
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Cross-validation of ELISA and a portable surface plasmon resonance instrument for IgG antibody serology with SARS-CoV-2 positive individuals†
Abdelhadi
Djaileb‡
ab,
Maryam
Hojjat Jodaylami‡
c,
Julien
Coutu
c,
Pierre
Ricard
c,
Mathieu
Lamarre
d,
Léa
Rochet
a,
Stella
Cellier-Goetghebeur
a,
Devin
Macaulay
d,
Benjamin
Charron
c,
Étienne
Lavallée
a,
Vincent
Thibault
c,
Keisean
Stevenson
 c,
Simon
Forest
c,
Ludovic S.
Live
b,
Nanouk
Abonnenc
e,
Anthony
Guedon
e,
Patrik
Quessy
e,
Jean-François
Lemay
e,
Omar
Farnós
f,
Amine
Kamen
f,
Matthew
Stuible
g,
Christian
Gervais
g,
Yves
Durocher
g,
François
Cholette
hi,
Christine
Mesa
h,
John
Kim
h,
Marie-Pierre
Cayer
j,
Marie-Joëlle
de Grandmont
j,
Danny
Brouard
j,
Sylvie
Trottier
k,
Denis
Boudreau
d,
Joelle N.
Pelletier
*a and
Jean-Francois
Masson
*c
c,
Simon
Forest
c,
Ludovic S.
Live
b,
Nanouk
Abonnenc
e,
Anthony
Guedon
e,
Patrik
Quessy
e,
Jean-François
Lemay
e,
Omar
Farnós
f,
Amine
Kamen
f,
Matthew
Stuible
g,
Christian
Gervais
g,
Yves
Durocher
g,
François
Cholette
hi,
Christine
Mesa
h,
John
Kim
h,
Marie-Pierre
Cayer
j,
Marie-Joëlle
de Grandmont
j,
Danny
Brouard
j,
Sylvie
Trottier
k,
Denis
Boudreau
d,
Joelle N.
Pelletier
*a and
Jean-Francois
Masson
*c
aDepartment of Chemistry, Department of Biochemistry and PROTEO, The Québec Network for Research on Protein Function, Engineering and Applications, Université de Montréal, CP 6128 Succ. Centre-Ville, Montreal, Québec H3C 3J7, Canada. E-mail: joelle.pelletier@umontreal.ca; Tel: +1-514-343-2124
bAffinité Instruments, 1250 rue Guy, Suite 600, Montréal, Québec H3H 2L3, Canada
cDepartment of Chemistry, Quebec Centre for Advanced Materials (QCAM), Regroupement Québécois sur les Matériaux de Pointe (RQMP), and Centre interdisciplinaire de recherche sur le cerveau et l'apprentissage (CIRCA), Université de Montréal, CP 6128 Succ. Centre-Ville, Montreal, Québec H3C 3J7, Canada. E-mail: jf.masson@umontreal.ca; Tel: +1-514-343-7342
dDepartment of Chemistry and Centre for Optics, Photonics and Lasers (COPL), Université Laval, 1045, av. de la Médecine, Québec City, Québec G1V 0A6, Canada
eCNETE and PROTEO, The Québec Network for Research on Protein Function, Engineering and Applications, Cégep de Shawinigan, 2263 Avenue du Collège, Shawinigan, Québec G9N 6 V8, Canada
fDepartment of Bioengineering, McGill University McConnell Engineering Building, 3480 University Street, Montreal, Québec H3A 0E9, Canada
gMammalian Cell Expression, Human Health Therapeutics Research Centre, National Research Council Canada, Montréal, Québec, Canada
hNational Laboratory for HIV Reference Services, National Microbiology Laboratory at the JC Wilt Infectious Diseases Research Centre, Public Health Agency of Canada, Winnipeg, Canada
iDepartment of Medical Microbiology and Infectious Diseases, University of Manitoba, Winnipeg, Canada
jHéma-Québec, Affaires médicales et innovation, 1070, avenue des Sciences-de-la-Vie, Québec City, G1V 5C3, Québec, Canada
kCentre de recherche du Centre hospitalier universitaire de Québec and Département de microbiologie-infectiologie et d'immunologie, Université Laval 2705, boulevard Laurier, Québec City, Québec, Canada G1V 4G2
First published on 1st July 2021
Abstract
We report on the development of surface plasmon resonance (SPR) sensors and matching ELISAs for the detection of nucleocapsid and spike antibodies specific against the novel coronavirus 2019 (SARS-CoV-2) in human serum, plasma and dried blood spots (DBS). When exposed to SARS-CoV-2 or a vaccine against SARS-CoV-2, the immune system responds by expressing antibodies at levels that can be detected and monitored to identify the fraction of the population potentially immunized against SARS-CoV-2 and support efforts to deploy a vaccine strategically. A SPR sensor coated with a peptide monolayer and functionalized with various sources of SARS-CoV-2 recombinant proteins expressed in different cell lines detected human anti-SARS-CoV-2 IgG antibodies in clinical samples. Nucleocapsid expressed in different cell lines did not significantly change the sensitivity of the assays, whereas the use of a CHO cell line to express spike ectodomain led to excellent performance. This bioassay was performed on a portable SPR instrument capable of measuring 4 biological samples within 30 minutes of sample/sensor contact and the chip could be regenerated at least 9 times. Multi-site validation was then performed with in-house and commercial ELISA, which revealed excellent cross-correlations with Pearson's coefficients exceeding 0.85 in all cases, for measurements in DBS and plasma. This strategy paves the way to point-of-care and rapid testing for antibodies in the context of viral infection and vaccine efficacy monitoring.
Introduction
In the event of a viral outbreak, it is of the utmost importance to rapidly test populations that are actively infectious, thereby offering the capacity to limit widespread contagion. Identification of individuals who are actively infected with SARS-CoV-2 mainly relies on real-time reverse transcription polymerase chain reaction (rRT-PCR) amplification of the viral genetic material collected in nasopharyngeal swabs.1,2 These assays show high sensitivity and can be highly specific. Due to high workload and to reagent shortages during the epidemic stage, PCR tests are mainly performed on individuals displaying COVID-19-related symptoms. Although hard numbers are only beginning to come to light,3 a significant fraction of infected individuals remain asymptomatic.4 Asymptomatic, contagious individuals often go undetected and are thought to contribute to the spread of the disease. To address this global pandemic, significant efforts are being rapidly deployed to adapt diagnostic tests to detect antibodies and provide a better indication of the spread of the disease.The immune system produces antibodies to SARS-CoV-2 within days to a few weeks following viral infection.5 Antibodies are expected to remain at a high level for months following infection, as previously shown following the 2003 outbreak of SARS-CoV-16,7 which has also been reported in sera and saliva of COVID-19-positive individuals.8 The immune reaction to coronaviruses generally provides acquired immunity via neutralizing antibodies9 in the event of a second exposure to the virus and also provides the basis for vaccine development. Vaccine development, clinical trials and immunity studies require assessing antibody titers or concentrations in animal and human subjects. As such, serological antibody testing is essential to assess the fraction of the population that is immune to a virus10 following infection or vaccination. On the longer term, the persistence of immunity to SARS-CoV-2 infections may need to be periodically assessed to ensure public health and prevent or monitor the resurgence of the virus.
Antibody detection is typically performed using serology immunoassays (IAs), with automated chemiluminescent IA (CLIA) and ELISA, and rapid lateral flow IA (LFIA) being the most prominent. In-house or commercially available diagnostic tests have been rapidly developed for SARS-CoV-2 antibodies (Table S1†).11–15 These IAs typically detect the immunoglobulin M (IgM) and immunoglobulin G (IgG) produced in response to SARS-CoV-2 infection or vaccination.16 While a highly valuable tool in the context of a viral epidemic, antibody tests, similarly to other tests, also have limitations such as false positives or false negatives related to technological or biological origin (for example, too early following an infection) that one should be aware of17 and the factors defining their performance.18,19
Central to the development of these tests is access to viral antigenic proteins. The SARS-CoV-2 recombinant proteins necessary for the development of the IAs assays are produced in prokaryotic or eukaryotic cell lines that are genetically modified to encode the viral proteins. The choice of production strain has an impact on capacity of the cognate human antibodies to bind to the recombinant proteins, depending on folding and glycosylation in different cell lines. In most cases, SARS-CoV-2 serology tests involve the detection of IgGs antibodies against nucleocapsid protein and the spike protein ectodomain, or its receptor binding domain (RBD). Persistence of the IgG antibodies in sera of COVID-positive individuals is more prolonged than IgM antibodies, such that IgG detection should be prioritized for immunity detection,20,21 although additional data may be needed to draw definitive conclusions. While antibodies against nucleocapsid and spike proteins are expected in sera of individuals who have been infected with SARS-CoV-2, only antibodies against spike are expected for individuals in whom immunity has been acquired by vaccination only. As such, developing tests for antibodies against nucleocapsid and spike protein are needed to gain knowledge on immunity of populations and whether this immunity was acquired following an infection or through vaccination.
Whereas ELISA offers high-throughput capacity, it requires several hour-long steps that lengthen overall assay time. Alternatively, faster and portable sensing technologies can decrease assay time and be employed at the point-of-care for infectious diseases.22 Lateral-flow assays have often been proposed to address this for IgM and IgG antibody detection and numerous are now commercially available for SARS-CoV-2 (Table S2†), but they can suffer from reliability issues and they are at best semi-quantitative. As such, a series of sensors will be needed to provide reliable and quantitative data on antibodies found in clinical samples of cohorts of individuals, to enable public health authorities to assess the evolution of the pandemic as well as vaccination efficacy.
Various platforms have been proposed for detection of SARS-CoV-2-related genetic material or viral load and for protein/antibody sensing,23–25,26 including nanophotonics,27 magnetic28 and electrochemical29 sensors. Among them, surface plasmon resonance (SPR) sensing is a label-free sensing technique30 that is particularly sensitive for large biomolecules such as antibodies. SPR sensing has been reported for the detection of antibodies to the first SARS-CoV,31 albeit in phosphate-buffered saline solution (PBS). Since then, SPR sensors have been reported to work in crude biofluids,32 illustrating their applicability potential for the direct detection of antibodies in clinical samples.33 Furthermore, portable SPR platforms have been reported and field-deployed.34
SPR sensing is thus well suited for quantitative analysis of SARS-CoV-2 protein–protein35,36 or protein–antibody37,38 interactions, detecting SARS-CoV-2 RNA39,40 or sensing antibodies associated to SARS-CoV-2.41,42 A recent study showed that the use of SPR imaging can be used to detect antibody isotypes in sera of clinical patients,43 paving the way for the use of SPR in clinical investigations. That initial study focused on the detection of antibodies only for the spike protein on an instrument confined to a centralized laboratory. A complete investigation of the clinical applicability of portable SPR for antibody detection for the nucleocapsid and spike proteins in sera of individuals and correlation to ELISA is necessary. In addition, the use of SPR has seldom been demonstrated with dried blood spots (DBS),44 and its use in the current context could be a game-changer due to the ease of collecting and shipping DBS. Here, we report the cross-validation of ELISA and SPR sensors for anti-nucleocapsid, anti-RBD, and anti-spike. We compare different sources of SARS-CoV-2 antigenic proteins and demonstrate their application to the detection of antibodies in COVID-19 positive individuals with control groups. We further demonstrate effective antibody detection in human plasma and DBS in addition to human serum (Scheme 1).
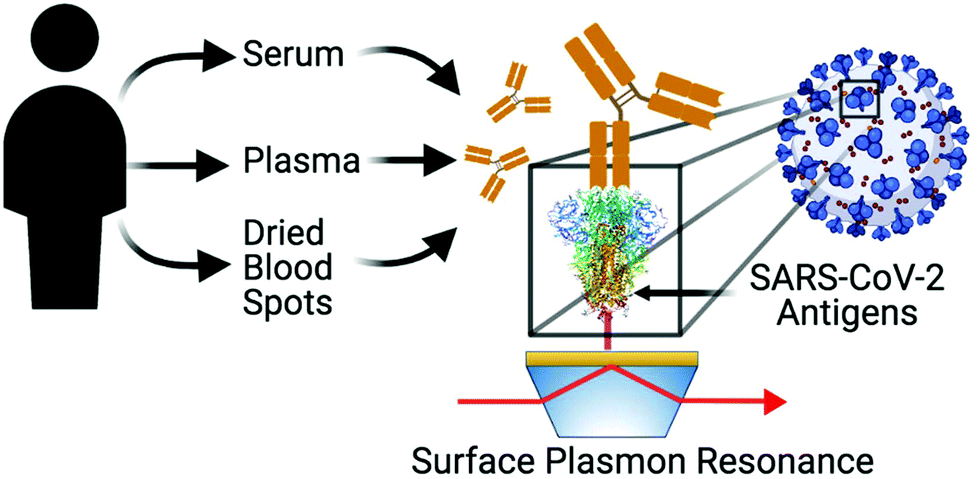 | ||
| Scheme 1 Different SPR sensors modified with a series of SARS-CoV-2 antigens (nucleocapsid, RBD or spike) are used for the detection of human antibodies from different blood products (serum, plasma or dried blood spots) collected from COVID positive or negative individuals. Scheme created with https://BioRender.com. | ||
Experimental section
Production of recombinant SARS-CoV-2 antigens
Alternatively, RBD was produced by Lemay and coworkers in Pichia pastoris SuperMan5. RBD (MN908947.3) was codon-optimized for this host and was C-terminally fused to the TEV cleavage site and to a hexa-His tag (see ESI† for detailed protocols). Briefly, the recombinant RBD protein was expressed in P. pastoris SuperMan5, harvested from the culture supernatant and purified using IMAC (Fig. S1†). The protein was aliquoted at a concentration of 0.16–0.17 mg mL−1 in storage buffer (PBS pH 7.4 with 10% glycerol) and stored at −80 °C.
ELISA assays
Presence of antibodies against SARS-CoV-2 was determined using a semiquantitative ELISA protocol adapted from the work led by Krammer and further work led by Finzi and by Bazin.47–49 Briefly, 96-well immunoassay plates were coated with the relevant SARS-CoV-2 antigen overnight, washed, blocked using 3% skim milk powder (w/v) and washed again prior to addition of serum samples (see ESI† for detailed protocols). For method development and calibration, commercial anti-SARS-CoV-2 antibodies from animal sources were diluted in commercial human serum. For assays of clinical human serum samples, samples were inactivated at 56 °C in a water bath for 1 h then diluted 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :50 or as specified and 100 μL was deposited on the assay plate. Following a 1 h incubation, appropriately diluted host-specific secondary antibody (100 μL) was added. Plates were incubated for 1 h, washed and incubated for 20 min with 100 μL of 3,3′,5,5′-tetramethylbenzidine (TMB). Color development was initiated by addition of 100 μL of 2 M HCl. Absorbance was immediately recorded at 450 nm.
:50 or as specified and 100 μL was deposited on the assay plate. Following a 1 h incubation, appropriately diluted host-specific secondary antibody (100 μL) was added. Plates were incubated for 1 h, washed and incubated for 20 min with 100 μL of 3,3′,5,5′-tetramethylbenzidine (TMB). Color development was initiated by addition of 100 μL of 2 M HCl. Absorbance was immediately recorded at 450 nm.
SPR measurements
A portable SPR instrument50 (Affinité Instruments, Canada) and a SPR surface modified with a monolayer of 3-mercaptopropionic-Leu-His-Asp-Leu-His-Asp-COOH51 (3-MPA-LHDLHD-COOH, AffiCoat, Affinité Instruments, Canada) were employed in the construction of the SPR sensor (Fig. S1†). After stabilization of the SPR signal in water, the surface was modified with a 1:1 aqueous solution of 100 mM N-hydroxysuccinimide (NHS) and 400 mM N-ethyl-N′-(3-dimethylaminopropyl)carbodiimide hydrochloride (EDC) for 2 min, rinsed for 20 seconds with the immobilization buffer as optimized and reacted for 20 min with SARS-CoV-2 proteins (concentrations optimized, see results). The sensor was rinsed for 20 s with the immobilization buffer and passivated with 1 M ethanolamine pH 8.5 for 10 min. The sensor was then equilibrated in the running buffer composed of PBS (137 mM NaCl, 10 mM phosphate, 2.7 mM KCl, pH 7.4) supplemented with 0.1% bovine serum albumin (BSA) and 0.005% Tween 20 or human serum depending on the analysis and as specified (Fig. S2†). SARS-CoV-2 antibodies (various sources, see details in the specific sections) were diluted in the running buffer or in whole human serum from human male AB plasma (cat. no. H4522, Sigma Aldrich) and 300 μL was injected with a syringe and measured for 15 min on the SPR sensor. The SPR shift was calculated from the RU difference between the beginning and the end of the measurement. In cases where the sensors were regenerated, a 10 mM glycine pH 2.2 solution was injected for a few seconds to remove antibodies from the previously immobilized proteins and restabilized in the running buffer or human serum. Most experiments were performed with a fluidic cell allowing an instrumental triplicate measurement, while some clinical samples were analyzed with a newly designed fluidic cell with 4 independent channels allowing the measurement of up to 4 samples in a single run.
Clinical samples
Adult volunteers (equal number of males and females) were recruited by the Centre Hospitalier Universitaire de Québec – Université Laval (CHUL) in Quebec City, Canada. The study was approved by the “Comité d’éthique de la recherche du CHU de Québec – Université Laval” (registration number 2021-5241). Patients were included after written informed consent. The volunteers were 18 years or older and had received a PCR-positive diagnostic for COVID-19 between 14 and 21 days prior to serum collection. The volunteers did not show symptoms of fever or dyspnea for at least 48 h prior to sampling, with little or no cough. Negative controls were collected from individuals having never received a COVID-positive test. Intravenous blood samples of 30 mL were collected and anonymized. Blood was collected in 6 mL tubes (BD Vacutainer 367815), the tubes were then gently inverted, held at room temperature for 15–30 min and spun at 1600G for 15 min. Serum (1 mL aliquots) was transferred in cryovials (Sarstedt Inc., product 72.694.006) and frozen in an upright position at −20 °C until a batch was sent to the Montreal labs on dry ice. The serum samples were then stored at −80 °C until use.For ELISA assays, heat inactivated sera were diluted 1:50 for use, unless otherwise specified. For SPR validation assays, no heat treatment was applied to the sera, which were diluted 1:5 with the running buffer before analysis for the detection of IgGs. The fluidic cell with 4 independent channels was used to collect this data and the SPR instrument was placed in a laminar flow cabinet in a biosafety Level 2 (BSL2) laboratory.
A panel of 20 contrived dried blood spots (DBS)52 and matching plasma were collected from different donors at Mount Sinai Hospital (Toronto, Canada) and at the National Microbiology Laboratory (Winnipeg, Canada). Ethical approval was obtained from the Health Canada and Public Health Agency of Canada Research Ethics Board (REB 2020-022P). Plasma samples were diluted 1:5 in running buffer for SPR analysis and 1:20 for the in-house ELISA tests. For the DBS, four 6 mm disks were punched and resuspended in 300 μL of Dulbecco's PBS (DPBS) supplemented with 0.5% BSA and 0.05% Tween20 overnight at 4 °C with agitation (400 rpm). The DBS samples were then diluted 1:5 in the same PBS buffer supplemented with BSA and Tween20. Of those 20 DBS samples, two positive and two negative samples, as determined by a commercial SARS-CoV-2 IgG ELISA for anti-spike (EuroImmun), served to optimize the dilution conditions with the anti-spike SPR assay (1:2.5 and 1:5 were tested), where 1:5 was found optimal. The remaining 16 samples were tested on the EuroImmun SARS-CoV-2 ELISA, the in-house ELISA and with SPR for anti-spike and anti-nucleocapsid. All clinical samples were diluted in the running buffer used for the experiments.
Results and discussion
Production of recombinant SARS-CoV-2 antigens and development of ELISA assays
Although early experiments were undertaken with commercial sources of SARS-CoV-2 spike ectodomain, S1 and RBD domains of spike protein and nucleocapsid protein, issues with international transportation early in the pandemic prompted us to secure locally-sourced recombinant SARS-CoV-2 antigens. The spike protein ectodomain, its RBD and the nucleocapsid (rN) were expressed in several hosts. All were hexa-histidine tagged for ease of purification and some included further tags for purification or for identification. Detailed production and purification protocols are provided in the ESI.†Antigen-down colorimetric ELISA assays were developed according to standard methods adapted for these antigens.47,49,53 Commercial sources of hexa-histidine-tagged rN antigen expressed in E. coli (MyBiosource, cat. no. MBS569934; SinoBiological, cat. no. 40588-V08B) served for initial method set-up but were rapidly substituted by the locally-sourced hexa-His-tagged rN antigen expressed in E. coli (Lemay and coworkers). Comparison of the two sources of hexa-His-tagged rN in an ELISA assay using commercial murine anti-rN diluted in human serum as a primary antibody and a murine HRP-conjugated secondary antibody gave indistinguishable results (Fig. S3A†), confirming the validity of the locally-sourced hexa-histidine-tagged rN antigen. Substitution with a different lot of the same murine anti-rN antibody yielded indistinguishable results, confirming robustness of the method (Fig. S3B†). The sensitivity of the ELISA assay was determined under the same conditions by performing a serial dilution of the murine anti-rN antibody; the limit of detection was calculated at 0.016 μg mL−1 using three standard deviations above the mean of blank measurements (Fig. S3C†). Hexa-histidine-tagged RBD expressed in HEK cells (Kamen and coworkers) was successful in ELISA assays. However, the ELISA signal was significantly lower than that obtained for rN, with a maximum absorbance (OD450) near 0.4 (Fig. S3D†). Expression of hexa-histidine-tagged RBD was also accomplished by Lemay in Pichia pastoris SuperMan5 strain in a bid to obtain faster production. Despite the modified glycosylation of that strain, it appears that RBD was not sufficiently humanized to afford reactivity in ELISA assays (not shown). Hexa-histidine-tagged Spike ectodomain expressed in CHO cells by Durocher was also successful in ELISA assays (Fig. 1). The sensitivity of the ELISA assay for the spike ectodomain was determined by performing a serial dilution of the rabbit anti-S1/RBD antibody; the limit of detection was calculated at 0.0125 μg mL−1 (Fig. S3E†), in the same range as that obtained for detection of murine anti-rN antibody (Fig. S3B†). However, it is important to note that the concentrations reported here are for a surrogate model using antibodies elicited in animals and detection limits or concentrations should be interpreted as indicative of a relative performance only.
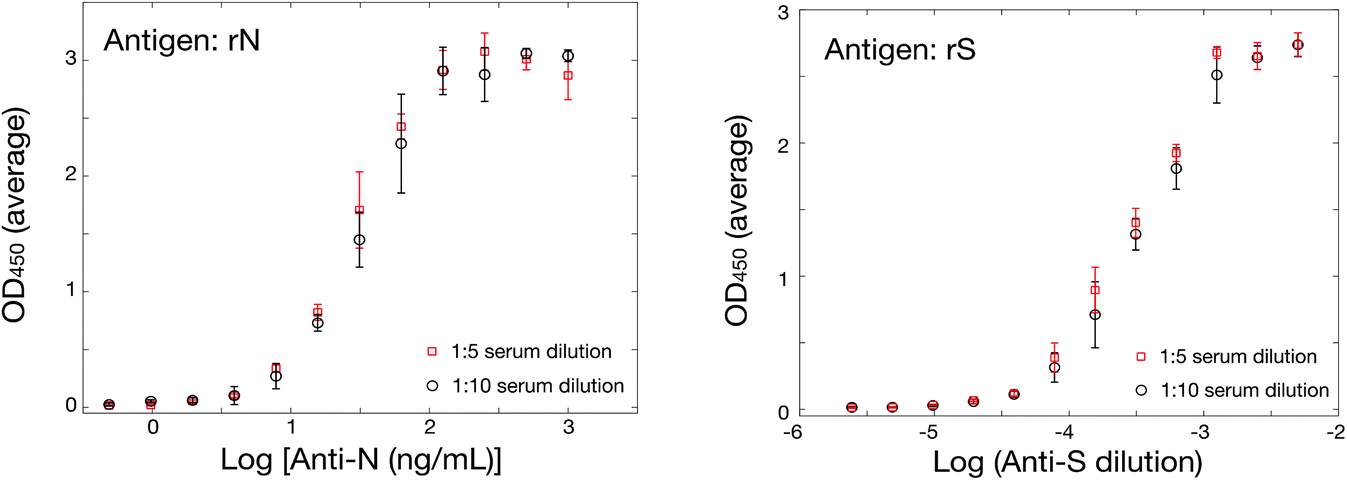 | ||
| Fig. 1 Validation of ELISA assays for the SARS-CoV-2 antigens with surrogate animal antibodies. (Left) ELISA using hexa-histidine-tagged rN antigen at different serum dilutions, 1:5 (red) and 1:10 (black). (Bottom) ELISA using hexa-histidine-tagged spike ectodomain expressed in CHO cells. | ||
SPR assay optimization using antibodies elicited in animals – nucleocapsid
The SPR instrument consists in a 4-channel wavelength interrogation system extensively described previously.50 Optimization of the SPR parameters was initially performed to maximize the response for anti-rN elicited in animal models (details in section 3 in ESI, Fig. S2, Tables S3 and S4†). As with ELISA, we compared two commercial sources of hexa-histidine-tagged rN antigen expressed in E. coli and our locally-sourced nucleocapsid proteins. All proteins were immobilized with the optimized conditions in acetate pH 4.5 buffer conditions and at 10 μg mL−1. The SinoBiological and both rN produced locally yielded immobilizations on the order of 1300 RU, higher than for the MyBiosource rN. Murine anti-rN antibody detection (10 μg mL−1) was lower for both commercial sources of rN proteins at approximately 400 RU, while the response for our two locally-produced rN (in E. coli or in CHO cells) was on the order of 800 to 950 RU in otherwise identical conditions. The reasons for the better performance of our locally-produced rN remains unknown. A potential explanation may be related to the cold chain management as both sources were produced within 150 km from the laboratory where the SPR experiments were performed and thus shipped and received within a maximum of 24 hours. The commercial sources were received with significantly longer delays as all required international or overseas shipping. We therefore solely used the locally expressed rN for the remaining experiments and results reported below were obtained with the hexa-histidine-tagged rN antigen expressed in E. coli, unless otherwise stated.One of the challenges for the design of antibody tests for human clinical samples is finding an appropriate source of antibodies to calibrate and optimize the sensor construction as human antibodies are not available and clinical samples difficult to obtain especially early on in a pandemic. We relied on the use of different antibodies elicited in animals as a surrogate (Table S4†). Only one source of surrogate murine antibody gave a nearly null signal, while others gave responses between 241 and 983 RU for 10 μg mL−1 anti-rN. This signal is significantly greater than the noise level of the instrument, which is on the order of 1–2 RU. The surrogate murine antibody with the highest SPR response (monoclonal, MyBiosource, MBS569903) was used for the remaining optimization experiments unless otherwise noted. Calibration was then performed in running buffer and in undiluted human serum to compare the performance of the SPR sensor in each condition (Fig. 2). While the SPR sensor performed similarly in both conditions, it was observed that the SPR signal was larger for lower antibody concentrations in running buffer than serum, but larger SPR signals were obtained at higher murine anti-rN concentrations in serum.
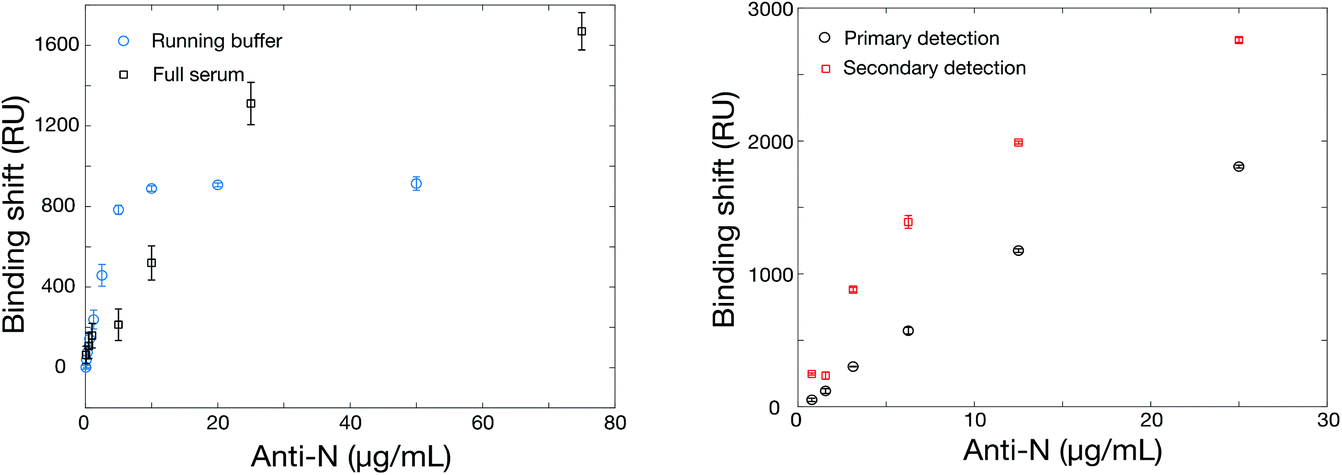 | ||
| Fig. 2 (Left) Calibration curves for murine anti-rN antibodies in PBS running buffer and in undiluted serum. (Right) Calibration in undiluted human serum of the SPR sensor with a secondary detection step using a goat anti-mouse IgG (H + L), as the surrogate antibody is a murine antibody. The rN was immobilized at 10 μg mL−1 in acetate pH 4.5 and the secondary antibody concentration was also 10 μg mL−1 dissolved in the running buffer. | ||
The increase in SPR signal for high antibody concentration in serum was also observed with rS (Fig. 4). While the phenomenon is under further investigation, some preliminary observations can be highlighted. The nonspecific adsorption or bulk refractive index effect of serum can be ruled out. All serum measurements were conducted on a surface passivated with a blank serum (serum containing no murine anti-rN antibody) prior to analysis, minimizing nonspecific adsorption. Finally, blank serum was injected into the reference channel of the SPR instrument at the same time as samples and the remaining background was subtracted from the measurement channels (Fig. S4†), confirming the enhancement of sensitivity in serum relative to PBS. We hypothesize that the enhancement results from adsorption of serum proteins on the captured antibodies, previously reported for abundant proteins such as albumin,54 increasing their mass and refractive index shift. We note that any remaining nonspecific adsorption on the SPR surface, albeit minimized, may help stabilize the surface-bound rN protein and improve binding of its cognate antibodies.
The matrix effect (refractive index, protein concentration, pH, etc.) can be significant in clinical sera and impede on the ability of SPR to perform direct detection of antibodies.55 As such, we implemented a secondary detection step with goat anti-mouse IgG (H + L), which is performed in running buffer following the detection of murine anti-rN in human serum (Fig. 2). In addition to being insensitive to the bulk RI variations of clinical sera, the secondary detection step improved the response by a factor of 2–3 times at lower concentrations and by about 50% at higher murine anti-rN concentrations. The limit of detection was calculated at 3 nM for the direct assay and at 2 nM for the secondary assay. The smaller gain by the secondary detection step at higher concentrations is likely due to steric effects. This is supported from the experiment performed with a secondary antibody with HRP (used for the ELISA test), which led to lower sensitivity (Fig. S5†). Henceforth, anti-mouse IgGs without HRP will be used for the secondary detection step in SPR sensing. The matching ELISA, of course, worked well with the HRP anti-IgG in serum diluted 1:5 and 1:10 (Fig. 1), and showed higher response than SPR with a dynamic range in the ng mL−1 compared to low μg mL−1 for SPR (Fig. 2). As a greater serum dilution is necessary with ELISA to avoid high background response, a different dilution factor was used for ELISA and SPR with clinical samples.
Finally, we also tested the use of humanized anti-rN as a surrogate antibody. However, we observed no signal in ELISA. The SPR calibration curve showed strong interaction of the humanized anti-rN (Fig. S6†), indicating that the lack of ELISA response is likely due to poor interaction of the secondary antibody with the humanized anti-rN. In summary, several sources of rN and murine anti-rN were evaluated to establish the ELISA and SPR tests for anti-rN antibodies. While both commercial and locally produced rN worked in SPR and in ELISA, we observed a stronger SPR signal for the locally produced rN. Murine anti-rN were a better surrogate to optimize the ELISA and SPR assays than a humanized anti-rN. As such, detection of murine anti-rN was achieved in diluted serum for ELISA with a dynamic range in the μg mL−1 range for both the direct and secondary detection assays with SPR in running buffer and in undiluted serum.
SPR assay optimization using antibodies elicited in animals – RBD
Contrary to rN, a small, non-glycosylated protein that is easily produced in E. coli, the spike protein ectodomain is large and heavily glycosylated rendering expression more challenging. Commercial sources were rare, prices high and supply low, at least at the beginning of the project. We therefore set out to produce spike protein ectodomain and its receptor-binding domain (RBD) in cell culture, locally. We also tested a commercial source of the S1 domain of spike protein (Beta Lifescience, cat. no. BLSN-0998P), with little success in SPR. Binding of the S1 domain led to low RUs in SPR and primary detection using rabbit anti-rS polyclonal antibody (Beta Lifescience, cat. no. BLSN-005P) gave almost no signal (Table S5†). Similar results were obtained at various pHs (acetate pH 4.5 and 5.5, PBS pH 6.5 and 7.4) and for rabbit anti-rS concentrations up to 25 μg mL−1. As a result, S1 domain was not further considered in this work.We then focused on the use of the RBD domain for the construction of the SPR sensors and ELISA tests for a rabbit anti-rS monoclonal antibody (Sino Biological, cat. no. 40150-R007). RBD expressed in HEK293SF cells and in Pichia pastoris SuperMan5 were compared. While the former led to excellent performance in SPR sensing, RBD expressed in Pichia pastoris SuperMan5 unfortunately did not lead to measurable signal in SPR or in ELISA. Immobilization of RBD expressed in HEK293SF led to SPR shifts of 1592 ± 222 RU for a concentration of 20 μg mL−1 in acetate pH 5.5 buffer. The SPR sensors provided promising outcome with responses of 1036 ± 96 RU and 1859 ± 96 RU respectively for the direct detection and secondary assay of rabbit anti-rS at a dilution of 1:125 (no concentration was provided by the commercial supplier, SinoBiological). Calibration for the rabbit anti-rS antibodies was then carried in running buffer, 10% serum and undiluted serum, with very similar performance for the direct and secondary detection in SPR (Fig. 3). In all cases, the SPR response was essentially within the error for the running buffer, diluted and undiluted serum. The dynamic range of the SPR sensor was in the 102–103 dilution range (Fig. 3), while the one for ELISA was in the 103–105 range (Fig. 1), providing assays for antibodies targeting the RBD domain of spike. Unfortunately, as the concentration of the antibody is unknown, calculation of a limit of detection was inappropriate.
 | ||
| Fig. 3 (Left) SPR calibration for the direct detection of rabbit anti-rS with RBD immobilized at 20 μg mL−1 in 10 mM acetate buffer pH 4.5 on the SPR chip and Right) secondary detection in identical conditions. | ||
SPR assay optimization using antibodies elicited in animals – recombinant spike (rS)
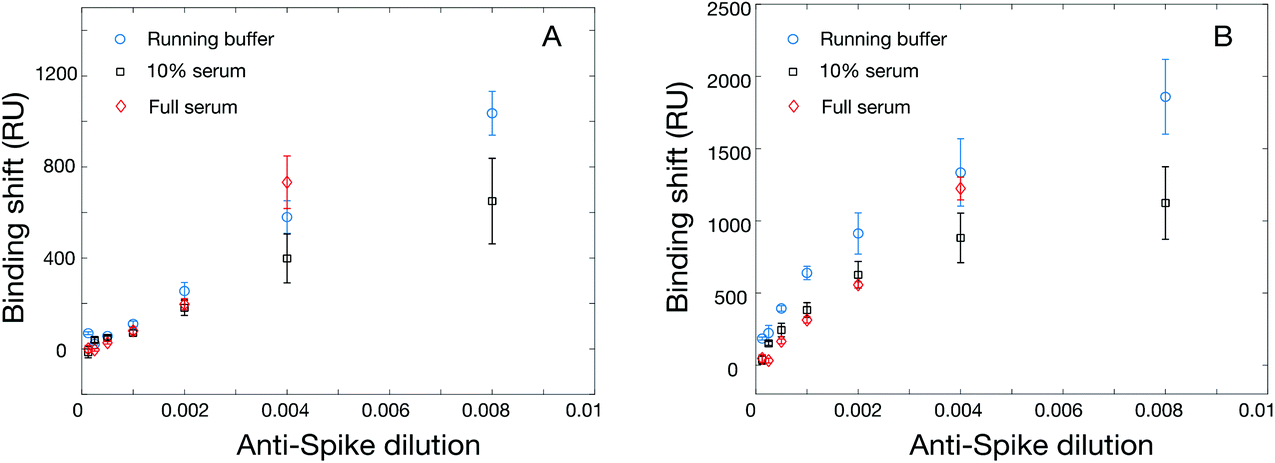
We next evaluated the use of the hexa-histidine-tagged Spike ectodomain expressed in CHO cells for the SPR assays. Two batches of rS from CHO cells were used for the initial optimization of the immobilization conditions in SPR, with similar sensitivity. The immobilization concentration was first optimized with acetate pH 5.5 buffer (Table S6 and Fig. S7†). While there was a correlation between immobilization concentration and SPR response (for the immobilization and the detection of rabbit anti-rS), the best compromise between sensitivity and reagent consumption was reached for 20 μg mL−1, which was used for the following experiments. We then optimized the regeneration of the SPR chip using glycine between concentrations. As shown in Fig. S8,† the SPR response was slightly smaller with the regeneration of the surface (about 70% sensitivity) between the measurement at each concentration, which is expected due to the accumulation of mass on the sensor without regeneration. The sensor calibrated without the regeneration was done on a single chip using an additive assay where all concentrations are successively injected on the chip. Interestingly, we were able to regenerate the sensor even after the detection of serum samples (Fig. S9†). These experiments show that the sensors can be regenerated at least 9 times before degradation of the response (denoted by a significant drift of the SPR baseline), providing higher throughput capability of the sensors for the measurement of clinical samples.Calibrations were then performed with the SPR assays for the detection of anti-rS, using the same surrogate rabbit antibody as for the RBD assay. The direct detection of rabbit anti-rS worked well in running buffer and in serum, and the detection in serum led to larger response (Fig. 4). This is might be due to the stabilization of rS in such a high concentration of human albumin, as the enhanced sensitivity in serum was not observed for this antibody with RBD. Secondary detection led to a significant improvement of the SPR response, especially for the detection in running buffer where the improvement was several folds. Comparatively, ELISA performed well in 1:5 and 1:10 serum (Fig. 1), with a 10× greater sensitivity than SPR (104 dilution for ELISA vs. 103 for SPR). Hence, the performance of the SPR and ELISA assays were demonstrated for murine anti-rN and rabbit anti-rS (both the ectodomain and the RBD domain) using these surrogate antibodies in human serum.
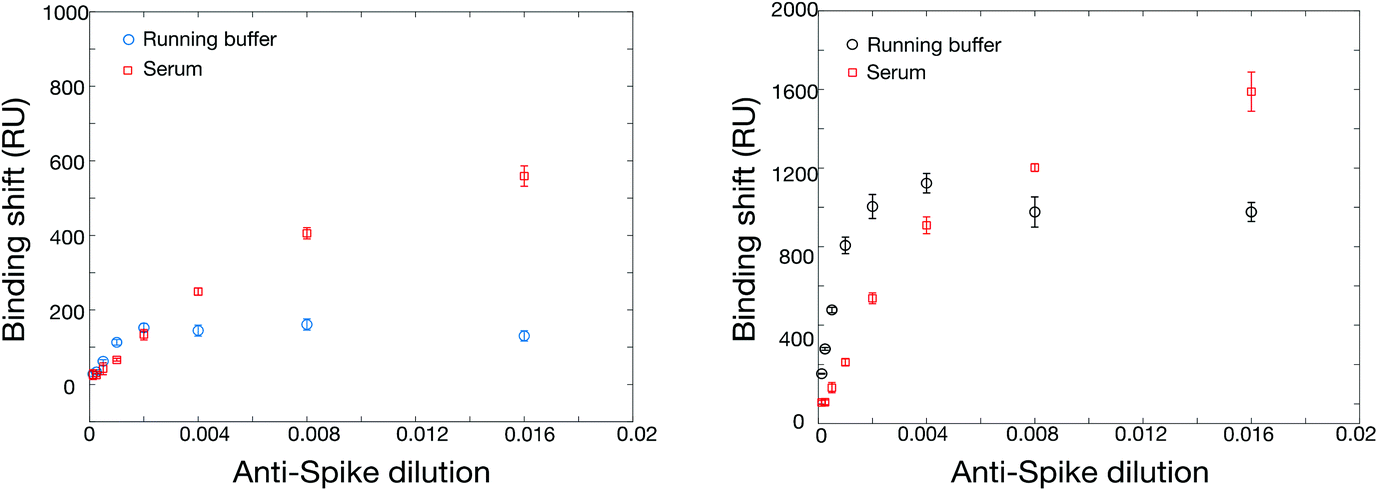 | ||
| Fig. 4 Calibration of the SPR sensor for rabbit anti-rS with direct detection (left panel, running buffer in blue circles and serum in red squares), with the secondary detection assay (right panel, running buffer in black circles and serum in red squares). Spike was immobilized at 20 μg mL−1 in 10 mM acetate buffer pH 4.5. | ||
Clinical samples – validation of the assays with human antibodies
Serum analysis from SARS-CoV-2 positive individuals
Serum samples were collected from an equal number of male and female individuals of 18 years and older in the Québec City region, Canada. Among those, three PCR-positive individuals (ID 4901, 5903 and 5907) and a negative individual (C001) were selected for the initial optimization of the assay parameters. In all cases, the response from a blank serum (without human anti-rN or human anti-rS) measured in the reference channel was subtracted from the response of the positive individuals. In the case of the clinical samples, we opted to dilute the sera as the volumes provided by the volunteers were relatively small and multiple analysis were planned with these samples. As such, measurement in undiluted sera were not possible as in the optimization steps with the murine models where ample source of sera was available.First, optimal conditions for secondary anti-human IgG antibodies were selected to increase the SPR response and to decrease the impact of the sample matrix, sera were first diluted 1:10. Using serum from individual 4901, we observed that using a goat polyclonal antibody to human IgG coupled to HRP (from Abcam) led to the highest amplification of the response between direct and secondary detection (Fig. S10†), but the AffiniPure goat anti-human IgG (H + L) was employed thereafter as the SPR response was nearly equivalent and a larger supply could be secured. This also shows that direct detection of human antibodies against spike could be detected using 1:10 serum dilution, with a signal between 200 and 500 RU, significantly larger than the background signal (mean = 13 RU, range from −40 to 110 RU). Secondary signal detection could exceed 1000 RU, providing the increased SPR response that is expected to be necessary for lower antibody concentration in individuals with a weaker immune response. Different serum dilutions were then tested to find the optimal dilution factor. It was found that the signal was relatively linear in direct detection with dilutions between 1:40 and 1:2.5, while the signal started to saturate at a dilution of 1:5 with the secondary detection (Fig. S11†). As such, a dilution of 1:5 was then used for the following experiments as it provides the largest signal in the secondary assay while using smaller volumes of the clinical samples. Error bars were also smaller with the secondary detection, which is anticipated as the background effect of refractive index and nonspecific adsorption from serum is minimal with secondary detection in a buffer. Thus, secondary detection will be used for screening purposes.
We further extended the validation of SPR sensing in serum samples with the measurement of human anti-rN, human anti-rS and human anti-RBD for a larger number of COVID-positive and negative individuals. For these experiments, human sera were collected 4 weeks post-infection from five PCR-positive adults (males and females). These samples were first analyzed with ELISA to confirm the presence of human IgG antibodies targeting rN and the ectodomain of rS (Table S7†). Four of the five positive samples showed a significantly stronger ELISA OD using rN antigen (range between 0.9 to 1.4 OD) for the PCR-positive individuals than the negative controls (range of 0.13 to 0.28 OD), and all 5 positive samples were correctly assigned in ELISA using rS antigen. As shown in Table 1, SPR sensing using rN, RBD or rS antigen has the sufficient sensitivity to detect human SARS-CoV-2 antibodies in all 5 positive clinical samples (diluted 1:5) targeting different proteins. Results from these positive sera were compared to 5 control sera from negative individuals (never tested positive for SARS-CoV-2), to ensure the response was specific. In some cases, the sera from the negative controls resulted in small negative shifts due to small refractive index mismatches. In all cases, the detection of human anti-rN, human anti-rS and human anti-RBD led to SPR responses at least one order of magnitude greater than those of the control samples, demonstrating the suitability of SPR sensing for human antibody detection in clinical samples.
:5). All values are SPR shifts are for secondary detection in running buffer and expressed in RU
| Sample ID | Anti-rN (RU) | Anti-RBD (RU) | Anti-rS (RU) | |
|---|---|---|---|---|
| Positives | 4907 | 1106 | 648 | 1370 |
| 4911 | 368 | 555 | 856 | |
| 5905 | 299 | 438 | 765 | |
| 6902 | 339 | 643 | 1604 | |
| 7001 | 421 | 1047 | 1189 | |
| Negatives | C002 | 1 | 36 | 52 |
| C005 | 10 | 96 | 110 | |
| C007 | 20 | 16 | −6 | |
| C008 | −3 | −27 | −4 | |
| C009 | −29 | −35 | −40 |
To confirm the validity of the SPR methods with a larger cohort of serum samples, the analysis of 32 SARS-CoV-2 positive and 8 SARS-CoV-2 negative control serum samples was undertaken with a point-mutated variant of the B.1.1.7 variant of concern (with the native D614 residue). ELISA response of the positive samples (OD450: 1.6 ± 0.5, n = 32) was significantly higher (p < 0.0001) than that of the controls (OD450: 0.14 ± 0.03, n = 8). Similarly, the SPR response of the positive samples (1.2 ± 0.5 kRU, n = 32) was significantly higher (p < 0.0001) than that of the controls (0.3 ± 0.2 kRU, n = 8), demonstrating the ability of SPR to reliably detect the seroconversion of SARS-CoV-2 individuals (Fig. 5).
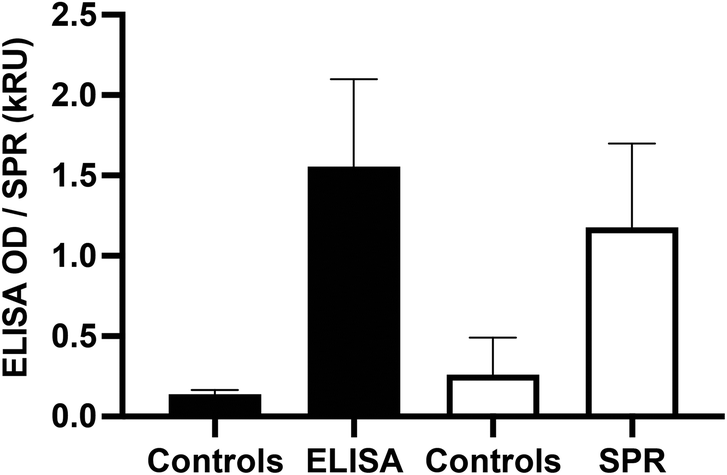 | ||
| Fig. 5 Validation of the SPR and ELISA methods with a point-mutated B.1.1.7 spike protein. The responses are expressed in OD450 for the ELISA and in kRU (1000 RU) for the SPR data respectively. | ||
Dried blood spot (DBS) panel from SARS-CoV-2 positive and negative individuals
We then moved to a different matrix to further validate the performance of SPR sensing in clinical samples. A series of DBS samples and matching plasma samples was collected from 10 negative and 10 positive individuals. The DBS were eluted with PBS with 0.5% BSA and 0.05% Tween20 at the National Microbiology Laboratories (NML) of the Public Health Agency of Canada (Winnipeg, Canada) and analyzed for IgG antibodies against the S1 domain of spike with the EuroImmun SARS-CoV-2 ELISA test. The samples were then shipped to Montreal and analyzed with the in-house ELISA and SPR assays for human anti-rS and anti-rN.The analysis of DBS was first optimized for human antibodies against rS, which is assessed more routinely than human antibodies against rN. The DBS samples were assayed at different dilution factors in the same elution buffer. Direct detection of human antibodies in DBS did not lead to conclusive results as the response from the positive and negative DBS samples was nearly equivalent, at approximately 700 RU. This is surely related to matrix effect from a mismatch of the refractive index of the DBS and running buffer, and from some nonspecific adsorption of the eluted biomaterial contained in DBS. DBS contains various proteins and cellular debris increasing the refractive index to a value above the ones of the running buffer. Dilution to 1:2.5 led to significant nonspecific response of the secondary detection step for negative controls, whereas a 1:5 dilution of the DBS gave essentially no response from the secondary antibody with the same negative DBS samples (RU slightly around or below 0). Positive DBS samples led to a response of the secondary detection step on the order of a few hundred RUs, clearly demonstrating the potential of detecting the human anti-spike antibodies in DBS with SPR (Tables S8 and S9,†Fig. 6). The shift values were calculated when equilibrium was reached for the secondary detection step. The slightly negative response of some of the controls was due to washing off nonspecifically adsorbed material from DBS samples. Larger SPR responses were obtained with secondary antibody at a concentration of 40 μg mL−1 (>300 RU for samples #1 and #3, both positive). The results from this dilution analysis were directly applied to human nucleocapsid antibodies, as the nonspecific adsorption will be identical for the same samples.
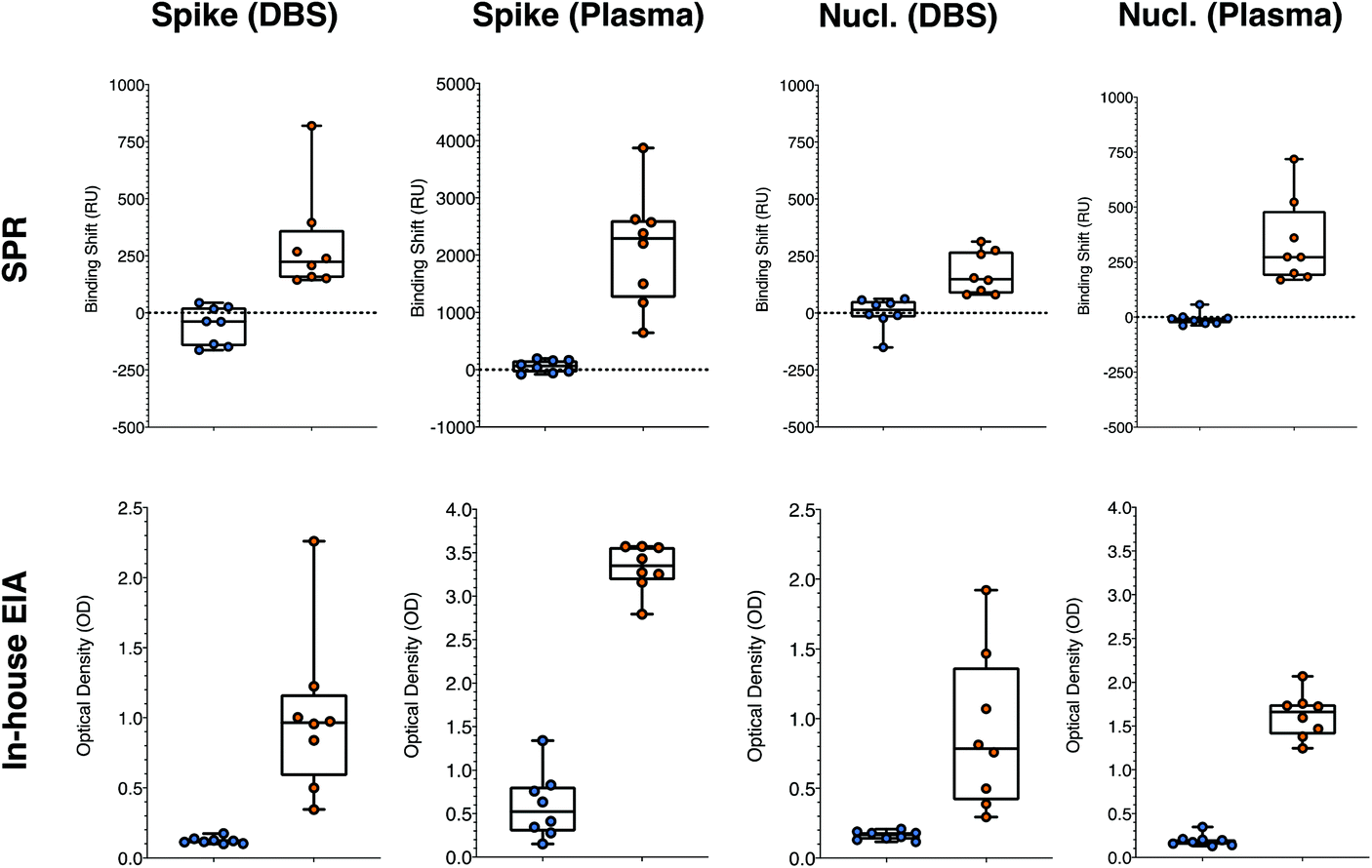 | ||
| Fig. 6 Statistical comparison of the responses obtained for the positive (right side, orange symbols) and negative (left side, blue symbols) for the SPR and in-house ELISA, for the detection of human anti-spike and anti-nucleocapsid antibodies in DBS and in plasma. All means were statistically different with p < 0.01. Both tests were performed on the same aliquots in Montreal by two independent users. SPR and ELISA were performed on 8 positive and 8 negative matching DBS and plasma samples. Nucl. = Nucleocapsid. (n = 3 for ELISA and n = 1 for SPR). | ||
The optimized SPR assays for human anti-rN and anti-rS in DBS with samples 1 to 4 reported above were then compared to ELISA on a panel of 16 samples (labeled #5 to #20 in Tables S8 and S9†), containing an equal number of SARS-CoV-2 positive and negative samples as assessed with the commercial EuroImmun ELISA platform run by the NML in Winnipeg. Applying a threshold value (RU or OD450 larger than the highest of the controls) of 100 RU and 0.2 OD for positive detection of human anti-rS in DBS, all negative and all positive samples were correctly assigned with SPR sensing and our in-house colorimetric ELISA (Table S8†). Thresholds of 75 RU and 0.2 OD applied for the detection of human anti-rN in DBS led to the correct assignation of all samples with SPR and with the in-house ELISA (Table S9†). It must be noted that the response for human anti-nucleocapsid was rather low in SPR and ELISA, such that the difference between the negative and the positive values was small (Table S9† and Fig. 6) and misassignment can be expected in larger sampling campaigns even though the sensitivity and the specificity was 100% in this data set. This is not really limiting, as human anti-spike antibody detection is more common in clinical monitoring.
Plasma panel from SARS-CoV-2 positive and negative individuals
Plasma from donors were collected and used to contrive DBS in parallel, providing matching samples for validation of the assays (Tables S8 and S9†). A secondary detection step was performed for the SPR experiments with anti-human IgG at 40 μg mL−1, as for the DBS samples. The plasma samples were diluted 1:5 in the running buffer prior to the SPR analysis of human anti-spike, as the SPR response in plasma for human anti-rS was rather large (range between 600 and 4000 RU). A larger dilution (1:10) for the SPR test in plasma for human anti-rN was necessary due to the higher background signal those conditions (up to about 200 RU), which decreased to below 60 RU at 1:10 dilution. Dilutions were greater for the in-house ELISA at 1:20 for both human anti-rS and human anti-rN. The higher background in plasma samples had no effect on the ability to correctly assign all positive and negative samples applying thresholds of 200 RU and 1.5 OD for the human anti-rS and 75 RU and 0.4 OD for human anti-rN (Tables S8 and S9† and Fig. 6).
Plasma being a more concentrated biofluid explains the relatively higher SPR response compared to DBS. In these tests, the SPR assay was performed with four 6 mm DBS punches, where the total capillary blood volume is estimated at about 65 μL. As the DBS samples were eluted in 300 μL, the DBS samples were already diluted approximately 1:5. Factoring the further dilution prior to analysis, the effective dilution of the DBS samples is about 1:25. Plasma was only diluted 1:5 or 1:10 in comparison, explaining the larger response in plasma.
The results are robust as the data presented here was collected on two instrumental platforms (SPR and ELISA), three different instruments (SPR and 2 different ELISAs), in two labs across Canada (Université de Montreal and PHAC in Winnipeg) and run by at least 4 independent users. The average response from the positive and negative samples were statistically significant with p-values of <0.01 in all cases. The response was also cross-correlated between the SPR, in-house ELISA and the commercial EuroImmun SARS-CoV-2 platforms and led to excellent collinearity of the different methods (Fig. 7). Pearson's coefficient exceeded 0.85 and were as high as 0.95 for the different collinearities measured for every cross-correlation with the various platforms, sample type (DBS and plasma), and antibodies (human anti-spike and human anti-nucleocapsid). This implies that the magnitude of the response for all three platforms was relatively proportional and quantitative.
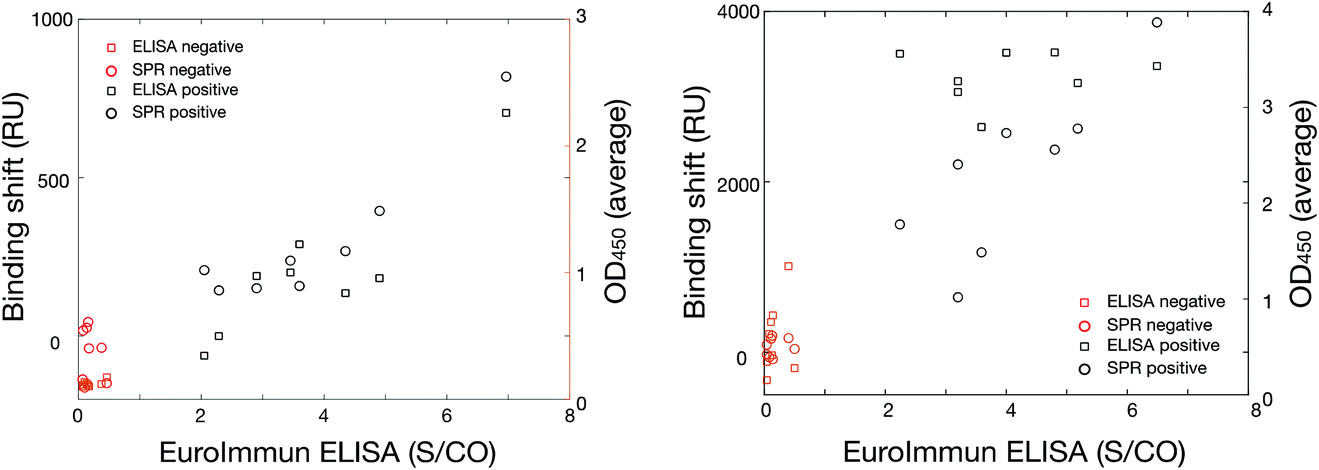 | ||
| Fig. 7 Cross-validation assays for DBS (left) and plasma (right) between the commercial EuroImmun SARS-CoV-2 anti-spike in the x-axis and SPR in the left y-axis (open circles, n = 1 per sample) and in-house ELISA (OD450, n = 3 per sample) in the right y-axis (open squares) for positive (black symbols) and negative (red symbols) samples. The Pearson's coefficients were all above 0.85, showing collinearity of the data. S/CO refers to the signal to cut-off of the ELISA measurements. | ||
These results demonstrate that SPR sensing on a portable platform performs equally well to ELISA for the detection of prior SARS-CoV-2 infections, with a detection time under 30 minutes. We therefore envision that the platform could be deployed to different locations as a consequence of instrument portability, in addition to use in centralized laboratories. Preliminary results suggest that the nucleocapsid chips can be conserved for at least one week in the freezer with excellent retention of activity towards anti-nucleocapsid (Fig. S14†), further providing evidence of the field-deployability of the SPR sensor. Detection can be carried in various blood-based products, including serum, plasma and DBS, providing versatility in the applications of the SPR sensor for SARS-CoV-2 antibody detection. The analysis of DBS is especially interesting as they can be easily collected by individuals at home, stored at room temperature (especially interesting for remote locations where a cold chain may not be accessible) and/or shipped to central laboratories using regular post. As such, the use of DBS should facilitate larger cohorts to be screened and access to individuals residing far from urban centers, providing a better picture of the epidemiologic situation in populations worldwide. The current study also constitutes one of the largest multi-center studies about the use of SPR sensors with clinical samples, further advancing the use of SPR towards clinical applications.
Conclusions
In conclusion, we demonstrate a SPR platform suited for the rapid detection of SARS-CoV-2-associated antibodies in a series of human blood products (serum, plasma and dried blood spots). The SPR test relied on the use of locally expressed SARS-CoV-2 proteins in different cell lines and the cross-validation with different in-house or commercial ELISA. For rN, it was observed that the locally produced proteins outperformed the commercial sources, likely due to a shorter transit time and better cold chain in transit between research institutes, but all sources of rN worked well. For the RBD and rS proteins, rS produced in CHO cells and RBD expressed in HEK293SF cells performed well in the assays. SPR and ELISA tests were optimized in human serum for anti-rN, anti-RBD and anti-rS, first with surrogate antibodies from animals, then with clinical samples of individuals with confirmed infections to COVID-19 (PCR positive) and with individuals with no prior infection to COVID-19. Then, cross-validation was undertaken with dried blood spots and plasma samples collected from individuals with confirmed infections to SARS-CoV-2 and a control group. All assays (SPR, in-house ELISA and a commercial ELISA kit), all sample types (dried blood spots and plasma) and proteins (spike and nucleocapsid) led to excellent discrimination of the positive and negative values (p < 0.01) and cross-correlation with Pearson's coefficients above 0.85, demonstrating the excellent performance of the various antibody screening methods. As antibody detection is needed for epidemiological surveys, to assist vaccine development and to provide data in the deployment of large-scale vaccination campaigns and follow-up studies, we demonstrate here that SPR sensing can be a significant tool in the several studies that will be undertaken. Since this detection method is generically applicable to other viral antigens or for detecting antibody cross-reactivity with antigens from variants of concern for SARS-CoV-2, the current report provides the blueprint for development of a series of antibody sensors for this virus and others, toward analysis of clinical samples. The portability of the SPR instrument could also allow deployment of the method in the field for rapid on-site measurements.Author contributions
Experimental analysis were done by A.D., M.H.J., J.C., P.R., M.L., L.R., S.C.-G., D.M., B.C., E.L., V.T., K.S., L.S.L, N.A., A.G., P.Q., J.-F.L, O.F., M.S., C.G., and C.M. Methodology was conceived by J.K., Y.D., and S.T. Conceptualization of the project was done by S.T., De.B., J.N.P. and J.-F.M. Funding was acquired by J.F.L, L.S.L, S.T., De.B., J.N.P. and J.-F.M. Writing of the original draft was done by J.-F.M. and was revised and edited by M.-P.C, M.-J.dG, Da.B., A.K., J.N.P, F.C., De.B., and Y.D. Software was created by L.S.L. Researchers were supervised by J.-F.L., A.K., Da.B., De.B., J.N.P. and J.-F.M. S.F. and F.C. curated the data.Conflicts of interest
J.-F. M., J. N. P. and L. S. L. have financial interest in Affinité Instruments.Acknowledgements
The authors thank Maxine Joly-Chevrier and Ève-Marline Joseph-Valcin and acknowledge financial support from the Canadian Institutes of Health Research (CIHR), the Natural Sciences and Engineering Research Council of Canada (NSERC) from the Discovery grant program and the College and Community Innovation Program – Applied Research Rapid Response to COVID-19 and the Canadian Foundation for Innovation (CFI).References
- K. S. Vashist, Diagnostics, 2020, 10(4), 202 CrossRef PubMed.
- C. Chen, G. Gao, Y. Xu, L. Pu, Q. Wang, L. Wang, W. Wang, Y. Song, M. Chen, L. Wang, F. Yu, S. Yang, Y. Tang, L. Zhao, H. Wang, Y. Wang, H. Zeng and F. Zhang, Ann. Intern. Med., 2020, 172(12), 832–834 CrossRef PubMed.
- M. Day, Br. Med. J., 2020, 368, m1165 CrossRef PubMed.
- H. Nishiura, T. Kobayashi, A. Suzuki, S.-M. Jung, K. Hayashi, R. Kinoshita, Y. Yang, B. Yuan, A. R. Akhmetzhanov, N. M. Linton and T. Miyama, Int. J. Infect. Dis., 2020, 94, 154–155 CrossRef CAS PubMed.
- W. Tan, Y. Lu, J. Zhang, J. Wang, Y. Dan, Z. Tan, X. He, C. Qian, Q. Sun, Q. Hu, H. Liu, S. Ye, X. Xiang, Y. Zhou, W. Zhang, Y. Guo, X.-H. Wang, W. He, X. Wan, F. Sun, Q. Wei, C. Chen, G. Pan, J. Xia, Q. Mao, Y. Chen and G. Deng, medRxiv, 2020. DOI: DOI:10.1101/2020.03.24.20042382.
- W.-C. Cao, W. Liu, P.-H. Zhang, F. Zhang and J. H. Richardus, N. Engl. J. Med., 2007, 357(11), 1162–1163 CrossRef CAS PubMed.
- P. C. Y. Woo, S. K. P. Lau, B. H. L. Wong, K.-h. Chan, C.-m. Chu, H.-w. Tsoi, Y. Huang, J. S. M. Peiris and K.-y. Yuen, Clin. Diagn. Lab. Immunol., 2004, 11(4), 665–668 CAS.
- B. Isho, K. T. Abe, M. Zuo, A. J. Jamal, B. Rathod, J. H. Wang, Z. Li, G. Chao, O. L. Rojas, Y. M. Bang, A. Pu, N. Christie-Holmes, C. Gervais, D. Ceccarelli, P. Samavarchi-Tehrani, F. Guvenc, P. Budylowski, A. Li, A. Paterson, F. Y. Yue, L. M. Marin, L. Caldwell, J. L. Wrana, K. Colwill, F. Sicheri, S. Mubareka, S. D. Gray-Owen, S. J. Drews, W. L. Siqueira, M. Barrios-Rodiles, M. Ostrowski, J. M. Rini, Y. Durocher, A. J. McGeer, J. L. Gommerman and A.-C. Gingras, Sci. Immunol., 2020, 5(52), eabe5511 CrossRef PubMed.
- N. Yuchun, W. Guangwen, S. Xuanling, Z. Hong, Q. Yan, H. Zhongping, W. Wei, L. Gewei, Y. Xiaolei, D. Liying, R. Lili, W. Jianwei, H. Xiong, L. Taisheng, D. Hongkui and D. Mingxiao, J. Infect. Dis., 2004, 190(6), 1119–1126 CrossRef PubMed.
- S. Funk, J. K. Knapp, E. Lebo, S. E. Reef, A. J. Dabbagh, K. Kretsinger, M. Jit, W. J. Edmunds and P. M. Strebel, BMC Med., 2019, 17(1), 180 CrossRef PubMed.
- F. Amanat, D. Stadlbauer, S. Strohmeier, T. H. O. Nguyen, V. Chromikova, M. McMahon, K. Jiang, G. A. Arunkumar, D. Jurczyszak, J. Polanco, M. Bermudez-Gonzalez, G. Kleiner, T. Aydillo, L. Miorin, D. S. Fierer, L. A. Lugo, E. M. Kojic, J. Stoever, S. T. H. Liu, C. Cunningham-Rundles, P. L. Felgner, T. Moran, A. Garcia-Sastre, D. Caplivski, A. C. Cheng, K. Kedzierska, O. Vapalahti, J. M. Hepojoki, V. Simon and F. Krammer, Nat. Med., 2020, 26(7), 1033–1036 CrossRef CAS PubMed.
- L. Grzelak, S. Temmam, C. Planchais, C. Demeret, L. Tondeur, C. Huon, F. Guivel-Benhassine, I. Staropoli, M. Chazal, J. Dufloo, D. Planas, J. Buchrieser, M. M. Rajah, R. Robinot, F. Porrot, M. Albert, K. Y. Chen, B. Crescenzo-Chaigne, F. Donati, F. Anna, P. Souque, M. Gransagne, J. Bellalou, M. Nowakowski, M. Backovic, L. Bouadma, L. Le Fevre, Q. Le Hingrat, D. Descamps, A. Pourbaix, C. Laouenan, J. Ghosn, Y. Yazdanpanah, C. Besombes, N. Jolly, S. Pellerin-Fernandes, O. Cheny, M. N. Ungeheuer, G. Mellon, P. Morel, S. Rolland, F. A. Rey, S. Behillil, V. Enouf, A. Lemaitre, M. A. Creach, S. Petres, N. Escriou, P. Charneau, A. Fontanet, B. Hoen, T. Bruel, M. Eloit, H. Mouquet, O. Schwartz and S. van der Werf, Sci. Transl. Med., 2020, 12(559), eabc3103 CrossRef CAS PubMed.
- N. M. A. Okba, M. A. Müller, W. Li, C. Wang, C. H. GeurtsvanKesse, V. M. Corman, M. M. Lamers, R. S. Sikkema, E. de Bruin, F. D. Chandler, Y. Yazdanpanah, Q. Le Hingrat, D. Descamps, N. Houhou-Fidouh, C. B. E. M. Reusken, B.-J. Bosch, C. Drosten, M. Koopmans and B. L. Haagmans, medRxiv, 2020, DOI:10.1101/2020.03.18.20038059.
- J. Zhao, Q. Yuan, H. Wang, W. Liu, X. Liao, Y. Su, X. Wang, J. Yuan, T. Li, J. Li, S. Qian, C. Hong, F. Wang, Y. Liu, Z. Wang, Q. He, Z. Li, B. He, T. Zhang, Y. Fu, S. Ge, L. Liu, J. Zhang, N. Xia and Z. Zhang, Clin. Infect. Dis, 2020, 71(16), 2021–2034 Search PubMed.
- A. Kruttgen, C. G. Cornelissen, M. Dreher, M. Hornef, M. Imohl and M. Kleines, J. Clin. Virol., 2020, 128, 104394 CrossRef PubMed.
- N. Ravi, D. L. Cortade, E. Ng and S. X. Wang, Biosens. Bioelectron., 2020, 165, 112454 CrossRef CAS PubMed.
- G. Liu and J. F. Rusling, ACS Sens., 2021, 6(3), 593–612 CrossRef CAS PubMed.
- A. Ghaffari, R. Meurant and A. Ardakani, Diagnostics, 2020, 10(7), 453 CrossRef CAS PubMed.
- M. A. C. Huergo and N. T. K. Thanh, Analyst, 2021, 146(2), 382–402 RSC.
- X. Guo, Z. Guo, C. Duan, Z. Chen, G. Wang, Y. Lu, M. Li and J. Lu, MedRxiv 2020, DOI: DOI:10.1101/2020.02.12.20021386.
- J. Sun, X. Tang, R. Bai, C. Liang, L. Zeng, H. Lin, R. Yuan, P. Zhou, X. Huang, Q. Xiong, J. Peng, F. Cui, B. Ke, J. Su, Z. Liu, J. Lu, J. Tian, R. Sun and C. Ke, Clin. Microbiol. Infect., 2020, 26(12), 1690 Search PubMed.
- H. Chen, K. Liu, Z. Li and P. Wang, Clin. Chim. Acta, 2019, 493, 138–147 CrossRef CAS.
- R. Antiochia, Microchim. Acta, 2020, 187(12), 639 CrossRef CAS PubMed.
- N. Bhalla, Y. W. Pan, Z. G. Yang and A. F. Payam, ACS Nano, 2020, 14(7), 7783–7807 CrossRef CAS PubMed.
- F. Y. Cui and H. S. S. Zhou, Biosens. Bioelectron., 2020, 165, 112349 CrossRef CAS PubMed.
- N. C. Cady, N. Tokranova, A. Minor, N. Nikvand, K. Strle, W. T. Lee, W. Page, E. Guignon, A. Pilar and G. N. Gibson, Biosens. Bioelectron., 2021, 171, 112679 CrossRef CAS PubMed.
- M. Soler, M. C. Estevez, M. Cardenosa-Rubio, A. Astua and L. M. Lechuga, ACS Sens., 2020, 5(9), 2663–2678 CrossRef CAS PubMed.
- K. Wu, R. Saha, D. Q. Su, V. D. Krishna, J. M. Liu, M. C. J. Cheeran and J. P. Wang, ACS Appl. Nano Mater., 2020, 3(10), 9560–9580 CrossRef CAS.
- A. K. Kaushik, J. S. Dhau, H. Gohel, Y. K. Mishra, B. Kateb, N.-Y. Kim and D. Y. Goswami, ACS Appl. Bio Mater., 2020, 3(11), 7306–7325 CrossRef CAS.
- J. Homola, Chem. Rev., 2008, 108(2), 462–493 CrossRef CAS PubMed.
- T. J. Park, M. S. Hyun, H. J. Lee, S. Y. Lee and S. Ko, Talanta, 2009, 79(2), 295–301 CrossRef CAS PubMed.
- C. Blaszykowski, S. Sheikh and M. Thompson, Chem. Soc. Rev., 2012, 41(17), 5599–5612 RSC.
- J. F. Masson, ACS Sens., 2017, 2(1), 16–30 CrossRef CAS PubMed.
- J. F. Masson, Analyst, 2020, 145(11), 3776–3800 RSC.
- J. H. Bong, T. H. Kim, J. Jung, S. J. Lee, J. S. Sung, C. K. Lee, M. J. Kang, H. O. Kim and J. C. Pyun, BioChip J., 2020, 14(4), 358–368 CrossRef CAS.
- P. Forssen, J. Samuelsson, K. Lacki and T. Fornstedt, Anal. Chem., 2020, 92(17), 11520–11524 CrossRef CAS PubMed.
- S. N. Walker, N. Chokkalingam, E. L. Reuschel, M. Purwar, Z. Y. Xu, E. N. Gary, K. Y. Kim, M. Helble, K. Schultheis, J. Walters, S. Ramos, K. Muthumani, T. R. F. Smith, K. E. Broderick, P. Tebas, A. Patel, D. B. Weiner and D. W. Kulp, J. Clin. Microbiol., 2020, 58(11), e01533–e01520 CrossRef CAS PubMed.
- S. Ravichandran, E. M. Coyle, L. Klenow, J. J. Tang, G. Grubbs, S. F. Liu, T. Wang, H. Golding and S. Khurana, Sci. Transl. Med., 2020, 12(550), eabc3539 CrossRef CAS PubMed.
- P. Moitra, M. Alafeef, K. Dighe, M. B. Frieman and D. Pan, ACS Nano, 2020, 14(6), 7617–7627 CrossRef CAS PubMed.
- G. G. Qiu, Z. B. Gai, Y. L. Tao, J. Schmitt, G. A. Kullak-Ublick and J. Wang, ACS Nano, 2020, 14(5), 5268–5277 CrossRef CAS PubMed.
- R. Funari, K. Y. Chu and A. Q. Shen, Biosens. Bioelectron., 2020, 169, 112578 CrossRef CAS PubMed.
- A. Djaileb, B. Charron, M. H. Jodaylami, V. Thibault, J. Coutu, K. Stevenson, S. Forest, L. S. Live, D. Boudreau, J. Pelletier and J. Masson, ChemRxiv, 2020, DOI: DOI:10.26434/chemrxiv.12118914.v1.
- R. B. M. Schasfoort, J. van Weperen, M. van Amsterdam, J. Parisot, J. Hendriks, M. Koerselman, M. Karperien, A. Mentink, M. Bennink, H. Krabbe, L. W. M. M. Terstappen and A. H. L. Mulder, Biosens. Bioelectron., 2021, 113165 CrossRef CAS PubMed.
- J. Lu, D. Spasic, F. Delport, T. Van Stappen, I. Detrez, D. Daems, S. Vermeire, A. Gils and J. Lammertyn, Anal. Chem., 2017, 89(6), 3664–3671 CrossRef CAS PubMed.
- O. Farnós, A. Venereo-Sánchez, X. Xu, C. Chan, S. Dash, H. Chaabane, J. Sauvageau, F. Brahimi, U. Saragovi, D. Leclerc and A. A. Kamen, Vaccines, 2020, 8(4), 654 CrossRef PubMed.
- M. Stuible, C. Gervais, S. Lord-Dufour, S. Perret, D. L'Abbé, J. Schrag, G. St-Laurent and Y. Durocher, J. Biotechnol., 2021, 326, 21–27 CrossRef CAS PubMed.
- J. Prevost, R. Gasser, G. Beaudoin-Bussieres, J. Richard, R. Duerr, A. Laumaea, S. P. Anand, G. Goyette, M. Benlarbi, S. Ding, H. Medjahed, A. Lewin, J. Perreault, T. Tremblay, G. Gendron-Lepage, N. Gauthier, M. Carrier, D. Marcoux, A. Piche, M. Lavoie, A. Benoit, V. Loungnarath, G. Brochu, E. Haddad, H. D. Stacey, M. S. Miller, M. Desforges, P. J. Talbot, G. T. Gould Maule, M. Cote, C. Therrien, B. Serhir, R. Bazin, M. Roger and A. Finzi, Cell Rep. Med., 2020, 1(7), 100126 CrossRef PubMed.
- J. Perreault, T. Tremblay, M. J. Fournier, M. Drouin, G. Beaudoin-Bussieres, J. Prevost, A. Lewin, P. Begin, A. Finzi and R. Bazin, Blood, 2020, 136(22), 2588–2591 CrossRef CAS PubMed.
- D. Stadlbauer, F. Amanat, V. Chromikova, K. Jiang, S. Strohmeier, G. A. Arunkumar, J. Tan, D. Bhavsar, C. Capuano, E. Kirkpatrick, P. Meade, R. N. Brito, C. Teo, M. McMahon, V. Simon and F. Krammer, Curr. Protoc. Microbiol., 2020, 57(1), e100 CAS.
- S. S. Zhao, N. Bukar, J. L. Toulouse, D. Pelechacz, R. Robitaille, J. N. Pelletier and J. F. Masson, Biosens. Bioelectron., 2015, 64, 664–670 CrossRef CAS PubMed.
- O. R. Bolduc, P. Lambert-Lanteigne, D. Y. Colin, S. S. Zhao, C. Proulx, D. Boeglin, W. D. Lubell, J. N. Pelletier, J. Fethiere, H. Ong and J. F. Masson, Analyst, 2011, 136(15), 3142–3148 RSC.
- F. Cholette, C. Mesa, A. Harris, H. Ellis, K. Cachero, P. Lacap, Y. Galipeau, M.-A. Langlois, A.-C. Gingras, C. Yansouni, J. Papenburg, M. Cheng, P. Chakraborty, D. Stein, P. V. Caeseele, S. Bartlett, M. Krajden, D. Goldfarb, A. McGeer, C. Osiowy, C. Hankins, B. Mazer, M. Drebot and J. Kim, Dried blood spot specimens for SARS-CoV-2 antibody testing: A multi-site, multi-assay comparison, Research Square, 2021. DOI:10.21203/rs.3.rs-366992/v1.
- J. Perreault, T. Tremblay, M.-J. Fournier, M. Drouin, G. Beaudoin-Bussières, J. Prévost, A. Lewin, P. Bégin, A. Finzi and R. Bazin, Blood, 2020, 136(22), 2588–2591 CrossRef CAS PubMed.
- J. Renard, C. Vidal-Madjar, B. Sebille and C. Lapresle, J. Mol. Recognit., 1995, 8(1–2), 85–89 CrossRef CAS PubMed.
- A. Aube, D. M. Charbonneau, J. N. Pelletier and J. F. Masson, ACS Sens., 2016, 1(11), 1358–1365 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Detailed experimental protocols for the expression of antigens and for the optimization of the SPR protocol. Supplementary tables on the different methods used for SARS-CoV2 antibody monitoring, for the optimization of the SPR sensors and of the SPR shifts of human blood-based products. Figures for the purification of the antigens, for the optimization of the ELISA tests and SPR sensors with antibodies elicited in animal models, for the optimization of the SPR sensor with human antibodies in serum samples and for the stability of the SPR sensor stored up to a week (PDF). See DOI: 10.1039/d1an00893e |
| ‡ These authors that contributed equally to the work. |
| This journal is © The Royal Society of Chemistry 2021 |