
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDetection of acquired radioresistance in breast cancer cell lines using Raman spectroscopy and machine learning†
Kevin Saruni
Tipatet
 *ab,
Liam
Davison-Gates
a,
Thomas Johann
Tewes
b,
Emmanuel Kwasi
Fiagbedzi
c,
Alistair
Elfick
a,
Björn
Neu
b and
Andrew
Downes
*a
*ab,
Liam
Davison-Gates
a,
Thomas Johann
Tewes
b,
Emmanuel Kwasi
Fiagbedzi
c,
Alistair
Elfick
a,
Björn
Neu
b and
Andrew
Downes
*a
aInstitute for BioEngineering, University of Edinburgh, UK. E-mail: andy.downes@ed.ac.uk; Fax: +44 (0)131 650 5661; Tel: +44 (0) 131 650 5660
bFaculty of Life Sciences, Rhine Waal University of Applied Sciences, Kleve, Germany
cWellcome Centre for Cell Biology, University of Edinburgh, UK
First published on 22nd April 2021
Abstract
Radioresistance—a living cell's response to, and development of resistance to ionising radiation—can lead to radiotherapy failure and/or tumour recurrence. We used Raman spectroscopy and machine learning to characterise biochemical changes that occur in acquired radioresistance for breast cancer cells. We were able to distinguish between wild-type and acquired radioresistant cells by changes in chemical composition using Raman spectroscopy and machine learning with 100% accuracy. In studying both hormone receptor positive and negative cells, we found similar changes in chemical composition that occur with the development of acquired radioresistance; these radioresistant cells contained less lipids and proteins compared to their parental counterparts. As well as characterising acquired radioresistance in vitro, this approach has the potential to be translated into a clinical setting, to look for Raman signals of radioresistance in tumours or biopsies; that would lead to tailored clinical treatments.
Introduction
In the UK, about 15% of all newly detected cancers are breast cancers, making it the most prevalent of all cancers.1,2 Risk factors include age, genetics, lifestyle and environmental factors.2 Immense progress in molecular analysis and genetic screening has led to the classification of breast cancers into different subtypes: luminal A, luminal B, normal breast like, human epidermal growth factor receptor-2 (HER-2+), basal and claudin-low tumours, and depending on the level of hormone receptors and HER-2 expressed on tumour cells,3–5 these subtypes are also referred to as either hormone receptor positive (HR+) or hormone receptor negative (HR−) tumours.6,7 Based on incidences in Scotland from 2009–2016 (N = 31![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 099), 85% of these were HR+/HER2− and HR−/HER2−, whereas 15% were of HR+/HER2+ and HR−/HER2+ subtypes;8 A similar trend is observed in the USA, where 78% of all breast cancer incidences between 2013 and 2017 were of HR+/HER2− and HR−/HER2− subtypes.9 Disease prognosis and responsiveness to radiotherapy in breast cancer has been shown to be subtype-specific.10–13 While molecular profiling of these subtypes is well-understood and achieves reasonable prognostic results,14–17 the link between the subtypes and their response to this particular therapy isn't well understood; therefore, it is of increasing importance to understand the relationship between individual tumours’ distinct molecular profiles and their differentiated response to extended (adjuvant) radiotherapy; this would help identify patients that would benefit most from this treatment.
099), 85% of these were HR+/HER2− and HR−/HER2−, whereas 15% were of HR+/HER2+ and HR−/HER2+ subtypes;8 A similar trend is observed in the USA, where 78% of all breast cancer incidences between 2013 and 2017 were of HR+/HER2− and HR−/HER2− subtypes.9 Disease prognosis and responsiveness to radiotherapy in breast cancer has been shown to be subtype-specific.10–13 While molecular profiling of these subtypes is well-understood and achieves reasonable prognostic results,14–17 the link between the subtypes and their response to this particular therapy isn't well understood; therefore, it is of increasing importance to understand the relationship between individual tumours’ distinct molecular profiles and their differentiated response to extended (adjuvant) radiotherapy; this would help identify patients that would benefit most from this treatment.
Small amounts of tumour biomolecules are present in the bloodstream, revealing changes related to the tumour; molecular profiling of these tend to give accuracies ranging between 80–95% for most cancer types tested.18 RS has been shown to diagnose stage II–IV breast cancer in blood plasma with accuracy >99%.19 In future studies, our approach could also be applied to monitor real-time biochemical changes in tumours during (radiation) treatment by analysing the corresponding biochemical changes in liquid biopsies of patients.
In genomics or molecular diagnostics, cancer cells can be screened for the presence and level of target molecules or genes (biomarkers); this method of molecular/genomic profiling achieves accuracies of between 75–78%,10 and 87.5% for predicting tumour recurrence after radiotherapy;10,20 improving these accuracies to above 95% would be a major contribution in personalised medicine.
Ionising radiation utilises X-rays or protons with the potential to cause damage to genetic material, which can result in the inhibition of cancer cell growth and eventually cell death.21 About 83% of breast cancer patients are treated with radiotherapy as part of their treatment regimen.22 Numerous studies have shown that adjuvant radiotherapy (after breast conserving surgery) can attain survival rates that are comparable to mastectomy, with the additional advantages of relatively mild toxic effects compared to other treatment options and favourable aesthetic outcome.23–25 Despite these successes, a number of breast cancer patients develop locoregional recurrences after their radiotherapy treatment course, and although cancer recurrence after radiotherapy can be as a result of residual untreated tumour cells,26 it can also be caused by cells that have survived the radiation treatment by developing either innate resistance (e.g. cancer stem cells) and/or acquired de novo resistance.27
Studies have shown that multiple factors are involved in highly complex mechanisms that result in the development of acquired radioresistance; key aspects include modification of signalling pathways that enhance DNA damage response, increased oncogenic miRNA production, cancer stem cells, epithelial-to-mesenchymal transition (EMT), metabolic alterations, and changes in the tumour microenvironment.28–32 Examples of approaches that are applied in cancer research to detect (acquired) radioresistance include DNA microarray tests, immunohistochemistry, and cell proliferation assays, in which the profiling of specific target molecules (biomarkers) is used to determine the prognosis of the disease and/or predict the outcome of an individual sample to a particular treatment or a combination of treatments.33–35
Raman spectroscopy (RS) provides a label-free and non-destructive approach of measuring the chemical composition of materials using a laser, resulting in light losing some energy to excite vibrations in molecules. A spectrum of red-shifted light reveals the molecular ‘fingerprint’ (i.e. chemical analysis) of the specimen; resulting in peaks at characteristic vibrational frequencies specific to chemical bonds, and can be evaluated to determine the concentration of specific molecules in the specimen.36 Despite the complexity of malignant tissue, which also comprises of a condensed network of normal cells, immune cells, blood vessels and a dense matrix of connective tissue, RS can discriminate cancerous vs. healthy tissue with accuracies above 90%,37–39 and can distinguish between benign, primary, and secondary tumours ex vivo.37 RS has also been extensively used in liquid biopsies for diagnosis, and to discriminate between cancerous and non-cancerous tissue in vivo intraoperatively.40,41
Most studies that utilised RS have investigated the short-term (e.g. up to 3 days) effect of radiation in human cancer cells and tissue.42–45 One particular study examined biochemical changes induced by radiation in an array of breast cancer cells using RS and multivariate analysis, it described changes in Raman profiles that show subtype-specific response to radiation.46 A recent study utilised surface enhanced Raman spectroscopy (SERS) using gold nanoparticles to improve Raman signals and was able to clearly discriminate between radiosensitive and radioresistant murine lymphoma cells by significantly enhancing subtle chemical differences between the two sublines.47
Fewer studies have applied RS to examine long-term radiation-induced biochemical changes in cells or tissue. One study that utilised RS in acquired radioresistant oral cancer phenotypes, described changes in proteins and nucleic acids, possibly due to alterations of the cell signalling cascades induced by radiation.48 A number of studies have looked at the immediate effects of radiation to cells and tissue of different cancers,43–46,48,49 but none has so-far characterised long-term acquired radioresistance in breast cancer using RS.
Radiation is mostly prescribed in small fractions of the total prescribed dose—usually about 60 Gy final dose administered over a number of days. The development of the radioresistant cell lines was carried out by weekly exposure of the wild-type cells to clinically permissible fractioned radiation doses, starting with an initial dose and cumulatively increasing it every week to a final radiation dose of 57 Gy as described in a previous study.34
The aim of the present study is to investigate possible common changes in chemical composition that occur after the development of acquired radioresistance in both HR+ and HR− breast cancer cells. This study is aimed at detecting changes induced by radiotherapy in vitro, but the ultimate aim of the research is to predict resistance to radiation at the earliest stage (with a tissue biopsy or blood plasma), and translate this to a clinical setting to improve tailoring of therapies and reduce deaths. In this study, we used in vitro cell lines to establish whether a clear Raman signal can be measured as cells develop radioresistance. It is likely that only a subset of cancer cells within a tumour can develop acquired radioresistance after treatment, and if we can detect clear differences between wild-type and radioresistant cells, then we may be able to detect changes within tissue and predict radioresistance at the diagnosis stage in a clinical setting. It is therefore necessary, before advancing into tissue and liquid biopsies, to understand the fundamental changes in a variety of cancer cell lines from the two most prevalent breast cancer subtypes according to their hormone-status (i.e. HR+/HER2− and HR−/HER2−).
Three breast cancer cell lines were utilised for this study, two HR+/HER2−: MCF-7 (luminal A), ZR-75-1 (luminal B), and one HR−/HER2−: WT-MDA-MB-231 (claudin-low) originating from different people;50 for each one a radioresistant (RR) phenotype was created: RR-MCF-7, RR-ZR-75-1 and RR-MDA-MB-231. Noteworthy is that both the two HR+-derived RR phenotypes (i.e. RR-MCF-7 and RR-ZR-75-1) lost their hormone-receptor expression but overexpressed epidermal growth factor receptor (EGFR) upon attainment of radioresistance. In addition to the changes observed with their receptors, our RR cell lines have been shown to significantly increase their migration and invasion capabilities with the attainment of radioresistance.34 While over 90% of breast cancer patients die as a result of metastatic rather than primary disease,51 understanding the progression of treatment-resistance and metastasis, and devising time-efficient, cost-efficient and highly accurate early-detection techniques, is of great importance and would save many lives.
To the best of our knowledge, no study has yet described matching spectral changes that are attributed to acquired radioresistance, and none has been able to describe these spectral differences by classifying acquired radioresistant cell lines from their wild-type in an array of both HR+ and HR− breast cancer cell lines with a validation score of 100%.
Materials and methods
Cell lines
The human breast cancer cell lines (MCF-7, ZR-75-1, MDA-MB-231, and the RR phenotypes) were all provided by Mark Gray, Roslin institute, University of Edinburgh. The radioresistant cells in our study were generated from their parental cells by exposing them to weekly radiation, starting with an initial dose of 2 Gy and increasing this dose by 0.5 Gy every week for 12 weeks, which equates to a cumulative radiation dose of 57 Gy as previously described.34Cell culture and preparation
Unless stated otherwise, all cell culture reagents were obtained from Gibco™, ThermoFischer Scientific. Cells were grown in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% foetal calf serum (FCS), 5 ml of pen–strep (50 U ml−1 penicillin, 50 mg ml−1 streptomycin) to 70–90% confluence before being harvested. They were then washed in phosphate buffered saline (PBS) before adding 3 ml TrypLE™ Express Enzyme and incubating for 5–10 minutes. TrypLE™ was diluted with growth media in the ratio 1:3. This mixture was then centrifuged at 300g for 5 minutes. The pellet was re-suspended in 10 ml PBS and centrifuged at 300g for 5 minutes. The pellet was re-suspended in 10% formalin for 10 minutes and centrifuged at 300g for 5 minutes. The pellet was again re-suspended in 10 ml PBS and centrifuged at 300g for 5 minutes. 10 μl of the pellet was pipetted onto the surface of a gold mirror (Thorlabs, Inc)52 and allowed to dry completely. Samples were then ready for Raman spectral acquisition.
Ionising radiation
Irradiation was performed 24 hours after seeding the cells in the 96-well plates. The cells were treated with doses ranging between 0.5–10 Gy using a Faxitron RX-650 (Faxitron X-ray Corporation, IL, USA).Sulforhodamine b (SRB) assay
An SRB assay is used to determine the density of surviving cells by measuring the amount of cellular protein bound by the SRB dye;53,54 the intensity of dye correlates to the amount of cellular protein, hence the cell density in that particular well. Breast cancer cells were seeded into 96-well plates and were routinely incubated. 144 h after irradiation, cultures were fixed by addition of 50 μl cold 25% TCA (trichloracetic acid) solution per well at 4 °C for 1 hour, after which they were washed with water and dried overnight. 50 μl SRB dye solution was added to each well, thereafter the plates were placed on a rocker for 30 minutes. They were then washed with 1% glacial acetic acid and dried overnight. 150 μl Tris buffer solution was added to each well and placed on a rocker for 1 hour. The samples were analysed at 540 nm by a plate reader (BP 800 Biohit).Raman spectroscopy
We used a Renishaw InVia Raman microspectroscopy system coupled to an upright microscope stage and a laser source with an excitation wavelength of 785 nm. A Leica 20× magnification lens (NA = 0.4) was under-filled to produce a spot diameter (∅) ∼10 μm. Spectra were acquired by performing repeat measurements on 100 different regions per pellet by scanning over a spectrum range of 380–1800 cm−1, and each of the 100 spots was illuminated with an exposure time of 200 s.Data analysis
Autofluorescence baseline was removed using an asymmetric least squares baseline correction protocol and Raman spectra were normalised to the amount of biological material within each sample.55 Thereafter, cosmic ray spikes were removed by averaging each wavenumber and isolating any samples which had exceeded the standard deviation of the remaining values by at least a factor of four. A Savitzky–Golay filter was applied for smoothing. Raman spectra were then split into training and test groups at the ratio of 2:1 respectively. Principal component analysis (PCA) was performed with the spectra from the training set. Next, linear discriminant analysis (LDA) was performed on the first 30 principal components. This process was repeated on the test data set using the eigenvectors and discriminant vectors obtained from the training data set for all transformations. The training set was used to construct machine learning models for classification of the data. Mann–Whitney–Wilcoxon test two-sided with Bonferroni correction was applied to compare the test-set values of the wild-type cell lines to their parental values. The program for this analysis was written in python 3.7, using the scikit learn module for machine learning analysis.
Results and discussion
Radiation
In the current work, we investigated the response to radiation of the parental cells (WT) and radioresistant phenotypes (RR) using SRB assays. Radioresistance of the paired cell clones was assessed by comparing the effects of radiation on cell proliferation in the RR cell lines and their parental (WT) clones. The WT and RR cell lines displayed varied radiation responses to increased radiation doses as illustrated in the concentration response curves in Fig. 1. | ||
| Fig. 1 The effects of radiation on proliferation assessed through SRB assays. Breast cancer cells were treated 24 hours after seeding, and cultured for 144 hours post-radiation (A: MCF7 cells. B: ZR-75-1 cells. C: MDA-MB-231 cells). Data expressed as mean ± standard error of mean (SEM); n = 3; 6 replicates per experiment. *P ≤ 0.03, **P ≤ 0.008, ***P ≤ 0.005, ****P ≤ 0.0001 (2-way ANOVA followed by Holm–Sidak's multiple comparison test performed, comparing each RR mean value to its parental mean value). | ||
Significant resistance to radiation was observed in the RR cells compared to the WT cells. The half-maximal inhibitory concentration (IC50), the measure of how much the function of a biological component is inhibited by half, were in the range of 2.86 to 5.42 Gy for the WT cells as compared to 8.08 to >10 Gy for the RR cells.
Raman spectroscopy
Raman spectra of 100 different spots on a mapped region of each cell pellet from the 6 cell lines were acquired. The spectra were averaged as depicted in ESI Fig. 1.† The difference spectra (RR–WT), plotted in Fig. 2, highlights the biochemical differences that have occurred between the parental and the RR phenotypes. An assignment of these changes in chemical composition is presented in Table 1, according to a database of biological molecules.36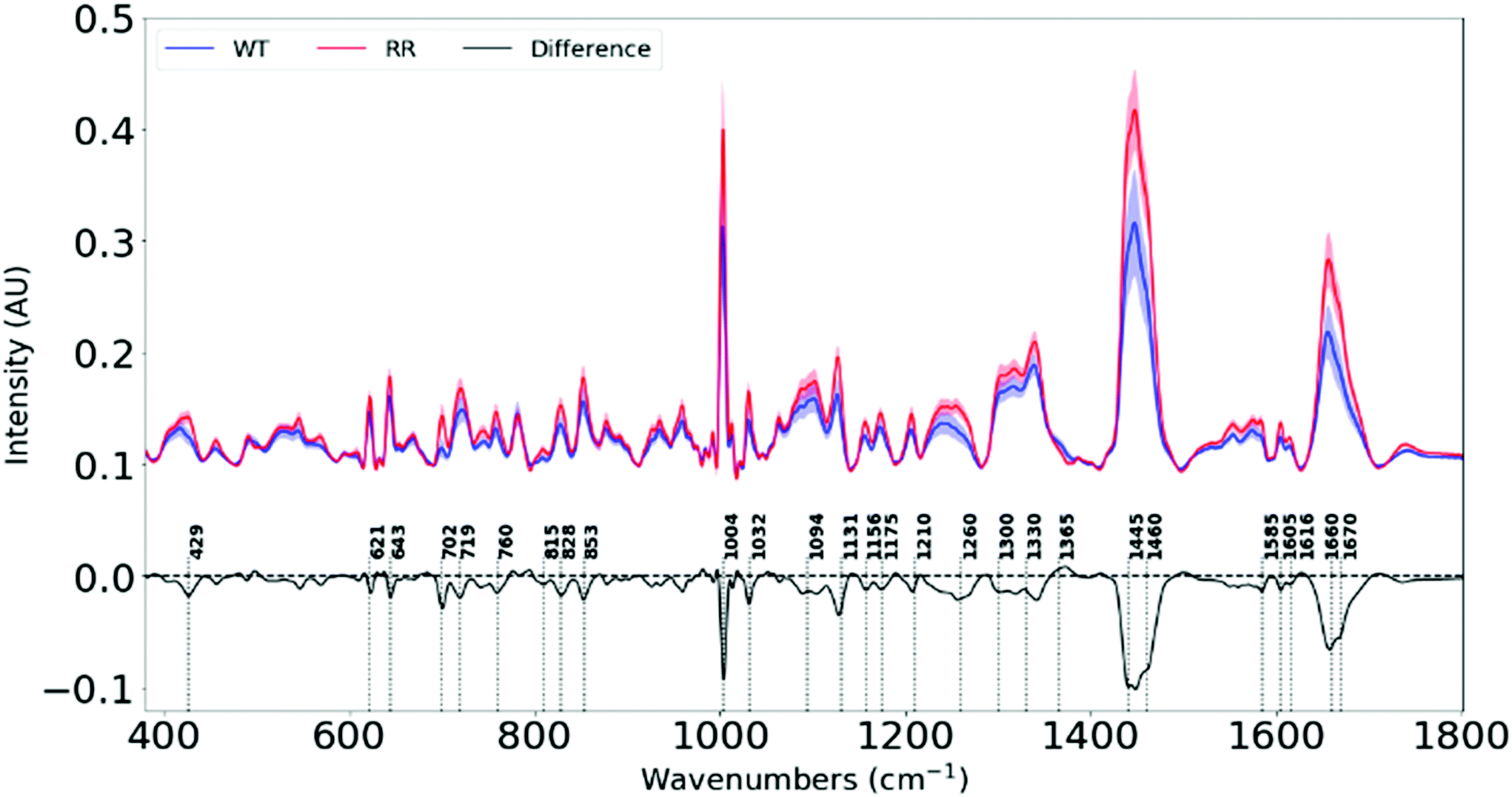 | ||
| Fig. 2 Average Raman spectra (offset for clarity) for wild-type and radioresistant cell lines. The difference spectrum is created by subtracting the average spectrum of the (WT) parental cells from the average spectrum of the radioresistant (RR) cells. | ||
| Raman shift (cm−1) | Variation from WT to RR | Biomolecules |
|---|---|---|
| p: protein, l: lipids, Phe: phenylalanine, Tyr: tyrosine, Pro: proline. | ||
| 429 | Decrease | Cholesterol |
| 621 | Decrease | Phe (p) |
| 643 | Decrease | Tyr (p) |
| 702 | Decrease | Cholesterol |
| 719 | Decrease | Phospholipids |
| 760 | Decrease | Try (p) |
| 815 | Decrease | Pro, Tyr (p) |
| 828 | Decrease | Tyr (p) |
| 853 | Decrease | Tyr (p) |
| 1004 | Decrease | Phe (p) |
| 1032 | Decrease | Phe (p) |
| 1094 | Decrease | Lipids |
| 1131 | Decrease | Lipids |
| 1156 | Decrease | C–C, C–N stretching (p) |
| 1175 | Decrease | C–H bending Tyr (p) |
| 1210 | Decrease | Tyr and Phe (p) |
| 1260 | Decrease | Amide III (p) |
| 1300 | Decrease | Lipids |
| 1445 | Decrease | Lipids |
| 1460 | Decrease | Lipids |
| 1585 | Decrease | Proteins |
| 1605 | Decrease | Phe, Tyr, C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C (p) C (p) |
| 1660 | Decrease | Amide I band (p) |
| 1670 | Decrease | CC stretch (l) |
Each measurement point in Fig. 3 is from a different region of a pellet – one pellet for each of the six cell lines. All samples were subject to the same culture conditions, so the variation between cell lines is attributed to changes in biochemical composition within cells lines from different patients. Variation between members of the same group will be partly attributed to the heterogeneity of the sample (with small sampling volumes) and partly due to the noise within individual spectra. A high degree of clustering and clear separation between wild type and radioresistant cells, repeated for all three RR cell types, mean that artefacts or variations in culture conditions do not explain the observed separations.
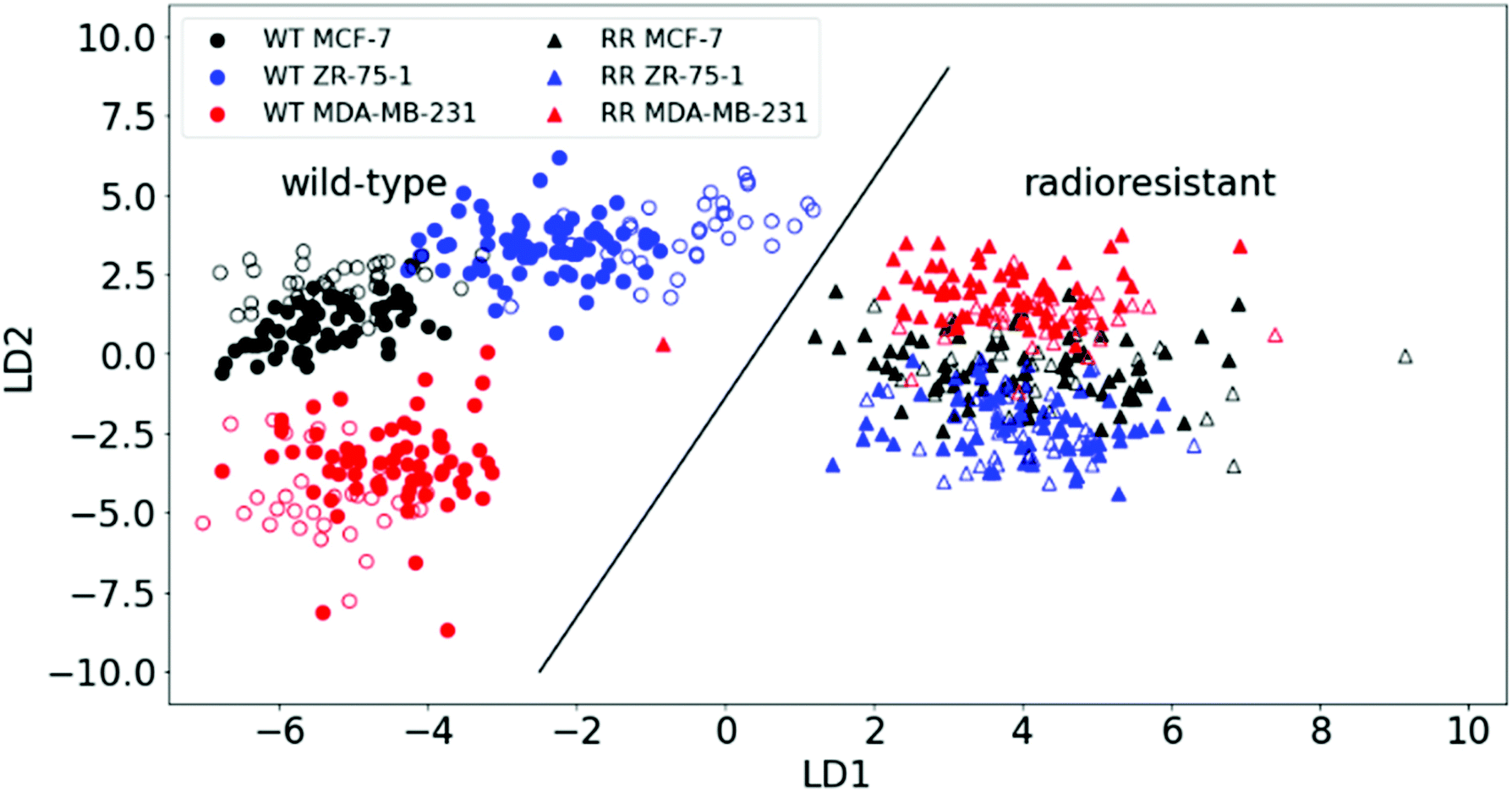 | ||
| Fig. 3 PCA–LDA scatter plot for Raman spectra of wild-type (WT) and radioresistant (RR) breast cancer cells. A classification accuracy of 100% is achieved in correctly classifying the radioresistant from the wild-type as depicted by the dividing line. The plotted points depict both 2/3rd of the training set (filled markers) and 1/3rd test set data (open markers). | ||
We see significant negative peaks at 1131 cm−1, 1300 cm−1, 1445 cm−1, 1460 cm−1 and 1670 cm−1 associated with lipids. Negative bands linked to lipids have been shown in irradiated human NSCLC xenografts49 and breast cancer cell lines.42 Lipid alteration is a well-known hallmark in breast cancer.56
Furthermore, negative peaks at 429 cm−1, 702 cm−1, 1445 cm−1 and 1670 cm−1 indicate lower cholesterol levels in our RR cells, and is shown in cancer cells that efflux cholesterol in order to enhance plasma membrane fluidity and epithelial to mesenchymal transition (EMT).57 Since our RR cells have been shown to overexpress EMT markers while downregulating epithelial markers, they also display increased metastatic traits. This signifies the possibility that they could have developed mechanisms to remove cholesterol from their plasma membranes in order to facilitate their newly-adopted aggressive and migratory characteristics associated with increased treatment resistance and metastasis.
The changes in the heights of spectral peaks associated with proteins observed in this study, correlate with changes observed in protein and functional assays with the same tumour models, which showed altered downstream signalling pathways related to cell survival, a more invasive EMT,34 and further work by this group has shown increased DNA damage repair.
Principal component analysis (PCA) of the two principal components with the highest spectral variation between the classes (as shown in ESI Fig. 3†) was used to simplify and visualise the differences in the Raman spectra of the two main classes (WT and RR). Each data point on the 2D scatter plot represents a spectrum acquired from one of 100 locations on a pellet. Just by observing their positions on the 2D plot, it is clear that the two main classes: WT and RR, are distinguishable by the formation of two separate clusters as depicted in ESI Fig. 2.†
PCA was able to show how well the RR phenotypes could be distinguished from the WT by RS, but it couldn't show differences within the cell lines (i.e. subclasses) with sufficient clarity. We therefore applied a supervised method: linear discriminant analysis (LDA) on the first 30 principal components, hence PCA–LDA. This method minimises the differences within (sub) classes while maximising their intra-class differences. Plotting the first two linear discriminant factors revealed clear separation between the two main classes (WT and RR) as well as within the WT subclasses as shown in Fig. 3.
The predictive power of the model was tested by applying an algorithm that randomly selects one-third of all spectra from all pellets as test data and plots the results together with the training set (two-thirds of data). A validation accuracy of 100% was achieved by correctly plotting the test set onto the main PCA–LDA training set plot significantly distinguishing the WT and RR cell lines (ESI Fig. 4;†p-value = 6.591 × 10−34). We also saw the formation of a distinct cluster of all three RR subclasses, indicating radiation-induced biochemical changes that are common to the RR phenotypes regardless of their original hormone status.
This finding is corroborated by gene-expression analysis and functional assays of a previous study that described a similar pattern highlighted by genetic markers and proteins that are known to regulate the transition from an epithelial to a mesenchymal phenotype of the HR+-derived cells, and is strongly linked to enhanced migratory capabilities with the development of acquired radioresistance in our RR cells.34 Individual spectra from all samples are plotted together in ESI Fig. 5.†
Conclusion
Raman spectroscopy, a label-free non-destructive technique, was used to examine changes in chemical composition associated with the development of acquired radioresistance in HR+ and HR− breast cancer cells. We show that RS together with machine learning can achieve high accuracies in the discrimination of all parental cell lines from their acquired radioresistant phenotype—cells that are associated with negative treatment outcomes (as shown in Fig. 1) and increased migration and invasion capabilities.34 Regardless of the hormone status and subtype of the parental cells, the RR phenotypes are shown to have similar difference spectra as well as the formation of a single cluster on the PCA–LDA scatter plot, suggesting that these cells undergo common biochemical changes in the process of acquiring radioresistance.A recent study34 with the same cell lines, which utilised a gene-expression assay routinely used in clinics to help predict treatment response, could not discriminate between the WT-MDA-MB-231 and its radioresistant phenotype, demonstrating the relevance of Raman spectroscopy and machine learning as a potential complementary approach to improving the detection accuracies currently achievable with the conventional assays utilised in many clinics. Compared to the gene expression profiling test, our results could discriminate between the WT and RR cell lines. Other conventional clinical assays like the antibody dependent ELISA, western blotting, immunohistochemistry might be effective in targeting specific molecules, but may also present significant shortcomings in profiling for acquired radioresistance, a process which requires sensing of multiple biomolecules simultaneously and with clinically-applicable accuracies. A major challenge with most clinical assays is however, the level of sensitivity and specificity achievable, which is a key decisive factor in advising suitable treatment options and improving survival rates of patients.
To our knowledge, this study is first of its kind to show similar changes in Raman spectra (and therefore chemical composition) in the process of acquired radiation resistance in both hormone-dependent and independent breast cancer cell lines using machine learning, and it is the first to accurately discriminate between the parental and radioresistant phenotypes (i.e., with 100% classification accuracy for 198 members of the test set). Our findings suggest that we may be able to observe similar differences between tumours not yet exposed to radiation, by acquiring spectra of patient biopsies. This would lead to personalised clinical treatments as a complementary tool with the potential to substantially improve accuracies in predicting radioresistance in individual tumours by applying it to tissue diagnosis before radiation. A Raman test on tissue biopsies could also help predict a patient's response to various other treatments as well as radiotherapy; this would personalise treatment by enabling the choice of treatments which gave positive patient outcomes in tissue with a similar chemical composition (Raman spectrum). Furthermore, this approach could also be used to investigate real-time changes in liquid biopsies (e.g. blood plasma) during radiotherapy treatment by detecting changes associated with the development of acquired radioresistance.
Conflicts of interest
No potential conflicts of interest were disclosed.Acknowledgements
We would like to thank Carol Ward, Simon Langdon, Mark Gray, Maria Bonello, and James Meehan at the division of pathology labs, IGMM, Edinburgh, for their kind assistance at the beginning stages of this study. Special thanks to Prof. Alan Murray for his mentorship and support together with the whole team of the Implantable Microsystems for Anticancer Therapy (IMPACT).All data used within this publication can be accessed at: https://doi.org/10.7488/ds/2981.
References
- J. Ferlay, M. Colombet, I. Soerjomataram, T. Dyba, G. Randi, M. Bettio, A. Gavin, O. Visser and F. Bray, Eur. J. Cancer, 2018, 103, 356–387 CrossRef CAS PubMed.
- CRUK, Together we will beat cancer, https://www.cancerresearchuk.org/about-cancer/breast-cancer/about.
- C. Sotiriou, S. Y. Neo, L. M. McShane, E. L. Korn, P. M. Long, A. Jazaeri, P. Martiat, S. B. Fox, A. L. Harris and E. T. Liu, Proc. Natl. Acad. Sci. U. S. A., 2003, 100, 10393–10398 CrossRef CAS PubMed.
- L. K. Dunnwald, M. A. Rossing and C. I. Li, Breast Cancer Res., 2007, 9, R6 CrossRef PubMed.
- D. A. Berry, C. Cirrincione, I. C. Henderson, M. L. Citron, D. R. Budman, L. J. Goldstein, S. Martino, E. A. Perez, H. B. Muss, L. Norton, C. Hudis and E. P. Winer, J. Am. Med. Assoc., 2006, 295, 1658–1667 CrossRef CAS PubMed.
- T. Sorlie, R. Tibshirani, J. Parker, T. Hastie, J. S. Marron, A. Nobel, S. Deng, H. Johnsen, R. Pesich, S. Geisler, J. Demeter, C. M. Perou, P. E. Lonning, P. O. Brown, A. L. Borresen-Dale and D. Botstein, Proc. Natl. Acad. Sci. U. S. A., 2003, 100, 8418–8423 CrossRef CAS PubMed.
- Z. Hu, C. Fan, D. S. Oh, J. S. Marron, X. He, B. F. Qaqish, C. Livasy, L. A. Carey, E. Reynolds, L. Dressler, A. Nobel, J. Parker, M. G. Ewend, L. R. Sawyer, J. Wu, Y. Liu, R. Nanda, M. Tretiakova, A. R. Orrico, D. Dreher, J. P. Palazzo, L. Perreard, E. Nelson, M. Mone, H. Hansen, M. Mullins, J. F. Quackenbush, M. J. Ellis, O. I. Olopade, P. S. Bernard and C. M. Perou, BMC Genomics, 2006, 7, 96 CrossRef PubMed.
- I. Mesa-Eguiagaray, S. H. Wild, P. S. Rosenberg, S. M. Bird, D. H. Brewster, P. S. Hall, D. A. Cameron, D. Morrison and J. D. Figueroa, Br. J. Cancer, 2020, 123, 852–859 CrossRef CAS PubMed.
- N. C. Institute, Cancer Stat Facts: Female Breast Cancer Subtypes, https://seer.cancer.gov/statfacts/html/breast-subtypes.html, (accessed 08/02/2021, 2021).
- E. K. Millar, P. H. Graham, S. A. O'Toole, C. M. McNeil, L. Browne, A. L. Morey, S. Eggleton, J. Beretov, C. Theocharous, A. Capp, E. Nasser, J. H. Kearsley, G. Delaney, G. Papadatos, C. Fox and R. L. Sutherland, J. Clin. Oncol., 2009, 27, 4701–4708 CrossRef PubMed.
- K. D. Voduc, M. C. Cheang, S. Tyldesley, K. Gelmon, T. O. Nielsen and H. Kennecke, J. Clin. Oncol., 2010, 28, 1684–1691 CrossRef PubMed.
- M. Kyndi, F. B. Sorensen, H. Knudsen, M. Overgaard, H. M. Nielsen and J. Overgaard, J. Clin. Oncol., 2008, 26, 1419–1426 CrossRef CAS PubMed.
- O. Metzger-Filho, Z. Sun, G. Viale, K. N. Price, D. Crivellari, R. D. Snyder, R. D. Gelber, M. Castiglione-Gertsch, A. S. Coates, A. Goldhirsch and F. Cardoso, J. Clin. Oncol., 2013, 31, 3083–3090 CrossRef CAS PubMed.
- M. Kittaneh, A. J. Montero and S. Glück, Biomarkers Cancer, 2013, 5, 61–70 Search PubMed.
- A. E. Cyr and J. A. Margenthaler, Surg. Oncol. Clin. North Am., 2014, 23, 451–462 CrossRef PubMed.
- K. A. Tendl and Z. Bago-Horvath, memo-Magazine of European Medical Oncology, 2020, 1–5 Search PubMed.
- S. Cleator and A. Ashworth, Br. J. Cancer, 2004, 90, 1120–1124 CrossRef CAS PubMed.
- J. D. Cohen, L. Li, Y. Wang, C. Thoburn, B. Afsari, L. Danilova, C. Douville, A. A. Javed, F. Wong, A. Mattox, R. H. Hruban, C. L. Wolfgang, M. G. Goggins, M. Dal Molin, T. L. Wang, R. Roden, A. P. Klein, J. Ptak, L. Dobbyn, J. Schaefer, N. Silliman, M. Popoli, J. T. Vogelstein, J. D. Browne, R. E. Schoen, R. E. Brand, J. Tie, P. Gibbs, H. L. Wong, A. S. Mansfield, J. Jen, S. M. Hanash, M. Falconi, P. J. Allen, S. Zhou, C. Bettegowda, L. A. Diaz Jr., C. Tomasetti, K. W. Kinzler, B. Vogelstein, A. M. Lennon and N. Papadopoulos, Science, 2018, 359, 926–930 CrossRef CAS PubMed.
- H. F. Nargis, H. Nawaz, A. Ditta, T. Mahmood, M. I. Majeed, N. Rashid, M. Muddassar, H. N. Bhatti, M. Saleem, K. Jilani, F. Bonnier and H. J. Byrne, Spectrochim. Acta, Part A, 2019, 222, 117210 CrossRef CAS PubMed.
- S. H. Cheng, C.-F. Horng, M. West, E. Huang, J. Pittman, M.-H. Tsou, H. Dressman, C.-M. Chen, S. Y. Tsai, J. J. Jian, M.-C. Liu, J. R. Nevins and A. T. Huang, J. Clin. Oncol., 2006, 24, 4594–4602 CrossRef CAS PubMed.
- S. P. Jackson and J. Bartek, Nature, 2009, 461, 1071–1078 CrossRef CAS PubMed.
- G. Delaney, S. Jacob, C. Featherstone and M. Barton, Cancer, 2005, 104, 1129–1137 CrossRef PubMed.
- A. A. Onitilo, J. M. Engel, R. V. Stankowski and S. A. Doi, Clin. Med. Res., 2015, 13, 65–73 CrossRef PubMed.
- J. Q. Cao, R. A. Olson and S. K. Tyldesley, Curr. Oncol., 2013, 20, e593–e601 CrossRef CAS PubMed.
- P. Poortmans, Radiother. Oncol., 2007, 84, 84–101 CrossRef PubMed.
- S. Tohme, R. L. Simmons and A. Tsung, Cancer Res., 2017, 77, 1548 CrossRef CAS PubMed.
- A. Ahmad, ISRN Oncol., 2013, 2013, 290568–290568 Search PubMed.
- M. Luo, L. Ding, Q. Li and H. Yao, Breast Cancer, 2017, 24, 673–682 CrossRef PubMed.
- T. Huang, L. Yin, J. Wu, J. J. Gu, J. Z. Wu, D. Chen, H. L. Yu, K. Ding, N. Zhang, M. Y. Du, L. X. Qian, Z. W. Lu and X. He, J. Exp. Clin. Cancer Res., 2016, 35, 188 CrossRef PubMed.
- M. Toulany, T. A. Schickfluss, W. Eicheler, R. Kehlbach, B. Schittek and H. P. Rodemann, Breast Cancer Res., 2011, 13, R28 CrossRef CAS PubMed.
- M. M. L. de Sousa, K. O. Bjoras, A. Hanssen-Bauer, K. Solvang-Garten and M. Otterlei, Sci. Rep., 2017, 7, 6322 CrossRef PubMed.
- J. Theys, B. Jutten, R. Habets, K. Paesmans, A. J. Groot, P. Lambin, B. G. Wouters, G. Lammering and M. Vooijs, Radiother. Oncol., 2011, 99, 392–397 CrossRef CAS PubMed.
- K. Fukuda, C. Sakakura, K. Miyagawa, Y. Kuriu, S. Kin, Y. Nakase, A. Hagiwara, S. Mitsufuji, Y. Okazaki, Y. Hayashizaki and H. Yamagishi, Br. J. Cancer, 2004, 91, 1543–1550 CrossRef CAS PubMed.
- M. Gray, A. K. Turnbull, C. Ward, J. Meehan, C. Martínez-Pérez, M. Bonello, L. Y. Pang, S. P. Langdon, I. H. Kunkler, A. Murray and D. Argyle, Radiat. Oncol., 2019, 14, 64 CrossRef PubMed.
- J. Choi, Y. N. Yoon, N. Kim, C. S. Park, H. Seol, I.-C. Park, H.-A. Kim, W. C. Noh, J.-S. Kim and M.-K. Seong, Sci. Rep., 2020, 10, 641 CrossRef CAS PubMed.
- A. C. S. Talari, Z. Movasaghi, S. Rehman and I. u. Rehman, Appl. Spectrosc. Rev., 2015, 50, 46–111 CrossRef CAS.
- A. Downes, Appl. Spectrosc. Rev., 2015, 50, 641–653 CrossRef CAS.
- J. Zhang, Y. Fan, M. He, X. Ma, Y. Song, M. Liu and J. Xu, Oncotarget, 2017, 8, 36824–36831 CrossRef PubMed.
- M. Jermyn, K. Mok, J. Mercier, J. Desroches, J. Pichette, K. Saint-Arnaud, L. Bernstein, M.-C. Guiot, K. Petrecca and F. Leblond, Sci. Transl. Med., 2015, 7, 274ra219 Search PubMed.
- L. A. Austin, S. Osseiran and C. L. Evans, Analyst, 2016, 141, 476–503 RSC.
- G. W. Auner, S. K. Koya, C. Huang, B. Broadbent, M. Trexler, Z. Auner, A. Elias, K. C. Mehne and M. A. Brusatori, Cancer Metastasis Rev., 2018, 37, 691–717 CrossRef CAS PubMed.
- Q. Matthews, A. Jirasek, J. J. Lum and A. G. Brolo, Phys. Med. Biol., 2011, 56, 6839–6855 CrossRef CAS PubMed.
- Q. Matthews, M. Isabelle, S. J. Harder, J. Smazynski, W. Beckham, A. G. Brolo, A. Jirasek and J. J. Lum, PLoS One, 2015, 10, e0135356–e0135356 CrossRef PubMed.
- M. S. Vidyasagar, K. Maheedhar, B. M. Vadhiraja, D. J. Fernendes, V. B. Kartha and C. M. Krishna, Biopolymers, 2008, 89, 530–537 CrossRef CAS PubMed.
- Q. Matthews, A. Jirasek, J. J. Lum and A. G. Brolo, Phys. Med. Biol., 2011, 56, 6839–6855 CrossRef CAS PubMed.
- P. Meksiarun, P. H. B. Aoki, S. J. Van Nest, R. G. Sobral-Filho, J. J. Lum, A. G. Brolo and A. Jirasek, Analyst, 2018, 143, 3850–3858 RSC.
- I. Aguilar-Hernández, D. L. Cárdenas-Chavez, T. López-Luke, A. García-García, M. Herrera-Domínguez, E. Pisano and N. Ornelas-Soto, Biomed. Opt. Express, 2020, 11, 388–405 CrossRef PubMed.
- M. Yasser, R. Shaikh, M. K. Chilakapati and T. Teni, PLoS One, 2014, 9, e97777–e97777 CrossRef PubMed.
- S. J. Harder, M. Isabelle, L. DeVorkin, J. Smazynski, W. Beckham, A. G. Brolo, J. J. Lum and A. Jirasek, Sci. Rep., 2016, 6, 21006 CrossRef CAS PubMed.
- D. L. Holliday and V. Speirs, Breast Cancer Res., 2011, 13, 215–215 CrossRef PubMed.
- C. DeSantis, R. Siegel, P. Bandi and A. Jemal, CA-Cancer J. Clin., 2011, 61, 408–418 CrossRef PubMed.
- Thorlabs, https://www.thorlabs.com/thorproduct.cfm?partnumber=PFSQ20-03-M01.
- V. Vichai and K. Kirtikara, Nat. Protoc., 2006, 1, 1112–1116 CrossRef CAS PubMed.
- E. A. Orellana and A. L. Kasinski, Bio-Protoc., 2016, 6, e1984 Search PubMed.
- Q. Matthews, A. Jirasek, J. Lum, X. Duan and A. G. Brolo, Appl. Spectrosc., 2010, 64, 871–887 CrossRef CAS PubMed.
- D. Hanahan and R. A. Weinberg, Cell, 2011, 144, 646–674 CrossRef CAS PubMed.
- N. Koundouros and G. Poulogiannis, Br. J. Cancer, 2020, 122, 4–22 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1an00387a |
| This journal is © The Royal Society of Chemistry 2021 |