
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceEnhanced hydrogen storage kinetics and air stability of nanoconfined NaAlH4 in graphene oxide framework†
Hyung Wan Do‡
,
HyeonJi Kim and
Eun Seon Cho *
*
Department of Chemical and Biomolecular Engineering, Korea Advanced Institute of Science and Technology (KAIST), Daejeon 34141, Republic of Korea. E-mail: escho@kaist.ac.kr
First published on 4th October 2021
Abstract
With a growing concern over climate change, hydrogen offers a wide range of opportunities for decarbonization and provides a flexibility in overall energy systems. While hydrogen energy is already plugged into industrial sectors, a physical hydrogen storage system poses a formidable challenge, giving momentum for safe and efficient solid-state hydrogen storage. Accommodating such demands, sodium alanate (NaAlH4) has been considered one of the candidate materials due to its high storage capacity. However, it requires a high temperature for hydrogen desorption and becomes inactive irreversibly upon air-exposure. To enhance sluggish reaction kinetics and reduce the hydrogen desorption temperature, NaAlH4 can be confined into a porous nanoscaffold; however, nanoconfined NaAlH4 with sufficient hydrogen storage performance and competent stability has not been demonstrated so far. In this work, we demonstrate a simultaneously enhanced hydrogen storage performance and air-stability for NaAlH4 particles confined in a nanoporous graphene oxide framework (GOF). The structure of the GOF was elaborately optimized as a nanoscaffold, and NaAlH4 was infiltrated into the pores of the GOF via incipient wetness impregnation. As a result of the nanoconfinement, both the onset temperature and activation energy for hydrogen desorption of NaAlH4 are significantly decreased without transition metal catalysts, while simultaneously achieving the stability under ambient conditions.
Introduction
Hydrogen has emerged as a clean and sustainable energy source capable of fulfilling growing global energy demand and potentially substituting traditional fossil fuels.1 Metal hydrides are suitable candidates for safe and high-density solid-state hydrogen storage, although several challenges in kinetic and thermodynamic requirements must still be overcome.2 Among metal hydrides, sodium alanate (NaAlH4) has been widely investigated owing to its high hydrogen storage capacity (7.4 wt%) and moderate decomposition temperature (∼180 °C).3 However, its slow hydrogen desorption kinetics and limited reversibility must be addressed for practical applications, along with a stability issue under ambient conditions in which NaAlH4 tends to lose its hydrogen storage capability similar to other metal hydrides. To overcome these issues, a variety of Ti- and Ce-based catalytic additives have been used to improve the hydrogen storage performance of NaAlH4.4–8 Rapid hydrogen desorption at temperatures below 100 °C and superior cycling properties have been demonstrated using high-performance transition metal–carbon composite catalysts.9,10 In addition to such transition metal-based catalysts, nanoconfinement of metal hydrides into a variety of nanoporous scaffolds has been employed. By effectively confining the metal hydrides inside nanopores of the scaffold, it is possible to achieve nanoparticles with dimensions below 10 nm.11,12 Nanoconfinement shortens the diffusion length of hydrogen atoms while increasing the number of grain boundaries and surface area, thereby improving hydrogen storage performance of metal hydrides. A wide range of host materials for NaAlH4 have been previously demonstrated, including mesoporous carbon,13–15 carbon aerogel,16 carbon nanofiber,17,18 carbon nanotube,19 mesoporous silica,20 metal–organic framework,21,22 and porous aluminium scaffold.23 Most of the prior works have focused on enhancing reaction kinetics and reversibility, and to our knowledge there have been no studies on increasing air stability of the nanoconfined NaAlH4.Here, we report a nanocomposite of NaAlH4 confined into graphene oxide framework (NaAlH4@GOF) via incipient wetness impregnation, and discuss its enhanced hydrogen desorption kinetics as well as air stability compared to those of bulk NaAlH4. A GOF is an expansion of GO layers by pillar molecules, and it has been shown as a promising material for gas storage,24 nanofiltration membrane,25 and ion-selective protective layer for redox-flow batteries26 because of a high surface area and adjustable interlayer spacing, simultaneously taking advantage of robustness and chemical versatility of GO. By virtue of such characteristics, GOF with a three-dimensional framework structure is an ideal host material for complex metal hydrides. Also, a gas-selective property of GO layers is expected to function as a protective layer for the confined metal hydrides.27
Experimental
Synthesis of graphene oxide framework (GOF)
To synthesize GOF, 200 mg of graphene oxide powder (Grapheneall Co., Ltd) was first dispersed in 20 mL of methanol via sonication at room temperature for 1 h. The dispersion was then transferred into a Teflon-lined autoclave, and 400 mg of benzene-1,4-diboronic acid (B14DBA, Alfa Aesar, 96% purity) was added to the dispersion. The solvothermal reaction was performed at 100 °C for 48 h in an autoclave reactor. After cooling down to room temperature, the mixture was centrifuged at 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 rpm for 20 min and washed with methanol three times to remove the unreacted excess B14DBA linker molecules. The resulting GOF was dried under vacuum at 120 °C for 24 h and stored inside an Ar-filled glovebox for the subsequent NaAlH4 infiltration process.
000 rpm for 20 min and washed with methanol three times to remove the unreacted excess B14DBA linker molecules. The resulting GOF was dried under vacuum at 120 °C for 24 h and stored inside an Ar-filled glovebox for the subsequent NaAlH4 infiltration process.
Synthesis of NaAlH4 impregnated GOF (NaAlH4@GOF)
The infiltration of NaAlH4 into the GOF was performed under an Ar atmosphere. NaAlH4 was purified by dissolving the as-purchased NaAlH4 (Sigma-Aldrich, 93% purity, hydrogen storage grade) in anhydrous tetrahydrofuran (THF) and removing metallic impurities using a PTFE syringe filter (CHMLAB Group) with a pore size of 0.2 μm. The 1 M solution of NaAlH4 in THF was prepared using the purified NaAlH4. The 50–100 mg of GOF was added to 10 mL of the NaAlH4 solution, and the mixture was magnetically stirred at room temperature for 15 h. To collect NaAlH4 impregnated GOF composite, the mixture was centrifuged at 10000 rpm for 20 min and washed with THF three times to remove bulk NaAlH4 that may have been recrystallized on the surface of the GOF. The resulting NaAlH4@GOF composite was vacuum-dried at room temperature overnight and stored in an Ar-filled glovebox until further analysis.
Material characterization
X-ray diffraction patterns for measuring interlayer spacing were acquired with a Rigaku SmartLab. N2 adsorption–desorption isotherms for specific surface area and pore size distribution were obtained with a Micromeritics 3Flex. Fourier-transform infrared spectroscopy (Bruker, Alpha), thermogravimetric analysis (Netzsch, TG 209 F1 Libra), and X-ray photoelectron spectroscopy (Thermo Scientific, K-Alpha) measurements were also performed to verify successful synthesis and characterize properties of the synthesized products. The NaAlH4 mass loading inside NaAlH4@GOF composite was measured via inductively coupled plasma-optical emission spectroscopy (Agilent, 720 ICP-OES).Measurement of hydrogen desorption performance
The hydrogen desorption performance of bulk NaAlH4 and NaAlH4@GOF was measured by temperature programmed desorption mass spectrometry (TPD-MS) using a MicrotracBEL BELCAT II catalyst analyser and BELMass quadrupole mass spectrometer. Calibration was performed prior to the measurements using a mixture gas of H2 and Ar with a known H2 concentration. The 50–100 mg of NaAlH4@GOF composite was loaded into a quartz sample holder and purged with Ar for 30 min. The sample was ramped from room temperature to 500 °C with a heating rate of 2.5–20.0 °C min−1 under a constant flow (30 mL min−1) of Ar carrier gas, and the final temperature was held at 500 °C for 30 min to ensure a complete desorption.Results and discussion
Synthesis and optimization of GOF
GOF was synthesized via solvothermal reaction of graphene oxide (GO) with benzene-1,4-diboronic acid (B14DBA) in methanol,24 as shown in Fig. 1a. B14DBA was chosen as the linker material based on good reactivity of boronic acids with hydroxyl functional groups of GO,28 and relatively short length of the B14DBA molecule which is composed of a single phenyl ring unit connected to the boronic acids. Alternative choice of linkers may be considered to readily modify a physical property of the resulting GOF as necessary. For example, d-spacing of the GOF can be slightly increased by using a linker with a longer length (e.g. 4,4′-biphenyldiboronic acid).29 To optimize physical properties of the framework before NaAlH4 infiltration, we synthesized a series of GOFs by varying the ratio of GO and B14DBA – referred as GOF (1:x) where x is the ratio of B14DBA relative to GO. Changes in d-spacing and Brunauer–Emmett–Teller (BET) specific surface area were measured via X-ray diffraction (XRD) and N2 adsorption–desorption isotherm experiments, respectively, to confirm successful synthesis of a three-dimensional nanoporous framework structure. Fig. 1b and S1a† show a monotonic increase in the d-spacing of GOF with increasing linker concentration. As more linker molecules are intercalated into the GO structures, the overall alignment angle between the linker and GO moves closer to being perpendicular and the diffraction peak shifts to a lower angle. The corresponding interlayer distance between the GO sheets is expanded from 0.86 nm to 1.06 nm, reaching a maximum value beyond GO to B14DBA ratio of 1:2. Furthermore, the XRD patterns with a single diffraction peak indicate a well-defined layer structure for the GOF as well as full removal of unreacted linkers. Fig. 1c and S1b† show type I (microporous) isotherms with a maximum BET specific surface area of 200.5 m2 g−1 for GO to B14DBA ratio of 1:2, comparable to previously reported values24,29 and much higher than 4.6 m2 g−1 for as-received GO. Average pore diameter is calculated to be around 2 nm (Fig. S2†), confirming a nanoporous structure. Once the maximum BET specific surface area is reached for GOF (1:2), an additional increase in the linker concentration leads to a decrease in the BET specific surface area with negligible increase in the d-spacing. Based on these results, we chose GOF (1:2) as the optimal scaffold for NaAlH4 impregnation.
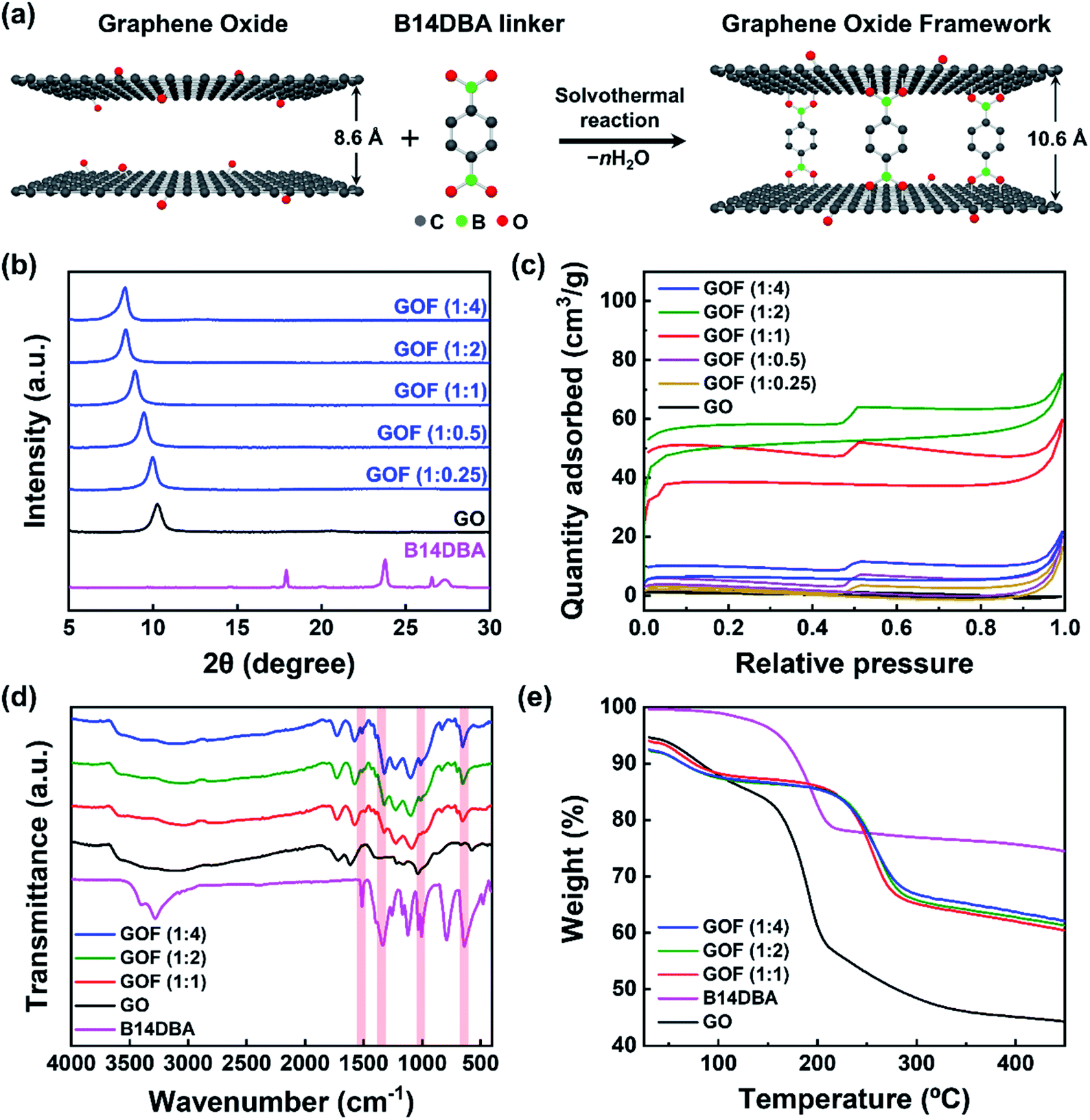 | ||
| Fig. 1 Synthesis and characterization of GOF. (a) A schematic diagram illustrating solvothermal reaction of GO and B14DBA linker to produce GOF. (b) XRD patterns of B14DBA, GO, and GOF synthesized using different ratios of GO and B14DBA. (c) N2 adsorption–desorption isotherms of GO and synthesized GOFs. (d) FT-IR spectra of B14DBA, GO, and synthesized GOFs. Characteristic bands of B14DBA are indicated in red. (e) TGA curves of GO, B14DBA, and synthesized GOFs. | ||
To further verify the formation of chemical bonding between GO and B14DBA, Fourier transform infrared spectroscopy (FT-IR) and thermogravimetric analysis (TGA) were performed. The FT-IR spectra (Fig. 1d) of GOF (1:1), GOF (1:2), and GOF (1:4) exhibit characteristic B–O deformation at ≈680 cm−1, B–C vibration at ≈1080 cm−1, B–O stretch at ≈1340 cm−1, and C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C stretch at ≈1520 cm−1,29 which are clearly observed in B14DBA but not in GO. A broad O–H stretch around 3200 cm−1 is also observed, indicating that some functional groups of GO remain in GOF after the solvothermal process. Moreover, the TGA curves (Fig. 1e) of GOF show a rapid mass loss around 220 °C due to decomposition of oxygen-containing functional groups,29 compared to a lower decomposition temperature around 150 °C for both GO and B14DBA. Combined with the increased d-spacing, enhanced BET specific surface area, and FT-IR measurements, this improved thermal stability suggests that B14DBA forms new chemical bonds with GO to evolve into a nanoporous framework, rather than simply being intercalated between the GO sheets.
C stretch at ≈1520 cm−1,29 which are clearly observed in B14DBA but not in GO. A broad O–H stretch around 3200 cm−1 is also observed, indicating that some functional groups of GO remain in GOF after the solvothermal process. Moreover, the TGA curves (Fig. 1e) of GOF show a rapid mass loss around 220 °C due to decomposition of oxygen-containing functional groups,29 compared to a lower decomposition temperature around 150 °C for both GO and B14DBA. Combined with the increased d-spacing, enhanced BET specific surface area, and FT-IR measurements, this improved thermal stability suggests that B14DBA forms new chemical bonds with GO to evolve into a nanoporous framework, rather than simply being intercalated between the GO sheets.
Preparation of NaAlH4@GOF nanocomposites
NaAlH4 was infiltrated into the nanoporous GOF via incipient wetness impregnation using solutions of purified NaAlH4 in tetrahydrofuran (THF), as illustrated in Fig. 2a. Wetness impregnation was chosen over melt infiltration as the method for preparing NaAlH4@GOF based on the thermal stability of GOF as well as the simplicity and low cost of the solution-based synthesis process. We used three different NaAlH4 concentrations (0.25 M, 1 M, and 3 M) for the impregnation process to determine the optimal process condition. The resulting NaAlH4 infiltrated GOFs were referred to as NaAlH4@GOF (x M) where x is the concentration of NaAlH4 in THF.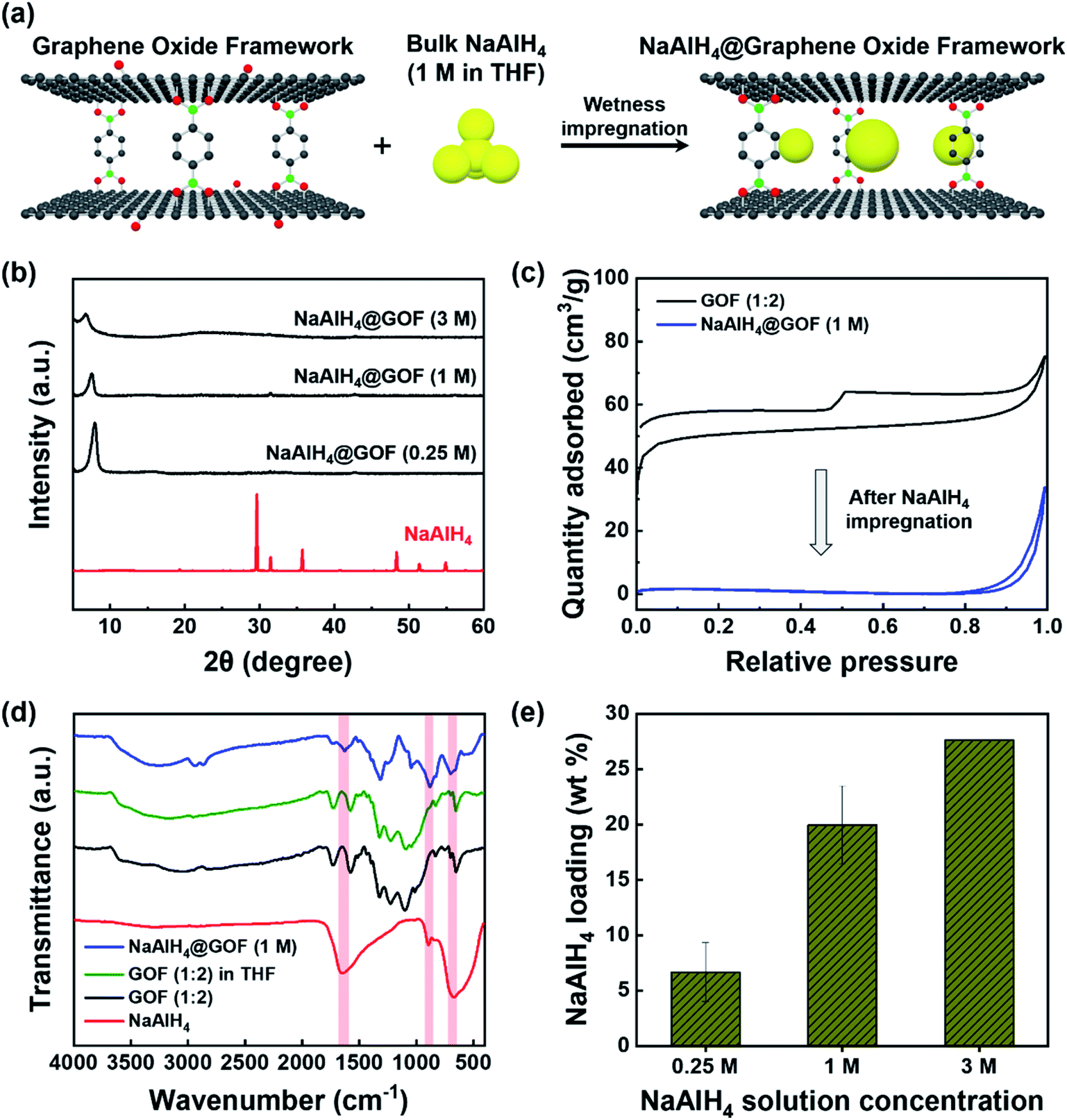 | ||
| Fig. 2 Synthesis and characterization of NaAlH4@GOF. (a) Schematic diagram illustrating incipient wetness impregnation of NaAlH4 into nanopores of GOF. (b) XRD patterns of as-received NaAlH4 and NaAlH4@GOF prepared using solutions with different NaAlH4 concentrations. (c) N2 adsorption–desorption isotherms of GOF (1:2) and NaAlH4@GOF (1 M). A significant decrease in BET specific surface area was observed upon NaAlH4 infiltration. (d) FT-IR spectra of NaAlH4, as-synthesized GOF (1:2), GOF (1:2) treated with THF for 15 h, and synthesized NaAlH4@GOF (1 M). Characteristic bands of NaAlH4 are indicated in red. (e) NaAlH4 loading inside NaAlH4@GOFs prepared using NaAlH4 solutions of various concentrations, obtained from ICP-OES analysis. | ||
As shown in the XRD patterns of bulk NaAlH4 and NaAlH4@GOFs (Fig. 2b), the sharp diffraction peaks from crystalline NaAlH4 are not observed for NaAlH4@GOFs. This implies a successful confinement of NaAlH4 inside the nanoporous scaffold resulting in nanocrystalline NaAlH4 and causing severe peak broadening, since amorphous NaAlH4 bulk phase has never been observed in NaAlH4 infiltrated nanoporous composites.22 Furthermore, a complete removal of recrystallized NaAlH4 that may have formed on the surface of the GOF is also verified from the XRD results. When a solution with a higher NaAlH4 concentration was used for wetness impregnation, the diffraction peak of NaAlH4@GOF is broadened and the d-spacing increases from 1.10 nm (0.25 M) and 1.17 nm (1 M) to 1.28 nm (3 M), suggesting that an extreme NaAlH4 loading may have a detrimental effect on the structural integrity of the framework (Fig. S3†). The surface morphology of GOF and NaAlH4@GOF from transmission electron microscopy (TEM) images do not show any significant differences upon NaAlH4 infiltration (Fig. S4†). In bright field scanning transmission electron microscopy with energy-dispersive X-ray spectroscopy (BF-STEM-EDS) measurements, the images of NaAlH4@GOF indicate the coexistence of GOF and confined NaAlH4; Na and Al mapping originated from NaAlH4 coincide with carbon and oxygen from GOF, which presents a successful confinement of NaAlH4 into GOF (Fig. S5†).
We additionally confirmed a successful infiltration of NaAlH4 inside the GOF through N2 adsorption–desorption isotherm and FT-IR measurements. Subsequent to the impregnation process, BET specific surface area of NaAlH4@GOF (1 M) is reduced to 6.6 m2 g−1, compared to 200.5 m2 g−1 for as-synthesized GOF (Fig. 2c). The FT-IR spectra of NaAlH4@GOF (1 M) shown in Fig. 2d clearly display Al–H bending modes at ≈670 cm−1 and 890 cm−1, and Al–H stretching modes at ≈1650 cm−1,30 which are not observed in GOF. A FT-IR spectrum of GOF treated with THF for 15 h without NaAlH4 was also measured as a control, which shows no change compared to before the THF treatment. Finally, NaAlH4 mass loading inside the NaAlH4@GOF composite was calculated for each NaAlH4 solution concentration (Fig. 2e) using Al content determined from inductively coupled plasma-optical emission spectroscopy (ICP-OES) measurements. The resulting NaAlH4 loadings were 6.7 wt% for NaAlH4@GOF (0.25 M), 20.0 wt% for NaAlH4@GOF (1 M), and 27.6 wt% for NaAlH4@GOF (3 M), demonstrating a comparable or higher mass loading of active materials relative to other existing porous scaffolds.12
Hydrogen desorption performance and air-stability of NaAlH4@GOF
The hydrogen desorption reaction of NaAlH4 consists of three steps, as shown in Fig. 3a. Hydrogen desorption properties of NaAlH4@GOF (1 M) were measured using temperature programmed desorption mass spectrometry (TPD-MS) and compared with those of bulk NaAlH4. The TPD-MS profiles at different heating rates and the corresponding Kissinger plots for bulk NaAlH4 and NaAlH4@GOF (1 M) are shown in Fig. 3b–e. The TPD-MS curves of bulk NaAlH4 (Fig. 3b) present an onset desorption temperature at 180 °C, close to the melting point of NaAlH4. However, NaAlH4@GOF (1 M) starts to release hydrogen at room temperature, in agreement with the previous reports that hydrogen desorption starts at ambient temperature for nanoconfined NaAlH4 with particle size below 10 nm.18,19,31 The three desorption peaks for bulk NaAlH4, which correspond to the desorption reaction of NaAlH4, Na3AlH6, and NaH, were 282 °C, 311 °C, and 401 °C, respectively, measured at a heating rate of 10.0 °C min−1. Above 400 °C, the dehydrogenation of NaAlH4@GOF occurs at a higher temperature than bulk NaAlH4. The redox reaction between oxygen in the remained THF and NaAlH4 – which likely make AlxOy nanoclusters-also releases hydrogen, and this is revealed as dehydrogenation at a higher temperature and higher activation energy of dehydrogenation in step 3 (Fig. 3a).31,32 We focused our investigation on the first two reaction steps, which are of interest for practical applications as these desorption reactions proceed at relatively lower temperatures and are known to be reversible. In the TPD-MS profile of NaAlH4@GOF (1 M) (Fig. 3d), the desorption temperatures for the first and second reaction step measured at a heating rate of 10.0 °C min−1 are significantly reduced to 108 °C and 231 °C, respectively. As some amount of confined NaAlH4 directly reacts with oxygen elements from GOF and THF, leading to the early decomposition of NaAlH4, it diminishes the hydrogen capacity in the first step compared to bulk NaAlH4.31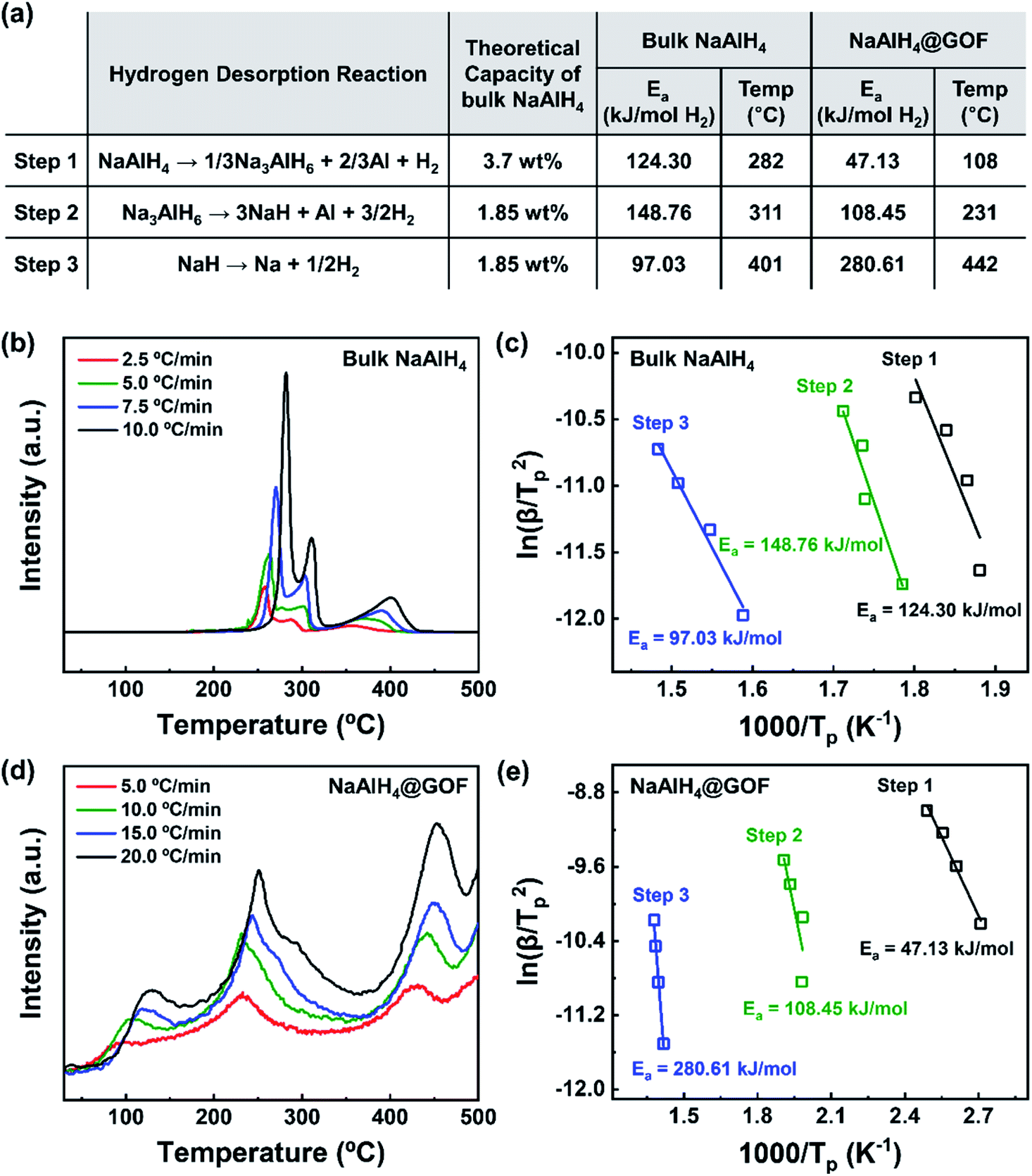 | ||
| Fig. 3 Hydrogen desorption performance of bulk NaAlH4 and NaAlH4@GOF (1 M). (a) Hydrogen desorption mechanism and corresponding theoretical capacity of bulk NaAlH4, along with activation energy and peak desorption temperature at a heating rate of 10.0 °C min−1 for bulk NaAlH4 and NaAlH4@GOF. (b) TPD-MS profiles of bulk NaAlH4 at heating rates of 2.5 °C min−1, 5.0 °C min−1, 7.5 °C min−1, and 10.0 °C min−1. (c) Kissinger plots with corresponding Ea for the three hydrogen desorption reactions of bulk NaAlH4. (d) TPD-MS profiles of NaAlH4@GOF (1 M) at heating rates of 5.0 °C min−1, 10.0 °C min−1, 15.0 °C min−1, and 20.0 °C min−1. (e) Kissinger plots with corresponding Ea for the three hydrogen desorption reactions of NaAlH4@GOF (1 M). | ||
We calculated the activation energy for the hydrogen desorption of both bulk and nanoconfined NaAlH4 using the Kissinger equation,33 and the results from each step are presented in Fig. 3a. The Kissinger plots corresponding to bulk NaAlH4 and NaAlH4@GOF (1 M) are shown in Fig. 3c and e, respectively. The activation energy of the hydrogen desorption process can be determined according to the following equation:
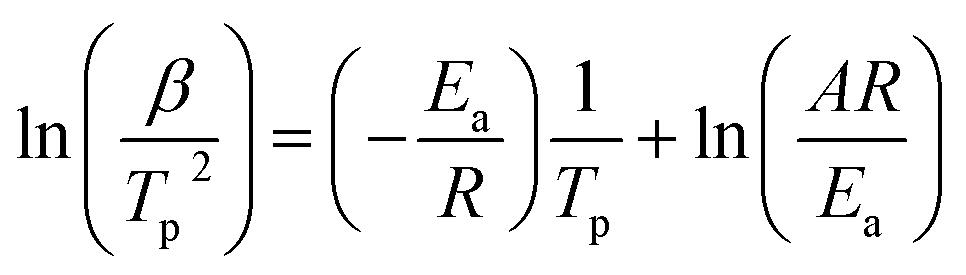
 versus
versus 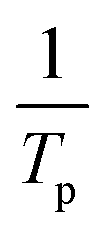 . The activation energy for the first and second desorption reaction is 124.30 kJ mol−1 and 148.76 kJ mol−1 for bulk NaAlH4, agreeing with the prior reports (Table S1†). Upon the confinement inside the nanoporous GOF, the activation energy decreases to 47.13 kJ mol−1 for the first reaction and 108.45 kJ mol−1 for the second reaction, demonstrating a remarkable improvement in the hydrogen desorption performance. The activation energy values for NaAlH4@GOF are comparable to or even smaller than the previous results in which NaAlH4 samples were doped with transition metal-based catalysts (Table S1†), presumably attributed to the well-defined nanoporous structure of GOF. It can be deduced that NaAlH4 particles are confined within 1–2 nm size of GOF pores – which is confirmed by BET measurements and d-spacing values – and this significantly boosts the nanoconfinement effect, resulting in the substantial kinetic enhancement without using transition metals as catalysts.
. The activation energy for the first and second desorption reaction is 124.30 kJ mol−1 and 148.76 kJ mol−1 for bulk NaAlH4, agreeing with the prior reports (Table S1†). Upon the confinement inside the nanoporous GOF, the activation energy decreases to 47.13 kJ mol−1 for the first reaction and 108.45 kJ mol−1 for the second reaction, demonstrating a remarkable improvement in the hydrogen desorption performance. The activation energy values for NaAlH4@GOF are comparable to or even smaller than the previous results in which NaAlH4 samples were doped with transition metal-based catalysts (Table S1†), presumably attributed to the well-defined nanoporous structure of GOF. It can be deduced that NaAlH4 particles are confined within 1–2 nm size of GOF pores – which is confirmed by BET measurements and d-spacing values – and this significantly boosts the nanoconfinement effect, resulting in the substantial kinetic enhancement without using transition metals as catalysts.
Interestingly, in addition to the decrease in the activation energies, we observed a loss in hydrogen capacity for NaAlH4@GOF. For bulk NaAlH4, the amount of hydrogen released was measured to be 6.10 wt% based on the area below the TPD-MS curve. For NaAlH4@GOF (1 M), the amount of hydrogen released was measured to be 1.01 wt% for the infiltrated NaAlH4 inside the composite, assuming 20.0 wt% NaAlH4 loading from ICP-OES results. The measured hydrogen capacity of NaAlH4@GOF corresponds to only 16.6% of that of bulk NaAlH4. We believe that this capacity loss is caused by oxygen functional groups in the GOF, which reacts with NaAlH4 and are reduced during the wetness impregnation process. To verify our hypothesis, we measured Raman spectra of GO and GOF (1:4). The Raman spectra (Fig. S6†) of GO and GOF (1:4) are nearly identical in position and intensity of the D-band peak around 1340 cm−1 and G-band peak around 1580 cm−1, suggesting that functional groups of GO remain in GOF even when a high linker concentration is used for synthesis. This observation agrees with the FT-IR spectra, where a broad O–H stretch is observed for all GOF samples (Fig. 1d). This issue may be addressed in the future by reacting the GOF with hydrogen or reducing oxygen functional groups in the GOF prior to wetness impregnation.34
On the other hand, the reduction of oxygen functional groups in the GOF during the wetness impregnation step leads to an improvement in air stability for NaAlH4@GOF. NaAlH4 is extremely reactive towards O2 and H2O, and oxidizes rapidly upon exposure to air.35 TPD-MS profiles of bulk NaAlH4 and NaAlH4@GOF (1 M) upon different durations of air exposure show a clear enhancement of air stability for the confined sample (Fig. 4a–c). The TPD-MS profile of air-exposed NaAlH4 showed the formation and disappearance of a desorption peak around 200 °C, which may be caused by destabilization from the reaction product between NaAlH4 and O2, H2O, etc. under ambient conditions.36 After 7 days of air exposure, bulk NaAlH4 retains only 1.8% of its initial hydrogen capacity whereas NaAlH4@GOF maintains 68.0% of its initial hydrogen capacity. To investigate the differences in chemical state of GOF caused by NaAlH4, we measured C 1s XPS spectra of GOF and NaAlH4@GOF (1 M) (Fig. 4d and e). The relative peak area corresponding to the CO chemical state decreases from 18.75% in GOF to 7.23% in NaAlH4@GOF. We believe that the improved air stability is caused by surface-reduced GOF acting as a barrier layer for H2O and O2, similar to selective gas permeation observed in reduced graphene oxide sheets.27,37,38
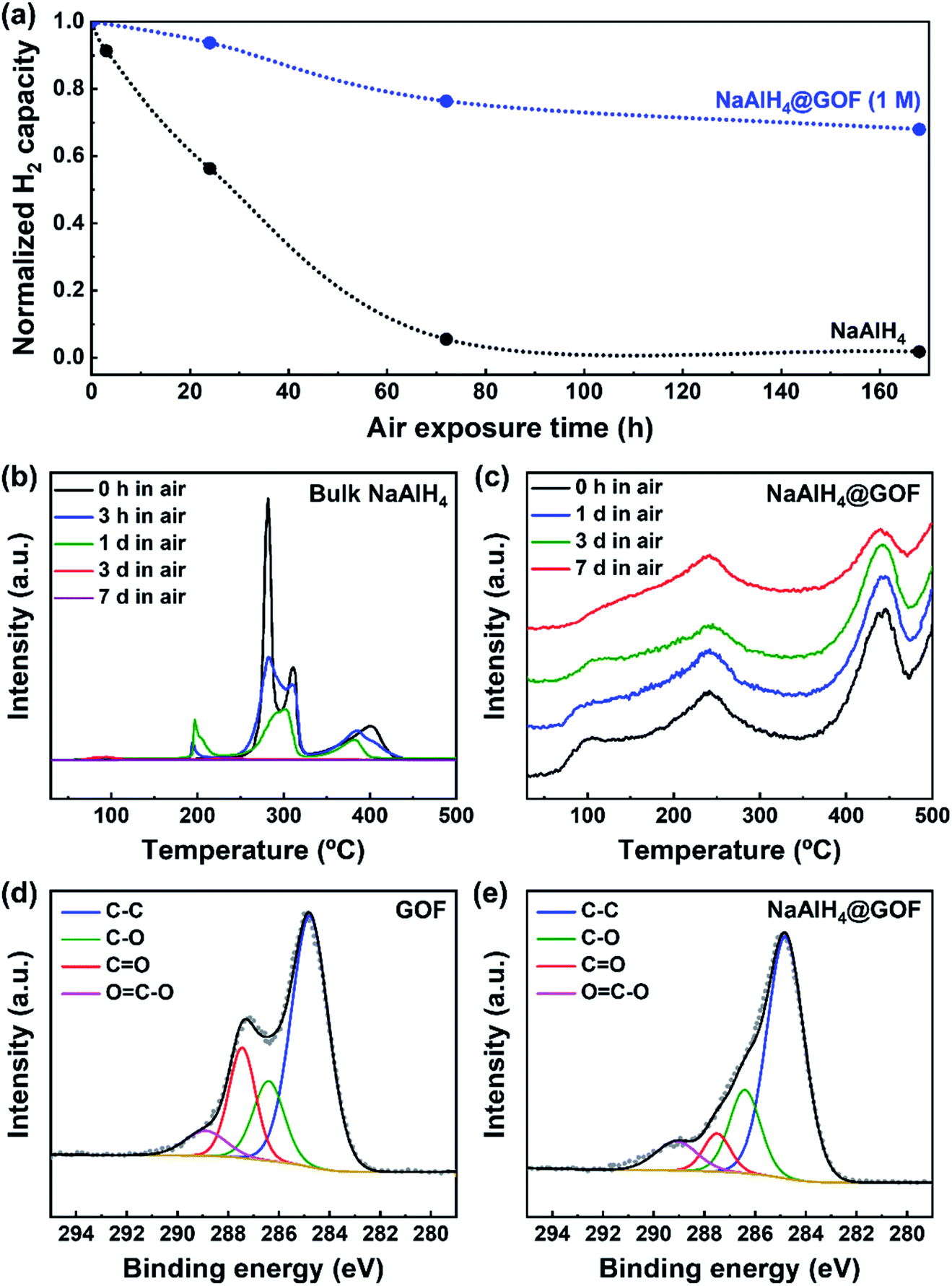 | ||
| Fig. 4 Hydrogen desorption performance of bulk NaAlH4 and NaAlH4@GOF (1 M) upon air exposure, and XPS spectra of GOF and NaAlH4@GOF (1 M). (a) Normalized hydrogen capacity of bulk NaAlH4 and NaAlH4@GOF exposed to air for 0 h, 3 h, 1 d, 3 d, and 7 d. (b) TPD-MS profiles of air-exposed bulk NaAlH4 at a heating rate of 10.0 °C min−1. (c) TPD-MS profiles of air-exposed NaAlH4@GOF (1 M) at a heating rate of 10.0 °C min−1. (d) C 1s XPS spectra of as-synthesized GOF. (e) C 1s XPS spectra of as-synthesized NaAlH4@GOF (1 M). | ||
Conclusions
In summary, this work demonstrates the viability of utilizing GOF as a nanoscaffold for confining hydrogen storage material for the first time. The framework structure with an optimized d-spacing results in a noticeable nanoconfinement effect for NaAlH4 in which both the onset hydrogen desorption temperature and the activation energy for the first two reversible hydrogen desorption reactions of NaAlH4 are significantly reduced without transition metal catalysts.However, hydrogen capacity loss is observed in the nanocomposite, possibly caused by the detrimental reaction between NaAlH4 and oxygen functional groups in the GOF. At the same time, the air stability of the confined NaAlH4 is dramatically improved as a result of the formation of surface-reduced GOF during NaAlH4 infiltration acting as a barrier layer. Although we focused our study on NaAlH4, the beneficial effect of GOF as a nanoporous host for confinement may be extended to other metal hydrides. Future work will involve optimizing the synthesis process to increase active NaAlH4 content, and studying the possible catalytic effect of boron atoms inside the GOF on the hydrogen storage behaviour of the GOF-based nanocomposites.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by Hyundai NGV and Hyundai Motor Company, and also by National R&D Program through the National Research Foundation of Korea (NRF) funded by Ministry of Science and ICT (2021M3H4A1A01079392).References
- A. Sartbaeva, V. L. Kuznetsov, S. A. Wells and P. P. Edwards, Energy Environ. Sci., 2008, 1, 79–85 RSC.
- A. Schneemann, J. L. White, S. Kang, S. Jeong, L. F. Wan, E. S. Cho, T. W. Heo, D. Prendergast, J. J. Urban, B. C. Wood, M. D. Allendorf and V. Stavila, Chem. Rev., 2018, 118, 10775–10839 CrossRef CAS PubMed.
- I. P. Jain, P. Jain and A. Jain, J. Alloys Compd., 2010, 503, 303–339 CrossRef CAS.
- B. Bogdanović and M. Schwickardi, J. Alloys Compd., 1997, 253–254, 1–9 CrossRef.
- C. P. Baldé, H. A. Stil, A. M. J. van der Eerden, K. P. de Jong and J. H. Bitter, J. Phys. Chem. C, 2007, 111, 2797–2802 CrossRef.
- B. Bogdanović, M. Felderhoff, A. Pommerin, F. Schüth and N. Spielkamp, Adv. Mater., 2006, 18, 1198–1201 CrossRef.
- Q. Gao, G. Xia and X. Yu, Nanoscale, 2017, 9, 14612–14619 RSC.
- Z. Yuan, Y. Fan, Y. Chen, X. Liu, B. Liu and S. Han, Int. J. Hydrogen Energy, 2020, 45, 21666–21675 CrossRef CAS.
- X. Zhang, Y. Liu, K. Wang, Y. Li, M. Gao and H. Pan, ChemSusChem, 2015, 8, 4180–4188 CrossRef CAS.
- Y. Liu, X. Zhang, K. Wang, Y. Yang, M. Gao and H. Pan, J. Mater. Chem. A, 2016, 4, 1087–1095 RSC.
- P. E. de Jongh and P. Adelhelm, ChemSusChem, 2010, 3, 1332–1348 CrossRef CAS.
- T. K. Nielsen, F. Besenbacher and T. R. Jensen, Nanoscale, 2011, 3, 2086–2098 RSC.
- Y. Li, G. Zhou, F. Fang, X. Yu, Q. Zhang, L. Ouyang, M. Zhu and D. Sun, Acta Mater., 2011, 59, 1829–1838 CrossRef CAS.
- C. L. Carr, W. Jayawardana, H. Zou, J. L. White, F. El Gabaly, M. S. Conradi, V. Stavila, M. D. Allendorf and E. H. Majzoub, Chem. Mater., 2018, 30, 2930–2938 CrossRef CAS.
- U. Jeong, H. Kim, S. Ramesh, N. A. Dogan, S. Wongwilawan, S. Kang, J. Park, E. S. Cho and C. T. Yavuz, Angew. Chem., Int. Ed., 2021, 60, 22478–22486 CrossRef CAS PubMed.
- T. K. Nielsen, M. Polanski, D. Zasada, P. Javadian, F. Besenbacher, J. Bystrzycki, J. Skibsted and T. R. Jensen, ACS Nano, 2011, 5, 4056–4064 CrossRef CAS PubMed.
- C. P. Baldé, B. P. C. Hereijgers, J. H. Bitter and K. P. de Jong, J. Am. Chem. Soc., 2006, 45, 3501–3503 Search PubMed.
- C. P. Baldé, B. P. C. Hereijgers, J. H. Bitter and K. P. d. Jong, J. Am. Chem. Soc., 2008, 130, 6761–6765 CrossRef PubMed.
- M. Christian and K.-F. Aguey-Zinsou, Nanoscale, 2010, 2, 2587–2590 RSC.
- S. Zheng, F. Fang, G. Zhou, G. Chen, L. Ouyang, M. Zhu and D. Sun, Chem. Mater., 2008, 20, 3954–3958 CrossRef CAS.
- R. K. Bhakta, J. L. Herberg, B. Jacobs, A. Highley, R. Behrens Jr, N. W. Ockwig, J. A. Greathouse and M. D. Allendorf, J. Am. Chem. Soc., 2009, 131, 13198–13199 CrossRef PubMed.
- V. Stavila, R. K. Bhakta, T. M. Alam, E. H. Majzoub and M. D. Allendorf, ACS Nano, 2012, 6, 9807–9817 CrossRef CAS PubMed.
- E. Ianni, M. V. Sofianos, M. R. Rowles, D. A. Sheppard, T. D. Humphries and C. E. Buckley, Int. J. Hydrogen Energy, 2018, 43, 17309–17317 CrossRef CAS.
- J. W. Burress, S. Gadipelli, J. Ford, J. M. Simmons, W. Zhou and T. Yildirim, Angew. Chem., Int. Ed., 2010, 49, 8902–8904 CrossRef CAS.
- Y. Zhang, S. Zhang and T.-S. Chung, Environ. Sci. Technol., 2015, 49, 10235–10242 CrossRef CAS PubMed.
- S. Kim, J. Choi, C. Choi, J. Heo, D. W. Kim, J. Y. Lee, Y. T. Hong, H.-T. Jung and H.-T. Kim, Nano Lett., 2018, 18, 3962–3968 CrossRef PubMed.
- E. S. Cho, A. M. Ruminski, S. Aloni, Y.-S. Liu, J. Guo and J. J. Urban, Nat. Commun., 2016, 7, 10804 CrossRef.
- K. Severin, Dalton Trans., 2009, 27, 5254–5264 RSC.
- G. Srinivas, J. W. Burress, J. Ford and T. Yildirim, J. Mater. Chem., 2011, 21, 11323–11329 RSC.
- S. Gomes, G. Renaudin, H. Hagemann, K. Yvon, M. P. Sulic and C. M. Jensen, J. Alloys Compd., 2005, 390, 305–313 CrossRef CAS.
- S. Chumphongphan, U. Filsø, M. Paskevicius, D. A. Sheppard, T. R. Jensen and C. E. Buckley, Int. J. Hydrogen Energy, 2014, 39, 11103–11109 CrossRef CAS.
- E. H. Majzoub, J. L. Herberg, R. Stumpf, S. Spangler and R. S. Maxwell, J. Alloys Compd., 2005, 394, 265–270 CrossRef CAS.
- H. E. Kissinger, Anal. Chem., 1957, 29, 1702–1706 CrossRef CAS.
- S. Pei and H.-M. Cheng, Carbon, 2012, 50, 3210–3228 CrossRef CAS.
- M. H. W. Verkuijlen, R. de Gelder, P. J. M. van Bentum and A. P. M. Kentgens, J. Phys. Chem. C, 2011, 115, 7002–7011 CrossRef CAS.
- D. E. Dedrick, R. Behrens and R. J. S. N. L. Bradshaw, Sandia Report, 2007, 4960, pp. 1–57 Search PubMed.
- E. S. Cho, A. M. Ruminski, Y.-S. Liu, P. T. Shea, S. Kang, E. W. Zaia, J. Y. Park, Y.-D. Chuang, J. M. Yuk, X. Zhou, T. W. Heo, J. Guo, B. C. Wood and J. J. Urban, Adv. Funct. Mater., 2017, 27, 1704316 CrossRef.
- D. J. Han, S. Kim and E. S. Cho, J. Mater. Chem. A, 2021, 9, 9875–9881 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1ra05111c |
| ‡ Current address: KIURI Institute, Yonsei University, Seoul 03722, Republic of Korea. |
| This journal is © The Royal Society of Chemistry 2021 |