radiotherapy.97,104–106 In general, a hypoxic environment is thought to interfere with the intracellular fate of free radicals caused by radiation. Briefly, the radiation interacts with intracellular molecules and one of the most abundant molecules is water, which leads to the formation of radical hydroxides (OH˙). These interact with DNA forming DNA-centered radicals (DNA˙). In the presence of oxygen, DNA˙ is oxidized to a DNA peroxide radical (DNA-OO˙), a process which is irreversible and leads to cytotoxicity via a DNA strand break. However, in the absence of oxygen (anoxia) or in severe hypoxia, this DNA-centered radical (DNA˙) can be repaired by endogenous thiols (RSH) thus reducing the efficacy of radiotherapy.107 This effect of oxygen is known as the oxygen fixation theory and radiation cytotoxicity is enhanced by oxygen by a factor of 2.5–3 in comparison to radiation in severely hypoxic conditions.106 In fact, increasing the oxygen level from 0% to up to 0.5% could double the effect of radiation, while reaching 5% oxygen could result in a three-fold radio-enhancement.106
Reversing the severe hypoxic environment of GBM by delivering oxygen could therefore enhance both radio- and chemotherapies. This could be applied not only to inoperable and recurrent GBM, but also to the surgical resection cavity which is often associated with hypoxia due to perioperative ischemia.108 Numerous efforts have been made to overcome this limitation by using hyperbaric oxygen therapy (HBOT) in which patients inhale 100% oxygen gas at higher pressure (∼2.8 atmospheres). Huang et al. summarized 11 clinical studies of radiotherapy with HBOT in GBM patients and showed that HBOT improved clinical outcomes in some cases.109 However, these clinical trials were conducted on a relatively small number of patients and many lacked a control group in the study design. Moreover, HBOT has some drawbacks including difficulties in the clinical application such as the requirement of a special radiation set-up, patient adherence (as some patients are claustrophobic), and possible increase in surrounding tissue damage due to non-specific oxygen elevation.110 To fully realize the therapeutic benefit of elevated tumor oxygenation, alternative approaches to generating oxygen in the tumor could be developed, as outlined below.
3.2 Localized oxygen-generating biomaterials
The development of oxygen-generating biomaterials, sometimes referred to as oxygen-producing biomaterials, is a rising field of research in tissue engineering and regenerative medicine to overcome hypoxia-induced tissue necrosis.111 Oxygen-generating biomaterials typically consist of an oxygen source that generates oxygen chemically and a polymeric material that entraps the oxygen source to hold it in position and prolong the release of oxygen over time. As oxygen-generating biomaterials generate the oxygen in situ, they are distinctly different from artificial oxygen carriers/blood substitutes or hemoglobin-based oxygen carriers (reviewed elsewhere112). For use as a radiotherapy adjuvant in glioblastoma therapies, local and sustained production of oxygen within the tumor or resection cavity microenvironment could be highly beneficial, so systemic strategies using general oxygen-carriers are not considered further herein.Oxygen-generating biomaterials have been reported to maintain the survival of engrafted engineered tissues113 as well as treatment of ischemic diseases,114 chronic wound healing115 and supporting high metabolic activity tissues such as pancreatic cells116 and muscle cells.117 Although oxygen-generating biomaterials have mostly been fabricated to maintain the survival of cells, they could provide an adjuvant effect to enhance the tumor killing capacity of radiotherapy by overcoming the tumor hypoxia.
So, despite the development of oxygen-generating biomaterials focusing on applications in tissue engineering and transplantation, the discussion below highlights some methods of oxygen generation and finally how these could be applied in GBM therapy.
3.3 Oxygen source materials
Peroxides are one of the most heavily investigated oxygen-generating materials which depend on the chemical decomposition of hydrogen peroxide into oxygen. They involve water-insoluble inorganic solid peroxides such as calcium peroxide (CaO2), Zinc peroxides (ZnO2), and magnesium peroxide (MgO2) and water-soluble hydrogen peroxide adducts such as sodium percarbonate [(Na2CO3)2·3H2O2]. In addition, H2O2 molecular complexation with polymers such as polyvinylpyrrolidone (PVP) have also been explored as oxygen source materials which is formed through hydrogen bonds.118 When in contact with water, solid peroxides dissociate to liquid H2O2 which then decomposes to water and oxygen in the presence of a catalyst (Fig. 5).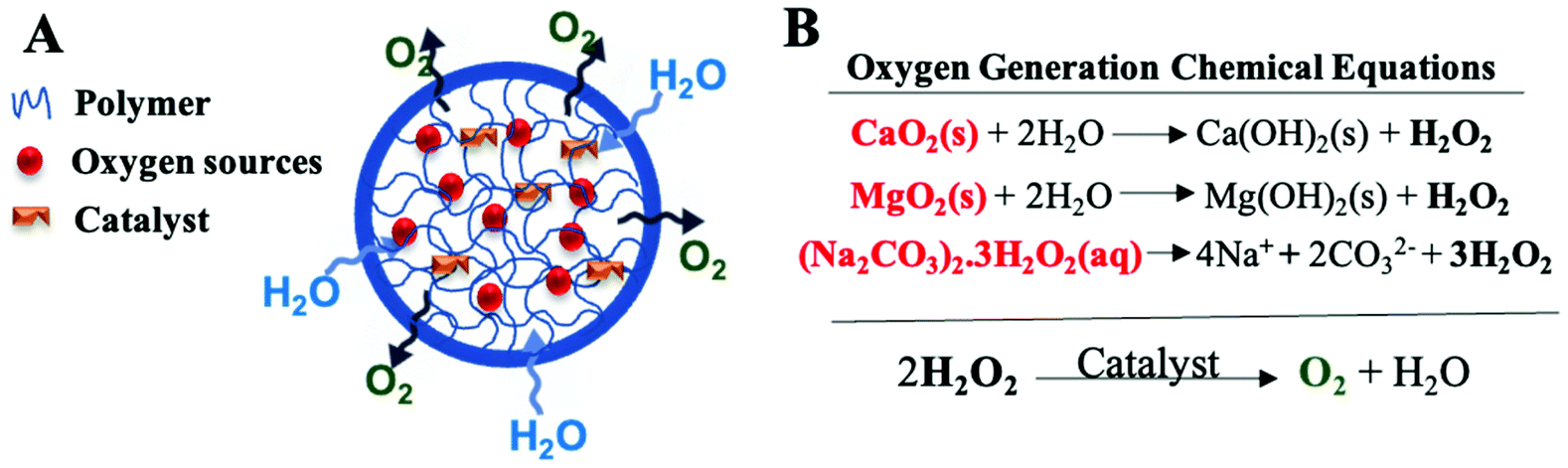 | ||
| Fig. 5 An overview of the principles behind the function of typical oxygen-generating materials. (A) Schematic representation of oxygen-generating materials which involve oxygen sources, such as solid peroxides, and a catalyst incorporated into a polymeric matrix to limit the inward diffusion of water and outward diffusion of hydrogen peroxides prior to conversion to oxygen and water. (B) Oxygen generation chemical equations which include the dissociation of solid peroxides into H2O2 and the decomposition of H2O2 into oxygen and water. Abbreviations: CaO2: calcium peroxides, MgO2: magnesium peroxide, (Na2CO3)2·3H2O2: sodium percarbonate. | ||
The chemical decomposition of H2O2 is slow and the chemical reaction rate depends on several factors such as pH,119 temperature,120 the solubility of peroxides, and presence of specific buffers or catalysts.119,121 The presence of a catalyst is important for accelerating the decomposition and preventing an accumulation of H2O2 which exerts cytotoxic effects.122 Catalase is a hemoprotein enzyme that is found in most cells and decomposes H2O2 to oxygen and water.111 It has therefore often been used with oxygen-generating biomaterials through conjugation with the carrier,123 embedding within the carrier,60,116 or being added to the surrounding medium,125 though in principle, other catalytic agents such as potassium iodide,126 platinum,116,127,128 palladium,128 rhodium,129 hemin130 and manganese dioxide131 could also be utilized.
The drawbacks of using solid peroxides as a source of oxygen include the potential for toxicity to surrounding tissue induced either by free radical toxicity of the peroxides, or by solid mineral depositions (mineral hydroxides such as Ca(OH)2) which remain post-oxygen generation.113 Peroxide toxicity might be overcome by using different delivery systems (carriers) or additives such as catalysts or antioxidants.
3.4 Polymeric carriers for peroxides
Oxygen-generating biomaterials can be incorporated into polymeric carriers to prolong the release of oxygen. Since the majority of oxygen-generating biomaterials synthesized to date have been for tissue engineering and regenerative medicine applications, a slow release of oxygen is desirable to sustain cell populations. However, the polymer coating also helps to avoid a burst release of H2O2 with the accompanying cytotoxicity that that would cause. Moreover, the carrier aids the incorporation of the catalyst which is needed for oxygen production. Furthermore, the use of carriers enables the use of multi-cargo agents such as chemotherapeutics to enhance the treatment of GBM.62 The carrier designs to date have included implantable devices, injectable microcarriers, and hydrogel delivery systems.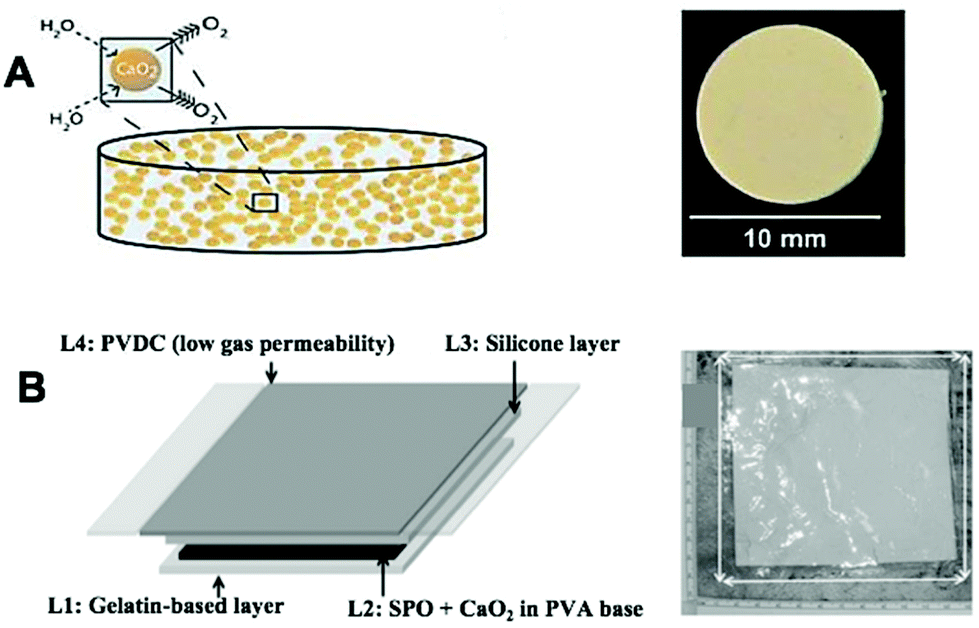 | ||
| Fig. 6 Bulk implantable oxygen-producing materials. (A) Schematic representation of CaO2 imbedded in a hydrophobic matrix of PDMS which hinders the inward diffusion of water and outward diffusion of oxygen. Reproduced from ref. 116 with permission from PNAS, copyright 2012. (B) Schematic representation of an oxygen-generating film showing four different layers as follows: layer 1 (L1) is a hydrophilic and oxygen permeable gelatin-based layer containing manganese chloride that is placed directly on top of the wound. L2 is an oxygen-generating layer that includes SPO and CaO2 in PVA-based matrix. L3 is a silicone layer that provides flexibility to the dressing. L4 is a thin PVDC-based layer that forms the outermost covering of the dressing that prevents the generated oxygen from leaking out of the dressing. Abbreviation: CaO2: Calcium peroxide, PDMS: polydimethylsiloxane, SPO: sodium percarbonate, PVA: polyvinyl alcohol, PVDC: polyvinylidene chloride. Reproduced from ref. 132 with permission from Wiley, copyright 2015. | ||
Chandra et al. synthesized an oxygen-generating film to improve the healing of chronic skin wounds.132 The film was made of four different layers, each adding functionality for the wound healing process. In terms of oxygen production, calcium peroxide and sodium percarbonate were incorporated in the second layer while a catalyst (manganese chloride; MnCl2) was embedded in the first layer (in contact with skin) to decompose H2O2 to O2 and minimize H2O2 leakage (Fig. 6B). This oxygen-generating film, with its gelatin underlayer, could be promising for reversing GBM hypoxia as it is flexible thus minimizing physical and mechanical mismatch. Furthermore, due to its flexibility, the film could be applied to inner lining of the surgical cavity while the protective layer of polyvinylidene chloride (PVDC) prevents unwanted release to the surrounding tissue. However, this system was only able to continuously release oxygen for three days, which is impractical for enhancing radiotherapy post-implantation and therefore improvement of the release duration is needed.
Whilst the aforementioned oxygen-generating biomaterials utilized solid peroxides, microparticles have utilized both solid and liquid peroxides as an oxygen source.114,123–126,133–137 Ng et al. encapsulated liquid H2O2 into PLGA microparticles via a double emulsion and solvent evaporation method.126 This encapsulation resulted in very modest sustained release of oxygen (5 hours). H2O2/PLGA microparticles have also been embedded in catalase-conjugated alginate via a hydrogel dripping method (to make microbeads) resulting in release for 24 hours (Fig. 7).134
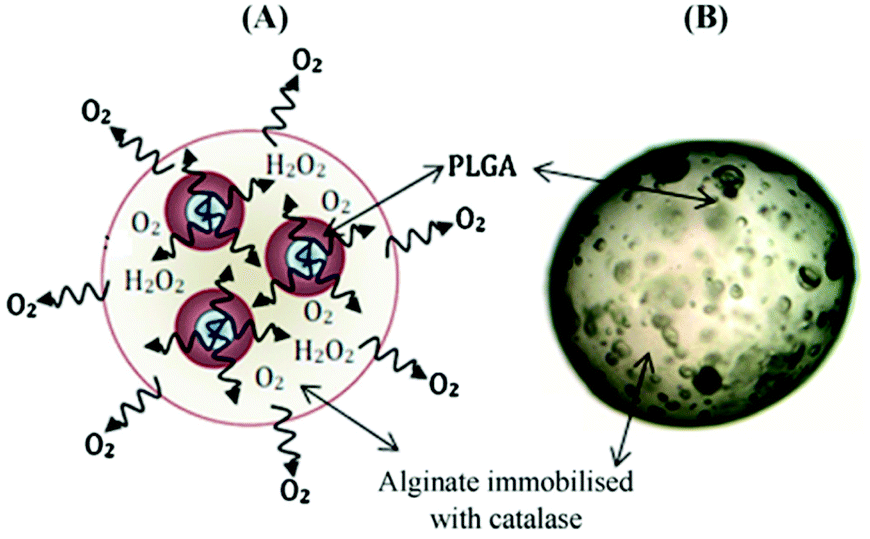 | ||
| Fig. 7 Example of oxygen-generating microbeads. (A) Representative depiction of decomposition route for H2O2 in a micro-system. (B) Optical microscope image of a PLGA-based micro-system encapsulated with alginate-catalase. Abbreviation: PLGA, poly(lactide-co-glycolide); H2O2, hydrogen peroxide. Reproduced from ref. 134 with permission from Elsevier, copyright (2011). | ||
To further improve the release, a complex of poly(vinylpyrrolidone) (PVP) and H2O2 (PVP-H2O2) incorporated into PLGA microparticles resulted in a prolonged release over 5 days.136 Moreover, Montazeri et al. have modified PVP-H2O2 PLGA microparticles by conjugating catalase onto the surface of the PLGA microparticles.123 Another interesting study injected PVP-H2O2 PLGA microparticles with a thermosensitive hydrophilic hydrogel to improve the retention of the microparticles at the injection site.114 This system resulted in a very favorable release of oxygen over a period of four weeks.
Solid peroxides such as CaO2 have also been encapsulated in microparticles. Newland et al. encapsulated CaO2 into polyethylene glycol diacrylate (PEGDA) polymer using an emulsion and UV crosslinking technique.125 Another two studies encapsulated CaO2 into polytrimthylene carbonate133 and PLGA138 by conventional oil/oil and oil/water emulsions and solvent evaporation methods and both resulted in the prolonged release of oxygen for over 21 days. Such prolonged release of oxygen could be beneficial to overcome GBM hypoxia and enhance radiation efficacy which is administered over six weeks in the current radiation protocol.9
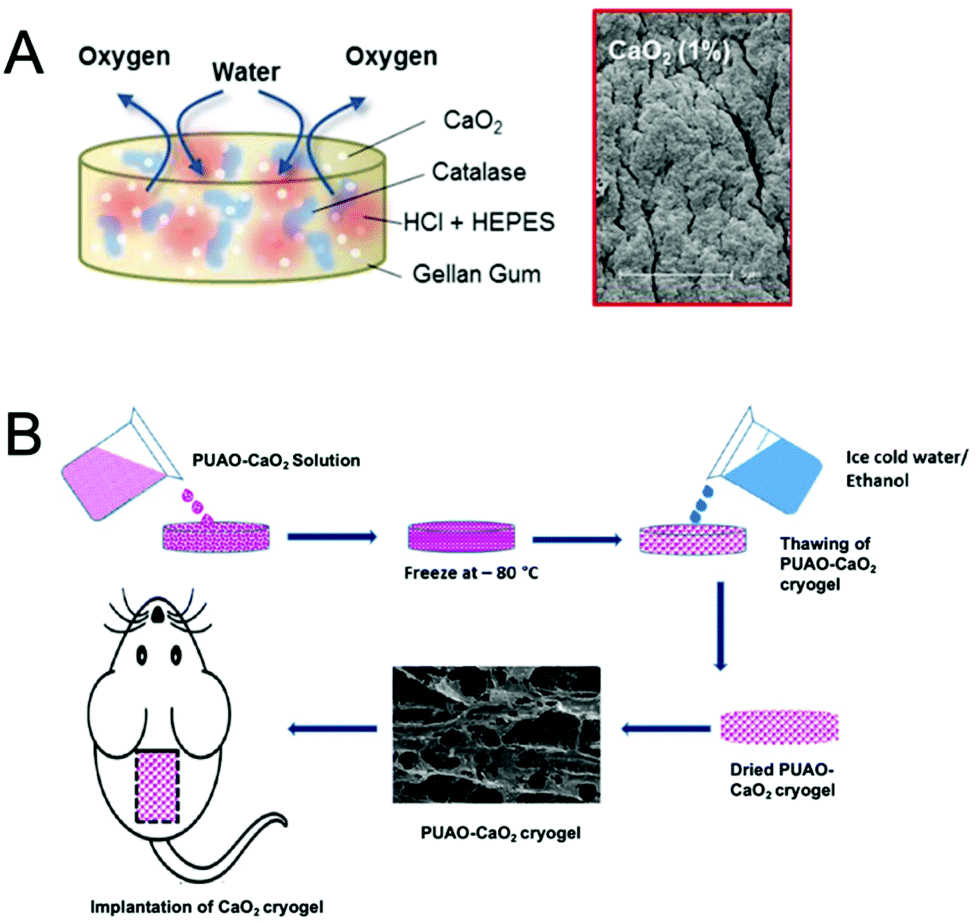 | ||
| Fig. 8 Hydrogel-based oxygen-generating biomaterials. (A) Schematic representation of a gellan gum hydrogel loaded with dual cargoes consist of calcium peroxide (CaO2) and doxorubicin. The scanning electron microscopy (SEM) image shows the morphology of the hydrogel after oxygen production. Reproduced from ref. 62 with permission from American Chemical Society, Copyright 2017. (B) Schematic representation of fabrication of CaO2-loaded polyurethane (PUAO) cryogel. Reproduced from ref. 142 with permission from American Chemical Society, copyright 2018. | ||
3.5 Nanoparticles for in situ oxygen generation
Unlike peroxide-based oxygen-generating biomaterials, this class of formulations depends on the high level of reactive oxygen species (ROS) within solid tumors to generate oxygen in situ.144 Manganese dioxide-based nanoparticles (MnO2 NPs) have been explored due to the high reactivity and specificity toward H2O2 to produce oxygen.145 Prasad et al. synthesized colloidally stable MnO2 NPs to generate oxygen and modify the microenvironment pH.145In vitro studies showed that MnO2 was able to generate oxygen when incubated with murine breast cancer EMT6 cells under hypoxic conditions. In terms of utilizing such nanoparticles as an adjuvant for radiation in GBM therapies, prior injection into the tumor would be required. Furthermore, a controlled release device or hydrogel which contains MnO2 NPs could be implanted into the surgical cavity for prolonging the nanoparticle release to last for the duration of the radiotherapy regimen. Moreover, it has been reported that cellular H2O2 plays a role in tumor migration146,147 and therefore MnO2 could potentially decrease the invasion of the GBM tumor to adjacent healthy tissue of the brain by reducing the levels of cellular H2O2.4. Future prospects
Whilst much of the early work in local delivery to GBM utilized large implantable wafers, there is a growing focus on micro/nanoscale delivery devices, soft hydrogels, or both in combination. Looking forward, for delivery systems to be safe, effective and be adopted by end users, we speculate that the following design criteria should be taken into consideration.Firstly, for bulk devices (i.e., not micro/nanoparticles) endeavor should be made at approximating the mechanical properties of the device to those of the surrounding tissue. In addition, space-filling properties, shown by hydrogels, cryogels and pastes are likely to be advantageous for both maximizing the interface with residual tumor cells and reducing dislodgment possibilities. Poor control over drug release must be overcome in order to sustain drug release to the order of months. If a synergistic mechanism with oral TMZ is desired then, 7–8 months of release would be favorable. This may be achieved through direct encapsulation though affinity-based methods of drug delivery may need to be investigated for long-term release.
In terms of oxygen-generating biomaterials, many studies with peroxides thus far fail to achieve the six weeks or so of release that would be required to act as an adjuvant for radiation therapy. An ideal local oxygen-generating system would exhibit a prolonged oxygen release that matches the radiation cycles. Hydrophobic polymers (such as PDMS) have thus far shown some of the most favorable release profiles, so the formation of a biodegradable hydrophobic polymer system (such as highly versatile PAE polymers148) could be a possible strategy towards sustained release. To avoid tissue damage due to a mismatch of mechanical properties, the idea system should be soft, yet easy to handle. Otherwise, an ideal local oxygen delivery system intended for use in the resection cavity would utilize in situ polymerization allowing cavity filling and dislodgment avoidance. Finally, an oxygen delivery system to GBM should demonstrate an excellent safety profile toward the brain tissue. To date, no such system exists that matches all these criteria concomitantly. The use of solid peroxides in the brain is questionable, due to the formation of their associated solid hydroxides (basic), thus prompting the thought that the use of endogenous tumor peroxides may be a safer route to oxygen generation.
The use of catalytic nanoparticles such as manganese dioxide (MnO2) nanoparticles take advantage of the endogenous hydrogen peroxide produced by tumor cells, thus negating the need for continual exogenous peroxide delivery. However, the stability (both too short or too long) and distribution parameters will need to be analyzed carefully to avoid unnecessary damage to healthy tissue. Furthermore, a whole range of host-response characteristics will need to be analyzed such as the nanoparticle toxicity, the host immune/foreign body response, and possible changes in vascularization (due to altered oxygen concentration). These will likely be material specific, so must be examined thoroughly for each material analyzed.
The rationale for using oxygen-generating biomaterials, such as those discussed herein, has largely revolved around improving the efficacy of radiotherapy for GBM. However, reversing tumor hypoxia, and therefore HIF-1α expression, may also have spill-over effects in reversing HIF-1α-mediated pathways such as neoangiogenesis, tumor growth and invasion. To reinforce this effect, one could envisage using a combination of oxygen-generating biomaterials to improve radiotherapy, and HIF-1α targeting drugs, such as acriflavine, to reduce tumor growth, and vascularization.149 Encapsulation of acriflavine in the biodegradable CPP and SA polymer has already yielded impressive results after local delivery to the rat 9L glioasarcoma model, even in the absence of radiotherapy.38
For all local delivery devices, factors such as storage, sterility and ease-of-use must also be considered. Whilst in situ forming hydrogels can be ideal in terms of mechanical and space-filling properties, they must be easy to use in a clinical setting. Delivery devices such as cryogelated hydrogels may prove beneficial in this aspect,150 where preformed and pre-loaded carriers can be shipped and stored in their dehydrated state and be rehydrated within the resection cavity. As tumor imaging improves and robotic surgical interventions become more widespread, the scope for using local interventions is increasing, provided that the innovation in delivery systems can keep pace.
5. Summary
Local delivery of chemotherapeutics and other adjuvant therapeutics shows promise as a strategy to improve the efficacy of GBM therapeutics. Such strategies include local injection or implantation of polymer-based biomaterials to sustainably deliver loaded therapeutics. Polymer-based biomaterials involve carriers such as implantable devices, hydrogel delivery systems and injectable nano/micro-carriers.Carmustine loaded wafers, the only clinically approved implantable delivery system for GBM, has thus far showed only a modest improvement in survival. In addition, the adverse reactions associated with wafers due to the physical mismatch between the rigid device and the soft surrounding tissues could last for weeks.
A wide range of novel delivery systems such as micro/nano materials or hydrogels are being developed to overcome these drawbacks. Hydrogel systems can be pre-formed and implanted into the surgical cavity of the tumor or form in situ upon injection into the tumor or resection cavity. Moreover, for GBMs where resection is impossible, stereotactic injection of small delivery systems may still be possible, to ensure a high local concentration of the therapeutic. Since tumor hypoxia negatively effects radiotherapy outcomes, one could also consider the use of oxygen-generating biomaterials as an adjuvant therapy. Whilst these are in their relative infancy, tailoring their use specifically for GBM therapeutics may result in more effective tumor ablation.
In sum, the rapid development of biomaterials in these research field provides promise for highly effective local intervention strategies for treating GBM.
Author contributions
CRediT roles: conceptualization – B.N.; writing – original draft – M.A.; writing – review &editing – M.A., M.G., and B.N.Conflicts of interest
There are no conflicts to declare.References
- T. A. Dolecek, J. M. Propp, N. E. Stroup and C. Kruchko, Neuro-Oncol., 2012, 14, v1–v49 CrossRef PubMed.
- Q. T. Ostrom, G. Cioffi, H. Gittleman, N. Patil, K. Waite, C. Kruchko and J. S. Barnholtz-Sloan, Neuro-Oncol., 2019, 21, v1–v100 CrossRef PubMed.
- D. N. Louis, H. Ohgaki, O. D. Wiestler, W. K. Cavenee, P. C. Burger, A. Jouvet, B. W. Scheithauer and P. Kleihues, Acta Neuropathol., 2007, 114, 97–109 CrossRef PubMed.
- H. Ohgaki and P. Kleihues, Am. J. Pathol., 2007, 170, 1445–1453 CrossRef CAS PubMed.
- H. Ohgaki and P. Kleihues, Clin. Cancer Res., 2013, 19, 764–772 CrossRef CAS.
- F. Hanif, K. Muzaffar, K. Perveen, S. M. Malhi and S. U. Simjee, Asian Pac. J. Cancer Prev., 2017, 18, 3–9 Search PubMed.
- A. Brodbelt, D. Greenberg, T. Winters, M. Williams, S. Vernon and V. P. Collins, Eur. J. Cancer, 2015, 51, 533–542 CrossRef.
- R. Stupp, S. Taillibert, A. A. Kanner, S. Kesari, D. M. Steinberg, S. A. Toms, L. P. Taylor, F. Lieberman, A. Silvani, K. L. Fink, G. H. Barnett, J. J. Zhu, J. W. Henson, H. H. Engelhard, T. C. Chen, D. D. Tran, J. Sroubek, N. D. Tran, A. F. Hottinger, J. Landolfi, R. Desai, M. Caroli, Y. Kew, J. Honnorat, A. Idbaih, E. D. Kirson, U. Weinberg, Y. Palti, M. E. Hegi and Z. Ram, JAMA, 2015, 314, 2535–2543 CrossRef CAS PubMed.
- R. Stupp, W. P. Mason, M. J. Van Den Bent, M. Weller, B. Fisher, M. J. B. Taphoorn, K. Belanger, A. A. Brandes, C. Marosi, U. Bogdahn, J. Curschmann and R. C. Janzer, et al. , N. Engl. J. Med., 2005, 352, 987–996 CrossRef CAS PubMed.
- I. Ciric, M. Ammirati, N. Vick and M. Mikhael, Neurosurgery, 1987, 21, 21–26 CrossRef CAS PubMed.
- M. Lacroix, D. Abi-Said, D. R. Fourney, Z. L. Gokaslan, W. Shi, F. DeMonte, F. F. Lang, I. E. McCutcheon, S. J. Hassenbusch, E. Holland, K. Hess, C. Michael, D. Miller and R. Sawaya, J. Neurosurg., 2001, 95, 190–198 CAS.
- R. L. Yong and R. R. Lonser, World Neurosurg., 2011, 76, 528–530 CrossRef PubMed.
- R. Lai, D. L. Hershman, T. Doan and A. I. Neugut, Neuro-Oncol., 2010, 12, 190–198 CrossRef PubMed.
- M. Geurts and M. J. Van Den Bent, Neuro-Oncol., 2018, 20, 868–869 CrossRef PubMed.
- J. Bernier, E. J. Hall and A. Giaccia, Nat. Rev. Cancer, 2004, 4, 737–747 CrossRef CAS PubMed.
- C. Gzell, M. Back, H. Wheeler, D. Bailey and M. Foote, Clin. Oncol., 2017, 29, 15–25 CrossRef CAS PubMed.
- J. Zhang, M. F. G. Stevens and T. D. Bradshaw, Curr. Mol. Pharmacol., 2011, 5, 102–114 CrossRef PubMed.
- R. Stupp, M. E. Hegi, W. P. Mason, M. J. van den Bent, M. J. Taphoorn, R. C. Janzer, S. K. Ludwin, A. Allgeier, B. Fisher, K. Belanger, P. Hau, A. A. Brandes, J. Gijtenbeek, C. Marosi, C. J. Vecht, K. Mokhtari, P. Wesseling, S. Villa, E. Eisenhauer, T. Gorlia, M. Weller, D. Lacombe, J. G. Cairncross and R. O. Mirimanoff, Lancet Oncol., 2009, 10, 459–466 CrossRef CAS PubMed.
- C. Bastiancich, P. Danhier, V. Préat and F. Danhier, J. Controlled Release, 2016, 243, 29–42 CrossRef CAS PubMed.
- K. R. Yabroff, L. Harlan, C. Zeruto, J. Abrams and B. Mann, Neuro-Oncol., 2012, 14, 351–359 CrossRef PubMed.
- A. Bregy, A. H. Shah, M. V. Diaz, H. E. Pierce, P. L. Ames, D. Diaz and R. J. Komotar, Expert Rev. Anticancer Ther., 2013, 13, 1453–1461 CrossRef CAS.
- K. E. Wallner, J. H. Galicich, G. Krol, E. Arbit and M. G. Malkin, Int. J. Radiat. Oncol. Biol. Phys., 1989, 16, 1405–1409 CrossRef CAS.
- A. Tabet, M. P. Jensen, C. C. Parkins, P. G. Patil, C. Watts and O. A. Scherman, Adv. Healthcare Mater., 2019, 8, 1–6 Search PubMed.
- C. Bastiancich, J. Bianco, K. Vanvarenberg, B. Ucakar, N. Joudiou, B. Gallez, G. Bastiat, F. Lagarce, V. Préat and F. Danhier, J. Controlled Release, 2017, 264, 45–54 CrossRef CAS PubMed.
- J. Siepmann, F. Siepmann and A. T. Florence, Int. J. Pharm., 2006, 314, 101–119 CrossRef CAS PubMed.
- D. M. Patel, N. Agarwal, K. L. Tomei, D. R. Hansberry and I. M. Goldstein, World Neurosurg., 2015, 84, 412–419 CrossRef PubMed.
- J. P. Fisher and D. C. Adamson, Biomedicines, 2021, 9, 324 CrossRef CAS PubMed.
- S. D. Wait, R. S. Prabhu, S. H. Burri, T. G. Atkins and A. L. Asher, Neuro-Oncol., 2015, 17, ii9–ii23 CrossRef CAS PubMed.
- A. J. Domb, Z. H. Israel, O. Elmalak, D. Teomim and A. Bentolila, Pharm. Res., 1999, 16, 762–765 CrossRef CAS.
- R. J. Tamargo, J. S. Myseros, J. I. Epstein, M. B. Yang, M. Chasin and H. Brem, Cancer Res., 1993, 53, 329–333 CAS.
- H. Brem, S. Piantadosi, P. C. Burger, M. Walker, R. Selker, N. A. Vick, K. Black, M. Sisti, S. Brem, G. Mohr, P. Muller, R. Morawetz and S. C. Schold, Lancet, 1995, 345, 1008–1012 CrossRef CAS.
- S. Valtonen, U. Timonen, P. Toivanen, H. Kalimo, L. Kivipelto, O. Heiskanen, G. Unsgaard and T. Kuurne, Neurosurgery, 1997, 41, 44–49 CrossRef CAS.
- K. A. Walter, M. A. Cahan, A. Gur, B. Tyler, J. Hilton, O. M. Colvin, P. C. Burger, A. Domb and H. Brem, Cancer Res., 1994, 54, 2207–2212 CAS.
- A. Olivi, M. G. Ewend, T. Utsuki, B. Tyler, A. J. Domb, D. J. Brat and H. Brem, Cancer Chemother. Pharmacol., 1996, 39, 90–96 CrossRef CAS.
- P. B. Storm, J. L. Moriarity, B. Tyler, P. C. Burger, H. Brem and J. Weingart, J. Neurooncol., 2002, 56, 209–217 CrossRef.
- J. L. Frazier, P. P. Wang, D. Case, B. M. Tyler, G. Pradilla, J. D. Weingart and H. Brem, J. Neurooncol., 2003, 64, 203–209 CrossRef PubMed.
- F. Dimeco, K. W. Li, B. M. Tyler, A. S. Wolf, H. Brem and A. Olivi, J. Neurosurg., 2002, 97, 1173–1178 CAS.
- A. Mangraviti, T. Raghavan, F. Volpin, N. Skuli, D. Gullotti, J. Zhou, L. Asnaghi, E. Sankey, A. Liu, Y. Wang, D. H. Lee, N. Gorelick, R. Serra, M. Peters, D. Schriefer, F. Delaspre, F. J. Rodriguez, C. G. Eberhart, H. Brem, A. Olivi and B. Tyler, Sci. Rep., 2017, 7, 1–13 CrossRef CAS PubMed.
- M. S. Lesniak, U. Upadhyay, R. Goodwin, B. Tyler and H. Brem, Anticancer Res., 2005, 25, 3825–3831 CAS.
- J. Perry, A. Chambers, K. Spithoff and N. Laperriere, Curr. Oncol., 2007, 14, 189–194 CrossRef CAS.
- M. Westphal, Z. Ram, V. Riddle, D. Hilt and E. Bortey, Acta Neurochir., 2006, 148, 269–275 CrossRef CAS PubMed.
- A. B. Fleming and W. M. Saltzman, Clin. Pharmacokinet., 2002, 41, 403–419 CrossRef CAS PubMed.
- S. H. Lin and L. R. Kleinberg, Expert Rev. Anticancer Ther., 2008, 8, 343–359 CrossRef CAS PubMed.
- J. P. Harris, J. R. Capadona, R. H. Miller, B. C. Healy, K. Shanmuganathan, S. J. Rowan, C. Weder and D. J. Tyler, J. Neural Eng., 2011, 8, 66011–66024 CrossRef CAS PubMed.
- K. A. Potter, A. C. Buck, W. K. Self and J. R. Capadona, J. Neural Eng., 2012, 9, 046020 CrossRef PubMed.
- M. Bhandari, A. K. Gandhi, B. Devnani, P. Kumar, D. N. Sharma and P. K. Julka, J. Clin. Diagn. Res., 2017, 11, XC04–XC08 CAS.
- S. H. Ranganath, Y. Fu, D. Y. Arifin, I. Kee, L. Zheng, H. S. Lee, P. K. H. Chow and C. H. Wang, Biomaterials, 2010, 31, 5199–5207 CrossRef CAS PubMed.
- S. H. Ranganath and C. H. Wang, Biomaterials, 2008, 29, 2996–3003 CrossRef CAS PubMed.
- C. V. Rahman, S. J. Smith, P. S. Morgan, K. A. Langmack, P. A. Clarke, A. A. Ritchie, D. C. Macarthur, F. R. Rose, K. M. Shakesheff, R. G. Grundy and R. Rahman, PLoS One, 2013, 8, 1–12 CrossRef.
- J. Lee, H. R. Cho, G. D. Cha, H. Seo, S. Lee, C. K. Park, J. W. Kim, S. Qiao, L. Wang, D. Kang, T. Kang, T. Ichikawa, J. Kim, H. Lee, W. Lee, S. Kim, S. T. Lee, N. Lu, T. Hyeon, S. H. Choi and D. H. Kim, Nat. Commun., 2019, 10, 1–9 CrossRef PubMed.
- Y. S. Zhang and A. Khademhosseini, Science, 2017, 356, eaaf3627 CrossRef PubMed.
- N. A. Peppas, P. Bures, W. Leobandung and H. Ichikawa, Eur. J. Pharm. Biopharm., 2000, 50, 27–46 CrossRef CAS PubMed.
- S. Naahidi, M. Jafari, M. Logan, Y. Wang, Y. Yuan, H. Bae, B. Dixon and P. Chen, Biotechnol. Adv., 2017, 35, 530–544 CrossRef CAS PubMed.
- C. C. Lin and A. T. Metters, Adv. Drug Delivery Rev., 2006, 58, 1379–1408 CrossRef CAS PubMed.
- S. R. Van Tomme, G. Storm and W. E. Hennink, Int. J. Pharm., 2008, 355, 1–18 CrossRef CAS PubMed.
- R. Hosseinzadeh, B. Mirani, E. Pagan, S. Mirzaaghaei, A. Nasimian, P. Kawalec, S. C. da Silva Rosa, D. Hamdi, N. P. Fernandez, B. D. Toyota, J. W. Gordon, S. Ghavami and M. Akbari, Adv. Ther., 2019, 2, 1900113 CrossRef CAS.
- U. Akbar, T. Jones, J. Winestone, M. Michael, A. Shukla, Y. Sun and C. Duntsch, J. Neurooncol., 2009, 94, 203–212 CrossRef CAS PubMed.
- B. Tyler, K. D. Fowers, K. W. Li, V. R. Recinos, J. M. Caplan, A. Hdeib, R. Grossman, L. Basaldella, K. Bekelis, G. Pradilla, F. Legnani and H. Brem, J. Neurosurg., 2010, 113, 210–217 CAS.
- A. K. Vellimana, V. R. Recinos, L. Hwang, K. D. Fowers, K. W. Li, Y. Zhang, S. Okonma, C. G. Eberhart, H. Brem and B. M. Tyler, J. Neurooncol., 2013, 111, 229–236 CrossRef CAS PubMed.
- M. H. Turabee, T. H. Jeong, P. Ramalingam, J. H. Kang and Y. T. Ko, Carbohydr. Polym., 2019, 203, 302–309 CrossRef CAS PubMed.
- P. Schiapparelli, P. Zhang, M. Lara-Velazquez, H. Guerrero-Cazares, R. Lin, H. Su, R. W. Chakroun, M. Tusa, A. Quiñones-Hinojosa and H. Cui, J. Controlled Release, 2020, 319, 311–321 CrossRef CAS PubMed.
- B. Newland, M. Baeger, D. Eigel, H. Newland and C. Werner, ACS Biomater. Sci. Eng., 2017, 3, 787–792 CrossRef CAS PubMed.
- Z. Zhao, J. Shen, L. Zhang, L. Wang, H. Xu, Y. Han, J. Jia, Y. Lu, R. Yu and H. Liu, Biomater. Sci., 2020, 8, 5306–5316 RSC.
- S. H. Ranganath, I. Kee, W. B. Krantz, P. K. H. Chow and C. H. Wang, Pharm. Res., 2009, 26, 2101–2114 CrossRef CAS PubMed.
- L. Ding, Q. Wang, M. Shen, Y. Sun, X. Zhang, C. Huang, J. Chen, R. Li and Y. Duan, Autophagy, 2017, 13, 1176–1190 CrossRef CAS PubMed.
- M. Zhao, F. Danhier, C. Bastiancich, N. Joudiou, L. P. Ganipineni, N. Tsakiris, B. Gallez, A. des Rieux, A. Jankovski, J. Bianco and V. Préat, Int. J. Pharm., 2018, 548, 522–529 CrossRef CAS.
- J. Il Kim, B. Kim, C. J. Chun, S. H. Lee and S. C. Song, Biomaterials, 2012, 33, 4836–4842 CrossRef PubMed.
- T. Fourniols, L. D. Randolph, A. Staub, K. Vanvarenberg, J. G. Leprince, V. Préat, A. Des Rieux and F. Danhier, J. Controlled Release, 2015, 210, 95–104 CrossRef CAS PubMed.
- T. Chen, T. Gong, T. Zhao, X. Liu, Y. Fu, Z. Zhang and T. Gong, Int. J. Pharm., 2017, 528, 127–132 CrossRef CAS PubMed.
- E. Allard, C. Passirani and J. P. Benoit, Biomaterials, 2009, 30, 2302–2318 CrossRef CAS PubMed.
- E. White, A. Bienemann, H. Taylor, K. Hopkins, A. Cameron and S. Gill, Contemp. Clin. Trials, 2012, 33, 320–331 CrossRef CAS PubMed.
- N. U. Barua, K. Hopkins, M. Woolley, S. Osullivan, R. Harrison, R. J. Edwards, A. S. Bienemann, M. J. Wyatt, A. Arshad and S. S. Gill, Drug Deliv., 2016, 23, 167–173 CrossRef CAS PubMed.
- W. G. B. Singleton, A. S. Bieneman, M. Woolley, D. Johnson, O. Lewis, M. J. Wyatt, S. J. P. Damment, L. J. Boulter, C. L. Killick-Cole, D. J. Asby and S. S. Gill, J. Neurosurg. Pediatr., 2018, 22, 288–296 Search PubMed.
- C. H. Heldin, K. Rubin, K. Pietras and A. Östman, Nat. Rev. Cancer, 2004, 4, 806–813 CrossRef CAS PubMed.
- A. I. Minchinton and I. F. Tannock, Nat. Rev. Cancer, 2006, 6, 583–592 CrossRef CAS PubMed.
- O. Benny, L. G. Menon, G. Ariel, E. Goren, S. K. Kim, C. Stewman, P. M. Black, R. S. Carroll and M. Machluf, Clin. Cancer Res., 2009, 15, 1222–1231 CrossRef CAS PubMed.
- M. Orunoğlu, A. Kaffashi, S. B. Pehlivan, S. Şahin, F. Söylemezoğlu, K. Karlı-Oğuz and M. Mut, Mater. Sci. Eng., C, 2017, 78, 32–38 CrossRef PubMed.
- J. Dong, G. Zhou, D. Tang, Y. Chen, B. Cui, X. Dai, J. Zhang, Q. Lan and Q. Huang, J. Cancer Res. Clin. Oncol., 2012, 138, 2079–2084 CrossRef CAS PubMed.
- C. Manaspon, N. Nasongkla, K. Chaimongkolnukul, P. Nittayacharn, K. Vejjasilpa, K. Kengkoom, A. Boongird and S. Hongeng, Pharm. Res., 2016, 33, 2891–2903 CrossRef CAS PubMed.
- H. L. Xu, K. L. Mao, C. T. Lu, Z. L. Fan, J. J. Yang, J. Xu, P. P. Chen, D. L. ZhuGe, B. X. Shen, B. H. Jin, J. Xiao and Y. Z. Zhao, Biomaterials, 2016, 107, 44–60 CrossRef CAS PubMed.
- D. F. Emerich, S. R. Winn, P. Snodgrass, D. LaFreniere, M. Agostino, T. Wiens, H. Xiong, R. T. Bartus, Y. Hu, J. Marsh, P. Snodgrass, D. LaFreniere, T. Wiens, B. P. Hasler and R. T. Bartus, Pharm. Res., 2000, 17, 767–775 CrossRef CAS PubMed.
- A. Kumari, S. K. Yadav and S. C. Yadav, Colloids Surf., B, 2010, 75, 1–18 CrossRef CAS PubMed.
- E. White, A. Bienemann, J. Malone, L. Megraw, C. Bunnun, M. Wyatt and S. Gill, J. Neurosci. Methods, 2011, 199, 87–97 CrossRef PubMed.
- R. Saito, M. T. Krauze, C. O. Noble, M. Tamas, D. C. Drummond, D. B. Kirpotin, M. S. Berger, J. W. Park and K. S. Bankiewicz, J. Neurosci. Methods, 2006, 154, 225–232 CrossRef CAS.
- J. A. MacKay, D. F. Deen and F. C. Szoka, Brain Res., 2005, 1035, 139–153 CrossRef CAS PubMed.
- J. H. Sampson, G. Archer, C. Pedain, E. Wembacher-Schröder, M. Westphal, S. Kunwar, M. A. Vogelbaum, A. Coan, J. E. Herndon, R. Raghavan, M. L. Brady, D. D. A. Reardon, A. H. Friedman, H. S. Friedman, M. I. Rodríguez-Ponce, S. M. Chang, S. Mittermeyer, D. Croteau, R. K. Puri, J. M. D. Markert, M. Prados, T. Chen, A. Mamelak, T. Cloughesy, J. Yu, K. Lillehei, J. Piepmeier, E. Pan, F. Vrionis, H. L. Moffitt, J. Olson, J. Chandler, N. Paleologos, R. W. Byrne, M. Lesniak, J. D. Weingart, P. Black, T. Mikkelsen, J. Uhm, R. Bucholz, L. Abrey, T. H. Schwartz, J. Bruce, A. Asher, S. Tatter, G. Barnett, A. E. Chiocca, J. B. Delashaw, K. Judy, S. Patel, B. Frankel, F. Lang, P. New, K. Fink, R. L. Jensen, M. Shaffrey, L. Taylor, W. Boling, B. Badie, A. Guha, V. Mehta, M. Hamilton, D. D. Eisenstat, F. Pirouzmand, D. Macdonald, R. Del Maestro, D. Fourney, M. Mehdorn, R. Goldbrunner, G. Schackert, A. Unterberg, Z. Ram, Z. Cohen, Z. Rappaport, J. J. Mooij, J. G. Wolbers, P. Warnke and V. Papanastassiou, J. Neurosurg., 2010, 113, 301–309 Search PubMed.
- A. J. Sawyer, J. K. Saucier-Sawyer, C. J. Booth, J. Liu, T. Patel, J. M. Piepmeier and W. M. Saltzman, Drug Delivery Transl. Res., 2011, 1, 34–42 CrossRef CAS PubMed.
- J. Zhou, T. R. Patel, R. W. Sirianni, G. Strohbehn, M. Q. Zheng, N. Duong, T. Schafbauer, A. J. Huttner, Y. Huang, R. E. Carson, Y. Zhang, D. J. Sullivan, J. M. Piepmeier and W. M. Saltzman, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 11751–11756 CrossRef CAS PubMed.
- G. M. Bernal, M. J. LaRiviere, N. Mansour, P. Pytel, K. E. Cahill, D. J. Voce, S. Kang, R. Spretz, U. Welp, S. E. Noriega, L. Nuñez, G. Larsen, R. R. Weichselbaum and B. Yamini, Nanomedicine, 2014, 10, 149–157 CrossRef CAS PubMed.
- A. Arshad, B. Yang, A. S. Bienemann, N. U. Barua, M. J. Wyatt, M. Woolley, D. E. Johnson, K. J. Edler and S. S. Gill, PLoS One, 2015, 10, e0132266 CrossRef.
- S. K. Hobbs, W. L. Monsky, F. Yuan, W. G. Roberts, L. Griffith, V. P. Torchilin and R. K. Jain, Proc. Natl. Acad. Sci. U. S. A., 1998, 95, 4607–4612 CrossRef CAS PubMed.
- A. Carreau, B. El Hafny-Rahbi, A. Matejuk, C. Grillon, C. Kieda, B. El Hafny-Rahbi, A. Matejuk, C. Grillon and C. Kieda, J. Cell. Mol. Med., 2011, 15, 1239–1253 CrossRef CAS.
- A. M. Shannon, D. J. Bouchier-Hayes, C. M. Condron and D. Toomey, Cancer Treat. Rev., 2003, 29, 297–307 CrossRef CAS.
- S. M. Evans, K. W. Jenkins, W. T. Jenkins, T. Dilling, K. D. Judy, A. Schrlau, A. Judkins, S. M. Hahn and C. J. Koch, Radiat. Res., 2008, 170, 677–690 CrossRef CAS PubMed.
- S. M. Evans, K. D. Judy, I. Dunphy, W. T. Jenkins, W. T. Hwang, P. T. Nelson, R. A. Lustig, K. Jenkins, D. P. Magarelli, S. M. Hahn, R. A. Collins, S. Grady and C. J. Koch, Clin. Cancer Res., 2004, 10, 8177–8184 CrossRef CAS PubMed.
- S. Seidel, B. K. Garvalov, V. Wirta, L. Von Stechow, A. Schänzer, K. Meletis, M. Wolter, D. Sommerlad, A. T. Henze, M. Nistér, G. Reifenberger, J. Lundeberg, J. Frisén and T. Acker, Brain, 2010, 133, 983–995 CrossRef PubMed.
- J. Overgaard and M. R. Horsman, Semin. Radiat. Oncol., 1996, 6, 10–21 CrossRef PubMed.
- A. J. Majmundar, W. J. Wong and M. C. Simon, Mol. Cell, 2010, 40, 294–309 CrossRef CAS.
- D. Zagzag, Y. Lukyanov, L. Lan, M. A. Ali, M. Esencay, O. Mendez, H. Yee, E. B. Voura and E. W. Newcomb, Lab. Invest., 2006, 86, 1221–1232 CrossRef CAS.
- J. Kim, I. Tchernyshyov, G. L. Semenza and C. V. Dang, Cell Metab., 2006, 3, 177–185 CrossRef.
- O. Méndez, J. Zavadil, M. Esencay, Y. Lukyanov, D. Santovasi, S. C. Wang, E. W. Newcomb and D. Zagzag, Mol. Cancer, 2010, 9, 133 CrossRef PubMed.
- K. M. Comerford, T. J. Wallace, J. Karhausen, N. A. Louis, M. C. Montalto and S. P. Colgan, Cancer Res., 2002, 62, 3387–3394 CAS.
- J. M. Heddleston, Z. Li, R. E. McLendon, A. B. Hjelmeland and J. N. Rich, Cell Cycle, 2009, 8, 3274–3284 CrossRef CAS.
- L. H. Gray, A. D. Conger, M. Ebert, S. Hornsey and O. C. Scott, Br. J. Radiol., 1953, 26, 638–648 CrossRef CAS PubMed.
- M. Höckel and P. Vaupel, J. Natl. Cancer Inst., 2001, 93, 266–276 CrossRef.
- D. R. Grimes and M. Partridge, Biomed. Phys. Eng. Express, 2015, 1, 045209 CrossRef PubMed.
- P. Vaupel, Semin. Radiat. Oncol., 2004, 14, 198–206 CrossRef PubMed.
- A. L. Thiepold, S. Luger, M. Wagner, N. Filmann, M. W. Ronellenfitsch, P. N. Harter, A. K. Braczynski, S. Dützmann, E. Hattingen, J. P. Steinbach, C. Senft, J. Rieger and O. Bähr, Oncotarget, 2015, 6, 14537–14544 CrossRef PubMed.
- L. Huang, W. Boling and J. Zhang, Med. Gas Res., 2018, 8, 24–28 CrossRef CAS PubMed.
- K. Ogawa, K. Kohshi, S. Ishiuchi, M. Matsushita, N. Yoshimi and S. Murayama, Int. J. Clin. Oncol., 2013, 18, 364–370 CrossRef CAS PubMed.
- M. Gholipourmalekabadi, S. Zhao, B. S. Harrison, M. Mozafari and A. M. Seifalian, Trends Biotechnol., 2016, 34, 1010–1021 CrossRef CAS PubMed.
- A. Mozzarelli, L. Ronda, S. Faggiano, S. Bettati and S. Bruno, Blood Transfus., 2010, 8, s59 Search PubMed.
- N. G. A. Willemen, S. Hassan, M. Gurian, J. Li, I. E. Allijn, S. R. Shin and J. Leijten, Trends Biotechnol., 2021, 1–16 Search PubMed.
- Z. Fan, Z. Xu, H. Niu, N. Gao, Y. Guan, C. Li, Y. Dang, X. Cui, X. L. Liu, Y. Duan, H. Li, X. Zhou, P. H. Lin, J. Ma and J. Guan, Sci. Rep., 2018, 8, 1371 CrossRef.
- P. A. Shiekh, A. Singh and A. Kumar, Biomaterials, 2020, 249, 120020 CrossRef CAS PubMed.
- E. Pedraza, M. M. Coronel, C. A. Fraker, C. Ricordi and C. L. Stabler, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 4245–4250 CrossRef CAS PubMed.
- C. L. Ward, B. T. Corona, J. J. Yoo, B. S. Harrison and G. J. Christ, PLoS One, 2013, 8, e72485 CrossRef CAS PubMed.
- E. F. Panarin, K. K. Kalninsh and D. V. Pestov, Eur. Polym. J., 2001, 37, 375–379 CrossRef CAS.
- A. Northup and D. Cassidy, J. Hazard. Mater., 2008, 152, 1164–1170 CrossRef CAS PubMed.
- H. Wang, Y. Zhao, T. Li, Z. Chen, Y. Wang and C. Qin, Chem. Eng. J., 2016, 303, 450–457 CrossRef CAS.
- T. Schmidtke, D. White and C. Woolard, J. Hazard. Mater., 1999, 64, 157–165 CrossRef CAS.
- C. J. Feeney, M. V. Frantseva, P. L. Carlen, P. S. Pennefather, N. Shulyakova, C. Shniffer and L. R. Mills, Brain Res., 2008, 1198, 1–15 CrossRef CAS PubMed.
- L. Montazeri, S. Hojjati-Emami, S. Bonakdar, Y. Tahamtani, E. Hajizadeh-Saffar, M. Noori-Keshtkar, M. Najar-Asl, M. K. Ashtiani and H. Baharvand, Biomaterials, 2016, 89, 157–165 CrossRef CAS PubMed.
- S. I. H. Abdi, J. Y. Choi, H. C. Lau and J. O. Lim, Tissue Eng. Regen. Med., 2013, 10, 131–138 CrossRef CAS.
- H. Newland, D. Eigel, A. E. Rosser, C. Werner and B. Newland, Biomater. Sci., 2018, 6, 2571–2577 RSC.
- S. M. Ng, J. Y. Choi, H. S. Han, J. S. Huh and J. O. Lim, Int. J. Pharm., 2010, 384, 120–127 CrossRef CAS PubMed.
- Z. Lu, J. Y. Gao, C. Fang, Y. Zhou, X. Li and G. Han, Adv. Sci., 2020, 7, 2001123 Search PubMed.
- J. Wei, J. Li, D. Sun, Q. Li, J. Ma, X. Chen, X. Zhu and N. Zheng, Adv. Funct. Mater., 2018, 28, 1706310 CrossRef.
- M. Ding, Z. Miao, F. Zhang, J. Liu, X. Shuai, Z. Zha and Z. Cao, Biomater. Sci., 2020, 8, 4157–4165 RSC.
- A. Sahu, K. Min, J. Jeon, H. S. Yang and G. Tae, J. Controlled Release, 2020, 326, 442–454 CrossRef CAS.
- T. Lin, X. Zhao, S. Zhao, H. Yu, W. Cao, W. Chen, H. Wei and H. Guo, Theranostics, 2018, 8, 990–1004 CrossRef CAS PubMed.
- P. K. Chandra, C. L. Ross, L. C. Smith, S. S. Jeong, J. Kim, J. J. Yoo and B. S. Harrison, Wound Repair Regen., 2015, 23, 830–841 CrossRef.
- H. Steg, A. T. Buizer, W. Woudstra, A. G. Veldhuizen, S. K. Bulstra, D. W. Grijpma and R. Kuijer, Polym. Adv. Technol., 2017, 28, 1252–1257 CrossRef CAS.
- S. I. H. Abdi, S. M. Ng and J. O. Lim, Int. J. Pharm., 2011, 409, 203–205 CrossRef CAS PubMed.
- R. R. Mallepally, C. C. Parrish, M. A. M. Mc Hugh and K. R. Ward, Int. J. Pharm., 2014, 475, 130–137 CrossRef CAS PubMed.
- Z. Li, X. Guo and J. Guan, Biomaterials, 2012, 33, 5914–5923 CrossRef CAS.
- B. S. Harrison, D. Eberli, S. J. Lee, A. Atala and J. J. Yoo, Biomaterials, 2007, 28, 4628–4634 CrossRef CAS PubMed.
- L. Daneshmandi and C. T. Laurencin, J. Biomed. Mater. Res., Part A, 2020, 108, 1045–1057 CrossRef CAS PubMed.
- N. Alemdar, J. Leijten, G. Camci-Unal, J. Hjortnaes, J. Ribas, A. Paul, P. Mostafalu, A. K. Gaharwar, Y. Qiu, S. Sonkusale, R. Liao and A. Khademhosseini, ACS Biomater. Sci. Eng., 2017, 3, 1964–1971 CrossRef CAS.
- S. Park and K. M. Park, Biomaterials, 2018, 182, 234–244 CrossRef CAS PubMed.
- J. Il Kang, K. M. Park and K. D. Park, J. Ind. Eng. Chem., 2019, 69, 397–404 CrossRef.
- P. A. Shiekh, A. Singh and A. Kumar, ACS Appl. Mater. Interfaces, 2018, 10, 18458–18469 CrossRef CAS.
- D. Eigel, C. Werner and B. Newland, Neurochem. Int., 2021, 147, 105012 CrossRef CAS.
- M. López-Lázaro, Cancer Lett., 2007, 252, 1–8 CrossRef PubMed.
- P. Prasad, C. R. Gordijo, A. Z. Abbasi, A. Maeda, A. Ip, A. M. Rauth, R. S. Dacosta and X. Y. Wu, ACS Nano, 2014, 8, 3202–3212 CrossRef CAS.
- W. T. Chiu, S. C. Shen, J. M. Chow, C. W. Lin, L. T. Shia and Y. C. Chen, Neurobiol. Dis., 2010, 37, 118–129 CrossRef CAS PubMed.
- F. Li, H. Wang, C. Huang, J. Lin, G. Zhu, R. Hu and H. Feng, Free Radic. Res., 2011, 45, 1154–1161 CrossRef CAS PubMed.
- D. G. Anderson, C. A. Tweedie, N. Hossain, S. M. Navarro, D. M. Brey, K. J. Van Vliet, R. Langer and J. A. Burdick, Adv. Mater., 2006, 18, 2614–2618 CrossRef CAS.
- K. A. Lee, H. Zhang, D. Z. Qian, S. Rey, J. O. Liu and G. L. Semenza, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 17910–17915 CrossRef CAS PubMed.
- B. Newland, C. Varricchio, Y. Körner, F. Hoppe, C. Taplan, H. Newland, D. Eigel, G. Tornillo, D. Pette, A. Brancale, P. B. Welzel, F. P. Seib and C. Werner, Carbohydr. Polym., 2020, 245, 116504 CrossRef CAS PubMed.
- J. B. Wolinsky, Y. L. Colson and M. W. Grinstaff, J. Controlled Release, 2012, 159, 14–26 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2021 |